
Abstract
The discovery in the early 1990s of the expansion of unstable simple sequence repeats as the causative mutation for a number of inherited human disorders, including Huntington’s disease (HD), opened up a new era of human genetics and provided explanations for some old problems. In particular, an inverse association between the number of repeats inherited and age at onset, and unprecedented levels of germline instability, biased toward further expansion, provided an explanation for the wide symptomatic variability and anticipation observed in HD and many of these disorders. The repeats were also revealed to be somatically unstable in a process that is expansion-biased, age-dependent and tissue-specific, features that are now increasingly recognised as contributory to the age-dependence, progressive nature and tissue specificity of the symptoms of HD, and at least some related disorders. With much of the data deriving from affected individuals, and model systems, somatic expansions have been revealed to arise in a cell division-independent manner in critical target tissues via a mechanism involving key components of the DNA mismatch repair pathway. These insights have opened new approaches to thinking about how the disease could be treated by suppressing somatic expansion and revealed novel protein targets for intervention. Exciting times lie ahead in turning these insights into novel therapies for HD and related disorders.
THE FELLOWSHIP OF THE REPEAT DISORDERS
An unexpected discovery: unstable DNA in fragile X syndrome
In May 1991, a remarkable series of papers began to appear that revealed an “unstable region of DNA” that was increased in size in individuals with fragile X syndrome (FXS) [1, 2] (see Fig. 1 for a timeline of events). Within a matter of a few weeks, this unstable region was further revealed as containing a polymorphic CGG repeat [3] that mapped precisely to the position of the larger genomic DNA restriction fragments observed in FXS patients [4]. These data were particularly exciting as they provided potential explanations for two particularly unusual features of FXS and revealed previously unimagined genetic instability in humans. Firstly, as evidenced by its name, FXS had long been defined by the association of intellectual disability with a fragile site on the X-chromosome [5]. The fragile site presents as a region near the tip of the long arm of the X-chromosome that fails to condense during metaphase in cultured cells, particularly when the cells are grown under conditions of reduced folate [6]. The presence of a CGG repeat, and associated changes in the methylation patterns in the region, suggested that chromosomal fragility in the region was in some way directly associated with the unusual sequence nature and epigenetic consequences of the CGG repeat expansion. Secondly, and of more direct relevance to Huntington’s disease (HD) and other inherited human disorders, it was also revealed that the enlarged region was highly unstable with different sizes present in individuals from the same family, consistent with intergenerational instability [1, 7]. This intergenerational instability was of particular interest, as it also offered a potential explanation for the unusual inheritance patterns observed in FXS. Even in the first description of the sex-linked nature of FXS by Martin and Bell in 1943, the authors noted there were two males, that by their position within the pedigree, were obligate carriers of the mutant X-chromosome, but did not present with FXS, but could nonetheless transmit the disease to affected grandsons through their carrier daughters [8]. The existence of these so called normal transmitting males were confirmed in multiple additional families [9, 10]. Additionally, it was shown that the penetrance of the disease increased in successive generations, with the penetrance appearing to be in some way associated with the number of female transmissions relative to a normal transmitting male [9, 10]. The existence of these unusual inheritance patterns were received with a good deal of skepticism and the phenomena became known as the Sherman paradox [11, 12]. The Sherman paradox was thus resolved, when it was demonstrated that: affected FXS males inherited a full mutation expansion of greater than approximately 210 CGG repeats from their carrier mothers; that normal transmitting males carried non-penetrant premutations of 50 to 200 CGG repeats; and that such premutations were biased toward expansion in subsequent generations when transmitted by carrier females [7]. The increasing penetrance thus being a product of the fraction of mutant X-chromosomes that expand intergenerationally from premutations to full mutations. Notably, the intergenerational mutation rate of premutation FXS alleles was near an astonishing 100%. At the time such mutation rates were unprecedented in human genetics. Up until that point the most unstable sequences that had been detected were the hypervariable minisatellite tandem repeats, that were first described in 1980 [13] and subsequently exploited as key genetic markers in DNA profiling and DNA fingerprinting [14, 15], and that had intergenerational mutation rates in the order of 1 to 5% [16]. Even these minisatellite “hypermutation” frequencies of 1 to 5%, were still orders of magnitude greater than anything else previously observed in the human germline.
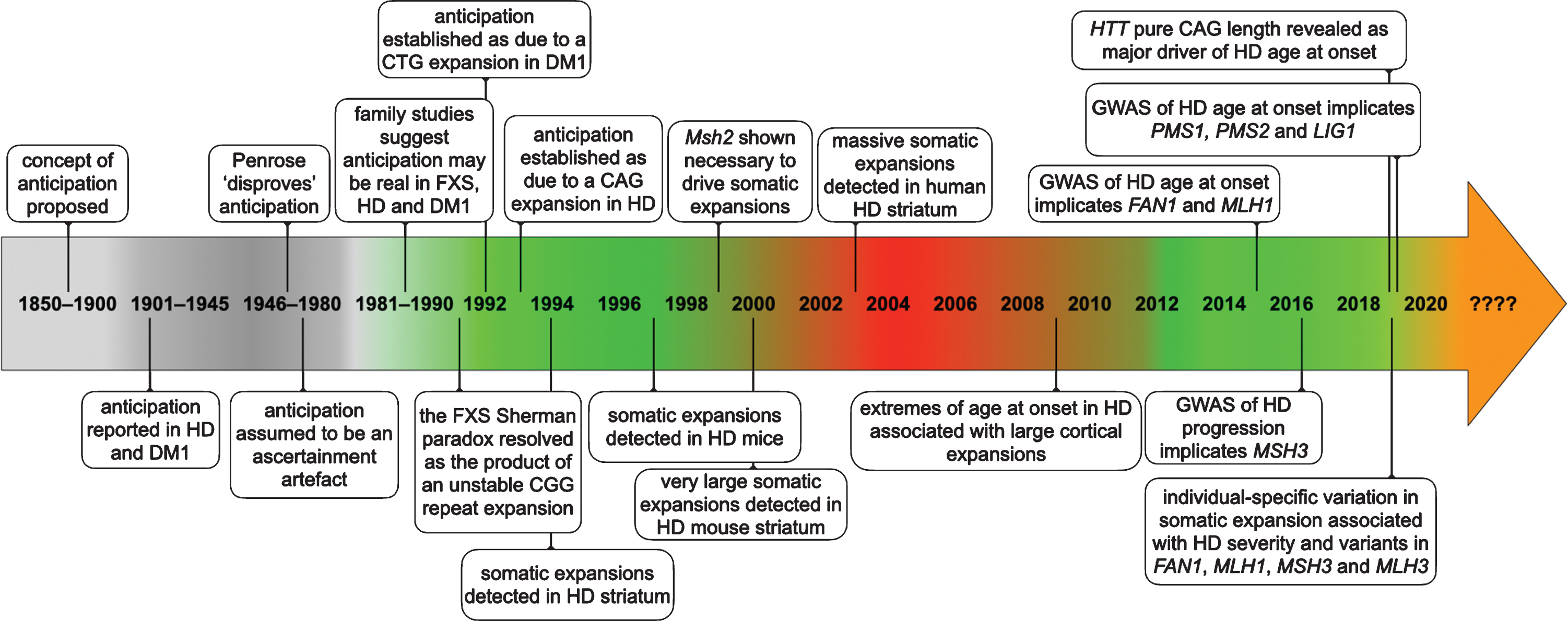
Timeline of some of the key events establishing anticipation as a genuine biological phenomenon and somatic expansion as contributing toward HD pathology.
Two is company: spinal and bulbar muscular atrophy is also caused by a repeat expansion
At about the same time these electrifying developments in FXS were reported, a short paper in Nature in July 1991 [17] described “androgen receptor (AR) gene mutations in X-linked spinal and bulbar muscular atrophy.” Specifically, the paper described the presence of an “enlargement of the CAG repeat” of 40 to 52 repeats in spinal and bulbar muscular atrophy (SBMA) patients, “roughly double” the 17 to 26 repeats observed in unaffected controls. The authors further speculated that “enlargement of the polyglutamine repeat may prevent the androgen receptor from performing an important regulatory activity in motor-neurons, thereby leading to the degeneration of these cells which is characteristic of the disease.” Of note however, the SBMA AR CAG expansion was not initially revealed as genetically unstable. Indeed, the SBMA AR CAG is only moderately unstable in the germline and this was only revealed a year or two later [18, 19]. Nonetheless, the existence of a second trinucleotide repeat expansion mediating another inherited human disorder, established repeat expansion as a novel mechanism of inherited disease in humans.
The shadow of the past: anticipation and eugenics
Whilst it is easy now to imagine unstable DNA as explaining a variety of different phenomena, such as for instance anticipation, it is important to remember that right up until the point at which the disease-causing mutations were identified, the very existence of anticipation as a genuine biological observation was not yet firmly established. The phenomena of antedating, or as it later became known anticipation, earlier age at onset observed in successive generations, was first proposed to occur in the mid-19th century [20, 21]. Unfortunately, these ideas were taken up by some, most notably, Frederick Mott, within the burgeoning eugenics movement of the early part of the 20th century who proposed anticipation to apply to “mental illness” and “insanity” in general and used this as justification for proposals to limit the reproductive rights not just of the “insane”, but also the “higher grade imbeciles” who continue to provide “fresh tainted stocks” [22]. Despite being an ardent eugenicist himself, Karl Pearson (an early pioneer of mathematical statistics and originator of the Pearson correlation coefficient) [23], and Pearson’s student, David Heron [24], noted the potential “fallacy” in the interpretation of the pedigree data used to support the existence of anticipation; namely, that disease-dependent effects on reproductive success would inevitably lead to the identification of a greater average age at onset in affected parents relative to their children. Notably, some descriptions of the inheritance of HD in the early part of the 20th century reported that this disorder also appeared to occur earlier in successive generations [25–27]. Nonetheless, it was recognised by Charles Davenport, another prominent eugenicist and advocate of the sterilisation of HD individuals, that the “law of anticipation” in HD might be “partly, if not wholly, illusory” due to the ascertainment biases as previously outlined by Pearson and Heron [28]. Around the same time anticipation was also noted in myotonic dystrophy type 1 (dystrophia myotonica, DM1) when apparently unrelated individuals with the adult onset form of the disease with a primarily neuromuscular presentation, were shown to be connected by prior generations with often cataracts as their only symptom [29]. Over the next few decades the existence of anticipation in DM1 appeared to be supported by additional family studies (e.g., [30, 31]) in some of which the authors at least partially considered the issue of ascertainment bias (e.g., [32–34]). Following the atrocities of the second world war, and the backlash against the eugenic ideals of the early 20th century, Lionel Penrose published a paper in 1947 in which he described in detail the many potential biases that could account for apparent anticipation in ascertained families [35]. Penrose considered the evidence for anticipation in a number of disorders, including HD, and concluded, that even in DM1 for which the apparent evidence was strongest, the confounding effects of several different ascertainment biases could not be excluded as having yielded the level of apparent anticipation observed. For a more detailed discussion of the history of anticipation and the eugenics movement see the works by Harper et al. [36] and Judith Friedman [37]. Penrose’s publication was widely interpreted, somewhat erroneously, as having proved that anticipation did not occur, and enhanced by the absence of a plausible genetic mechanism that could yield anticipation, the existence of anticipation as a genuine biological phenomena was widely dismissed. Indeed, as late as 1989 a paper describing a detailed analysis of the inheritance patterns in DM1 was entitled “Anticipation in myotonic dystrophy: fact or fiction?” and conservatively concluded “that anticipation may be inherent in the transmission of myotonic dystrophy” [38]. In this study, all of the potential, and very real, biases described by Penrose, were carefully taken into account and corrected for, and still evidence for striking anticipation of 20 to 30 years per generation persisted. The neurologist, Chris Höweler, who conducted most of this work for his PhD, recounts how difficult it was finding two external examiners who would approve his thesis, with one expert apparently commenting “I’m not going to read it, that’s bull****, I’m not going to spend my time reading about bull***” [39]. Even the two examiners who eventually approved the thesis commented “‘we don’t know whether you are right, but you have good arguments”. Notably, Höweler et al. considered several possible explanations for how anticipation might occur, even suggesting that “a gradual change of the mutation itself in successive generations might be assumed” [38].
A short cut to the mutation: unstable DNA as an explanation for anticipation?
Even prior to the identification of the disease causing mutations, the concept that the anticipation in DM1 might be analogous to the increasing penetrance observed in FXS was posited [38, 40]. Thus, building on the observations of extreme genetic instability observed in FXS and how this could resolve the Sherman paradox, it was quickly suggested that the anticipation observed in DM1 could possibly be explained by an unstable DNA fragment increasing in length from one generation to the next [41].
A mutation unmasked: a CTG repeat expansion in myotonic dystrophy type 1
With the discovery of the FXS mutation, the lack of a genetic mechanism to explain anticipation evaporated. Moreover, the presence of a putative expansion provided a rapid route toward identification of the causative mutation; cloned fragments from the critical region delineated by traditional linkage mapping could be used as probes on Southern blot hybridisation analyses of restriction digested genomic DNA in the absence of any knowledge of their sequence or gene content. And, within months of the reports of the identification of the FXS mutation, reports appeared in February 1992 of the presence of unstable DNA in DM1 [42–44], which was quickly confirmed as the expansion of a CTG repeat in the DMPK gene [45–47]. The size of the CTG repeat inherited was shown to be inversely correlated to age at onset, and highly unstable and biased toward expansion in the germline, providing a simple molecular explanation for anticipation [36].
The old chestnut: anticipation in HD?
As alluded to above, HD had long been posited to display anticipation [25–27, 35]. By the molecular era of human genetics in the 1980s, it was broadly accepted that the juvenile form of HD was strongly associated with paternal transmission of the mutation [48–51]. Various explanations for the excess of transmitting fathers of juvenile HD were posited, including mitochondrial effects, the existence of X-linked modifiers, or genomic imprinting of the HD locus or modifier genes [50, 52–56]. Of note, in rejecting a mitochondrial DNA effect and positing instead the action of an imprinted modifier gene, Irwin et al. noted in 1989 that, even before the HTT gene had been identified, “The identification of a putative modifying gene that might be altered to retard disease onset is appealing as a possible therapeutic stratagem” [57]. Nonetheless, at this point the evidence for a broader degree of anticipation operating more generally within HD families was largely discounted as attributable to the very real biases detailed by Penrose [48–50, 59]. However, by mid 1992 with three disorders now associated with the expansion of trinucleotide repeats, and the molecular basis for anticipation in DM1 established, there was much speculation that other disorders, especially those with unusual inheritance patterns, were likely to share a similar genetic basis. Indeed, with anticipation suddenly a fact, many of the old arguments were put aside and anticipation in HD was assumed to be real and direct predictions were made that it would be explained by the expansion of a trinucleotide repeat [36, 60].
Farewell to ignorance: HD is caused by a repeat expansion and anticipation is real
Contrary to expectations, the large expansions of hundreds of repeats that facilitated the identification of the mutations in DM1 and FXS were not found in HD. Nonetheless, within the year the predictions of unstable DNA in HD were borne out with the identification of the HD causing polyglutamine encoding CAG repeat expansion in the HTT gene in March 1993 [61]. The initial report of the identification of the CAG expansion immediately revealed that the number of CAG repeats was inversely associated with age at onset, and that the repeat was intergenerationally unstable with a bias toward expansions [61]. These insights were quickly verified in additional cohorts, and it was rapidly established that anticipation was a genuine biological observation in HD and was particularly associated with expansions in the male germline [62–66].
In from the cold: anticipation legitimised
With the discovery of unstable DNA as the basis for the conspicuous anticipation observed in DM1, the concept of anticipation was very much “legitimised” as a genuine biological phenomenon [67]. A striking consequence of this apparent liberation from the shackles of Penrose’s deconstruction of the concept of anticipation, was that over the next few years apparent anticipation was reported in numerous conditions. Whilst some of these did indeed later pan out to be associated with unstable DNA (e.g., [68–70]) (see below), many, including claims for anticipation in, for example, rheumatoid arthritis [71], uni- and bi-polar affective disorder [72, 73], schizophrenia [74], rolandic epilepsy and speech dyspraxia [75], familial primary pulmonary hypertension [76], and Meniere disease [77], have not (or have not yet) been unequivocally associated with expanded simple sequence repeats. It thus seems likely that many of these reports of apparent anticipation, were indeed artefacts of the very real ascertainment biases first delineated by Pearson [23] and later expounded upon by Penrose [35]. Indeed, even after the discovery of unstable DNA, a lively debate ensued about how to fully correct for these ascertainment biases, in particular with regard to reports of anticipation in bi-polar affective disorder and schizophrenia (e.g., [78–85]). Of course, it also remains possible that there are other causes of genuine anticipation, such as for instance might be mediated by time dependent changes in environmental exposures, as has been proposed to account for the apparent anticipation observed in familial amyloidotic polyneuropathy type I for which the mutation is known to be a very static missense variant in the transthyretin gene [86]. Whilst there are many lessons to be learned from the convoluted history of genetic anticipation, some that may be of particular relevance to unravelling the role of somatic expansion in HD are that: i) we should follow where the data leads with an open mind and consider alternative explanations, even when they challenge our preconceptions or nominally established facts and mechanisms; and, most directly, ii) unstable DNA can explain previously unexplained phenomena.
Inside information: polyglutamine encoding CAG repeat expansions cause an array of inherited neurological disorders
In contrast to the large non-coding expansions of hundreds of repeats observed in DM1 and FXS, HD was found to be associated with a more moderate polyglutamine encoding expansion mostly in the range of 40 to 50 CAG repeats, similar to that observed in SBMA. Following the series of remarkable breakthroughs in FXS, DM1, SBMA and HD, and using the insights gained to accelerate the search for the causative mutations, over the next few years additional similarly moderately sized genetically unstable polyglutamine encoding CAG repeat expansions were detected in spinocerebellar ataxia type 1 (SCA1) (1993) [87], Machado Joseph disease/SCA3 (1994) [88, 89], dentatorubral pallidoluysian atrophy (1994) [90, 91], SCA2 (1996) [92, 93], SCA7 (1997) [94] and SCA17 (2001) [95]. In addition to a likely shared pathogenic mechanism mediated by a gain of function of the expanded polyglutamine containing protein [96], in each disorder there is an inverse relationship between inherited repeat length and age at onset, and in each case the repeats are intergenerationally unstable, particularly during male transmission. These factors combined with an intergenerational expansion bias, leads to anticipation in each disorder, very similar to that observed in HD. The SCA8 mutation was identified in 1999 as the expansion of a CTG•CAG repeat [97]. Interestingly, the SCA8 repeat is expressed as a polyglutamine encoding CAG tract on one strand, and as a CUG repeat as part of a non-coding transcript on the other strand, suggesting pathology may involve gain-of-function at both the protein and RNA levels [98]. SCA8 is also unusual in that disease-causing expansions are typically larger than observed in the other polyglutamine encoding CAG repeat disorders (∼ 80 to 250 repeats), yet expanded alleles show variable penetrance and are observed at relatively high frequency in the general population and/or are associated with atypical phenotypes (e.g., [97, 99–103]). Additionally, although expanded alleles are prone to expansion in the female germline, they are heavily biased toward contraction in the male germline [97, 104] and consequently anticipation is not a prominent feature of SCA8 families.
Many repeats: expanded simple sequence repeats cause a variety of disorders
Over the ensuing decade additional disorders were associated with genetically unstable expanded trinucleotide repeats, including: a CCG expansion in fragile X E (FRAXE) (1993) [105]; a GAA expansion in Friedreich ataxia (FA) (1996) [106]; a non-coding CAG expansion in SCA12 (1999) [107]; and a CTG repeat expansion in an alternatively spliced exon of the JPH3 gene in Huntington disease like 2 (HDL2) (2001)[108, 109]. The expansion of unstable simple sequence repeats in disease was also shown to extend beyond triplets including: a CCCCGCCCCGCG dodecamer repeat in progressive myoclonus epilepsy (1997) [110]; an ATTCT pentanucleotide expansion in SCA10 (2000) [111]; and, a CCTG tetranucleotide expansion in myotonic dystrophy type 2 (DM2) (2001) [112]. More latterly, at least partially facilitated by whole genome sequencing, additional disease-associated expansions have been discovered, including: a TGGAA pentanucleotide in SCA31 (2009) [113]; a GGGGCC hexanucleotide in frontotemporal dementia and amyotrophic lateral sclerosis (2011) [114, 115]; a GGCCTG hexanucleotide in SCA36 (2011) [116]; a CTG in Fuchs corneal dystrophy (2012) [117]; a CCCTCT hexanucleotide in X-linked dystonia-parkinsonism (2017) [118]; an ATTTC pentanucleotide in SCA37 (2017) [119]; a GGC expansion in Baratela-Scott syndrome GGC (2019) [120]; a GGC expansion in neuronal intranuclear inclusion disease-related disorders (2019) [121]; an AAGGG pentanucleotide in cerebellar ataxia, neuropathy, vestibular areflexia syndrome (2019) [122]; and, TTTCA/TTTTA pentanucleotide expansions in at least six different genes in benign adult familial myoclonic epilepsy (2018–2020) [123–127]. All of these disorders are associated with intergenerational instability of the repeat expansions, and many display associated atypical inheritance patterns such as anticipation.
At the sign of the fuzzy band: somatic mosaicism in fragile X syndrome and myotonic dystrophy type 1
In addition to intergenerational instability, it was noted in the primary FXS and DM1 studies, before the repeat expansion had even been characterised, that in many affected individuals, the enlarged fragment in blood DNA presented not as a discrete band on the Southern blot hybridisations of restriction digested DNA used to reveal the presence of the mutation, but rather as broad “fuzzy”, “smeary” or “blurred” bands [1, 42–44]. These observations suggested that the intergenerationally unstable region of DNA was also somatically unstable and varied in length between cells within the individual. Indeed, in FXS, many individuals present with two or more relatively discrete pre- or full-mutation alleles (in addition to a small non-disease associated allele in females), that are present in multiple tissues, consistent with very early embryonic mutation events [7]. Indeed, it would appear that the FXS full mutation is somatically unstable during very early development, but is then somatically stabilised, likely as a result of methylation of the region [128–132]. In DM1, however, it was quickly established that there were differences in repeat size not just within tissues, but even more dramatically between tissues [133–136]. Highly consistent with a possible role for somatic expansion in contributing toward the tissue specificity of the symptoms, it was rapidly established (1993–1994) that the repeat length observed in skeletal muscle was often thousands of repeats longer than that observed in blood DNA [133–136]. Moreover, in further contrast to FXS, whilst there is evidence for embryonic instability in DM1, this is usually only observed in congenital cases inheriting very large expansions, and in most individuals, the repeat appears to be relatively stable during embryogenesis with most somatic expansions arising postnatally [134, 137–145]. Indeed, there is clear evidence that somatic expansions in blood DNA continue to accrue throughout the lifetime of the individual [141, 146–149]. Given that larger alleles are associated with earlier onset and more severe disease in DM1, it seems logical to assume that somatic expansion contributes towards the disease process. Indeed, it is now apparent that the individual-specific rate of somatic expansion is associated with both disease severity and progressive DM1 phenotypes (i.e. individuals in whom the repeat expands more rapidly somatically, have an earlier onset and more rapid disease course) [148, 151].
A pattern mirrored: somatic mosaicism in HD?
Possible evidence for somatic instability was likewise reported in the primary study defining the CAG expansion in HD, with the authors noting a “diffuse fuzzy PCR product” in the blood DNA of at least one HD patient [61]. However, early follow-up studies using polyacrylamide gel electrophoresis analysis of radiolabelled PCR products revealed that in contrast to clear differences between blood and sperm DNA, the length of the primary PCR product derived from the mutant HD chromosome did not change in lymphoblastoid cell lines and primary blood DNA samples collected many years apart, between a limited number of peripheral tissues, or between different regions of the brain, leading to premature claims of “mitotic stability” and “gametic, but not somatic instability” [65, 152]. Although repeat length variation around the primary allele was noted, these variants were conservatively interpreted as most likely representing the PCR “chatter” that is well known to generate repeat length heterogeneity even for small non-disease associated alleles [65, 152]. Whilst retrospectively it is possible to point to a greater spread of larger fragments above the primary disease associated allele in the striatal samples in particular, these data highlighted that the very large shifts in modal repeat length observed in the somatic tissues of FXS and DM1 patients were not replicated in the majority of HD patients inheriting moderately sized expansions (40 to 50 CAG repeats) [65, 152]. The following year however, a more detailed analysis of possible somatic variation was undertaken by Telenius et al. and came to a very different conclusion providing compelling evidence for “somatic mosaicism in HD which is seen predominantly in the basal ganglia and other regions of the brain selectively involved in HD” [153]. Using a wide array of peripheral tissues and different brain regions from five adult onset HD patients, the authors convincingly demonstrated that whilst a tail of PCR slippage products smaller than the primary PCR product was detected for both disease and non-disease associated alleles, a tail of larger fragments, up to approximately +5 CAG repeats, was only prominently observed in specific brain regions for the disease associated allele [153]. Notably, these putative somatic expansions were observed at a much lower level in the cerebellum and peripheral tissues and were likewise not observed using a cloned PCR template. Even more convincingly, the authors demonstrated that in two juvenile HD cases the modal length was 13 CAG repeats bigger in other brain regions relative to the cerebellum (78 versus 65 CAGs repeats respectively in one case with onset at 6 years, and 86 versus 78 CAGs repeats respectively in a second case with onset at 4 years). Unfortunately, blood DNA was not available to help identify the inherited progenitor allele length and thus determine if the HTT CAG repeat was particularly prone to expansions in most brain regions and/or was simply stable or actively prone to contraction in the cerebellum. Nonetheless, these data confirmed that somatic expansions were observed in the affected brain regions and were observed at a much lower level in the cerebellum, a brain region that is relatively spared from degeneration in adult-onset HD, at least in early in the disease course. This study thus provided the first direct evidence that somatic expansion represented a very plausible explanation for at least some of the regional specificity of downstream neuropathology in HD. However, whilst the authors noted that these observations were consistent with somatic expansion contributing toward regional selectivity of neuronal loss, they themselves noted that they were not able to ascertain in which cell types (e.g., neurons versus glia) that the expansions occurred and noted that it remained possible that somatic expansions occurred predominantly in glia as a secondary by-product of neuronal death and active gliosis [153]. Such an interpretation was likely at least partly driven by the assumption at the time that expansion events occurred via DNA replication slippage [154–156] (see below), and thus could arise in mitotically dividing cells, but not in post-mitotic neurons. A further independent study also concluded that there was indeed evidence for a greater preponderance of somatic expansions in various brain regions relative to those observed in blood and cerebellum, but the authors noted these were not restricted to the primary affected brain regions, and concluded also that the differences were “too small to make this mechanism an obvious candidate for the cause of differential neuronal degeneration in HD” [157]. Another important observation from around this time was the demonstration, that not only was the mutant HTT protein expressed in HD brains, but that it appeared more diffuse in its size relative to the non-disease associated allele, with a greater spread in the cortex relative to the cerebellum [158]. These differences were most apparent in juvenile cases where they appeared to directly reflect the spread of somatic expansions detectable at the DNA level. The authors again noted that although largely absent in the cerebellum, HTT proteins with apparently somatically expand glutamine repeats were detected in other regions of the brain that are less affected in HD, and that their cellular origin (i.e., glia versus neuron) remained unknown [158]. A role for somatic expansion was further supported by the observation that, at least in one patient there was a correlation between the degree of regional somatic mosaicism in the brain and regional pathological severity as assessed by a qualitative assessment of neuronal loss [159].
THE TWO TOWERS
The taming of Mus domesticus: mouse models
In the years after the identification of the disease-causing mutations, much effort in the triplet repeat field, as it had become known, was expended generating cell and organismal models that could be used to further understand the pathologic processes. Key to these developments were the generation of various mouse models. The first mouse model of a repeat expansion disorder, an SBMA transgenic model incorporating 45 CAG repeats in an androgen receptor cDNA construct failed to replicate either disease pathology or genetic instability [160]. Excitingly however, a SCA1 transgenic incorporating a much larger 82 CAG repeat allele in an ataxin 1 cDNA transgene with high levels of expression specifically in cerebellar Purkinje cells did display a neurodegenerative ataxic phenotype [161]. However, the repeat was not detectably unstable. The first HD repeat model, an HTT cDNA transgenic with 44 CAG repeats unfortunately contained a frameshift mutation that likely contributed to the lack of a phenotype in these animals [162]. Notably though, these mice did not display any detectable genetic instability either.
The parting of the ways: downstream pathology versus the mechanism of expansion
At about this point in time, in the mid 1990s, after the identification of the disease-causing mutations, the triplet repeat expansion field (at this time none of the non-triplet repeat expansions had yet been identified) split broadly into two camps: those assailing the pathogenic pathways downstream of the repeat expansion; and those that concentrated more on surmounting the mechanisms of repeat instability per se. In a bizarre twist of fate however, the first transgenic model to display a robust HD-like phenotype was actually generated by Mangiarini et al. to model genetic instability [163]. The R6 lines were based on a human HTT exon 1 transgene that expressed a truncated HTT protein containing an expanded polyglutamine tract encoded by ∼130 CAG repeats, and this proved sufficient to generate a progressive neurological phenotype. The R6/2 mice in particular have become established as one of the most widely used HD animal models to investigate a wide range of aspects of the polyglutamine pathology. Fortunately though, the original hypothesis was also proved correct and high levels of both intergenerational and somatic instability were also revealed, in particular in the R6/1 and R6/2 lines [164]. A notable feature of the R6 lines, like two series of DM1 mouse transgenics that were generated in parallel, and that also displayed genetic instability [165, 166], was the low copy number of the integrants that facilitated mutation detection relative to the large multicopy inserts that characterised previous transgenic repeat models [160–162]. Notably, the R6 transgenics revealed a pattern of age-dependent, tissue-specific somatic expansion that was most prominent in the striatum and virtually absent in the cerebellum. However, consistent with the cell division-dependent replication slippage models that predominated in mechanistic thinking at the time, the authors suggested that the somatic expansions in brain were derived from mitotically dividing glial cells [164]. These tissue-specific patterns of somatic expansion were also later replicated in knock-in mouse models in which the expanded CAG was targeted into the endogenous mouse Htt gene [167, 168] (see also Wheeler and Dion, this issue [169] for additional mouse models).
The small pool: very large striatal-specific somatic expansions in HD mice
With the exception of a very limited single molecule analysis of blood DNA used essentially as controls for single sperm analyses of male germline dynamics [170], up until the year 2000, all the previous analyses of somatic mosaicism in HD patients or animal models had been conducted using bulk DNA PCR analyses (i.e., PCR using large amounts of input DNA, typically >10 ng, equivalent to > >1,000 cellular equivalents). Although these studies revealed clear evidence for somatic instability, the size of the acquired somatic expansions beyond the length of the inherited progenitor allele detected were relatively small. In human brain samples, even in juvenile cases, the largest acquired somatic expansions detected were in the order of +13 repeats, and much less than this in patients with more typical HD germline expansions in the range of 40 to 50 CAG repeats [152, 159]. Somatic expansions in HD mice with much larger germline alleles of 100 + repeats were shown to have acquired somatic expansions in the order of about 20 repeats greater than the inherited progenitor allele [164, 167]. These bulk DNA analyses by standard PCR, were known to have limitations including Taq polymerase slippage that generates a tail of shorter products [171], thus masking any potential contractions, and, the preferential amplification of smaller alleles [172]. The PCR bias is further confounded by the detection method in which usually only a single radionucleotide or fluorescent moiety is incorporated into the PCR product, independent of the total fragment length. Moreover, using either radiolabelled products and polyacrylamide gel electrophoresis, or fluorescently labelled products and capillary gel electrophoresis, yields non-zero background signals that mask low frequency variants. Thus, these approaches were unlikely to accurately detect amplification of large low frequency somatic variants. In October 2000 Kennedy et al. reported the application of the sensitive small pool PCR approach [146, 173] to investigating somatic instability in two knock-in HD mouse lines with inherited progenitor alleles of either 72 or 80 CAG repeats [168]. In small pool PCR, multiple replicate PCRs are performed with very small amounts of input DNA (typically 1 to 50 molecules per reaction) for a limited number of PCR cycles such that PCR competition between small and large alleles is reduced. Moreover, the PCR products are detected by Southern blot hybridisation using a radiolabelled repeat unit probe that binds more efficiently to larger alleles containing more repeats. Using this approach, it is thus possible to partially compensate for the amplification bias of smaller alleles, and to detect the products of individual input molecules containing up to at least 1,000 trinucleotide repeats [146, 174]. Using this small pool PCR approach Kennedy et al. revealed that a subset of cells in the striatum of their knock-in HD mice had acquired somatic expansions of up to at least 250 repeats, some three times larger than the inherited progenitor allele. Somatic instability in these mice was revealed to be age-dependent and highly tissue-specific with the striatum clearly displaying the highest frequency of large expansions relative to other brain regions. These sensitive single molecule analyses, free of the confounding effects of Taq polymerase slippage that blight bulk DNA analyses, revealed that net somatic contractions were essentially absent and somatic mosaicism was highly biased toward further expansion. Notably, the absence of overt neurodegeneration in these mice argued against the concept that somatic mosaicism was a by-product of active gliosis [168].
The window on the brain: ultra large striatal-specific somatic expansions in HD individuals
The detection of such large somatic expansions in the primary affected brain region of a mouse model at least partially reignited the debate that somatic expansion might be a key driver of regional pathology in HD. Skepticism remained though, as the initial data presented by Kennedy et al. were derived not just from mouse models, but from mouse models with germline alleles of 72 or 80 CAG repeats, i.e., almost double the size of an allele inherited by a typical HD patient. There was considerable doubt that expansions anywhere near so large would be detected in humans. However, three years later, in 2003, Kennedy et al. reported the application of the same sensitive small pool PCR approach to investigating regional instability in the brains of HD individuals [175]. Astoundingly, they revealed that a subset of cells in the brain contained massive somatic expansions of many hundreds of repeats. Indeed, in two individuals who inherited either 41 or 51 repeats, some striatal and/or cortical cells were detected with more than 1,000 CAG repeats. Critically, these two individuals did not die of end-stage HD and received a neuropathological classification of Vonsattel grade 0 [176], i.e., no microscopic evidence of pathological cell loss in the striatum. In the individual with the smallest germline allele (41 repeats), who died at age 40, ∼13 years prior to their predicted age at motor onset, large somatic expansions were only detected in the striatum and were absent in the cortex and hypothalamus. In the second individual who inherited a larger germline allele (51 repeats), and who died at age 27, ∼6 years prior to their predicted age at motor onset, massive somatic expansions were detected in the striatum, and to a lesser extent in the cortex, but were absent in the cerebellum. Notably, analysis of another individual who inherited a much larger allele (∼75 repeats) and died 10 years after diagnosis, revealed a much less obviously regional-specific pattern of somatic expansion. Indeed, in this end-stage patient, the largest expansions were observed in the cortex rather than the striatum. These data thus revealed that large somatic expansions occur in early in the disease time-course, before the onset of overt motor symptoms. These data also suggested that the regional specificity of somatic expansion may more closely follow the regional specificity of the disease earlier in the disease course, and in individuals with smaller inherited alleles. These data also suggested that neurodegeneration may selectively target striatal neurons with large expansions, rather than precipitating large expansions as a secondary endophenotype of active gliosis [175].
Of glia and neurons: HTT somatic expansions accumulate in non-dividing neurons
Given the clear association between CAG repeat size and age at onset in humans, and CAG repeat size and cellular toxicity, and the rapidity with which germline expansions had been accepted as an explanation for anticipation, it is hard in retrospect to understand why the potential significance of somatic expansion, and in particular the significance of the detection of such large somatic expansions early in the disease course of HD [175] were not more widely considered. It is thus worth contemplating what some of the drivers for this were. As alluded to above, by the early 2000s, the basic science field had broadly gone in two main directions: one focussed on downstream pathology, which in HD was essentially polyglutamine toxicity (and the vexed question of whether aggregates are causative); and, the repeat instability field that was more focussed on the mechanisms of instability. As a consequence, relatively little attention was paid to the in vivo consequences of somatic expansion. More directly, questions were raised as to the reliability of the observations of such large expansions in HD brains and the possibility that such large expansions may be technical artefacts of PCR. However, pre-PCR size fractionation of genomic DNA restriction fragments containing the HTT repeat confirmed that such large expansions were not technical artefacts [177]. The other question that was resolved was whether expansions could occur in non-dividing striatal neurons. Using laser capture microdissection in both human HD brains and knock-in animal models, it was demonstrated that: although unstable in glia, expansions were typically larger in striatal neurons; there were more expansions in striatal, rather than cortical, neurons in less advanced HD cases; and, that in mice, smaller somatic expansions occurred in the relatively well spared nitric oxide synthase-positive interneurons compared with the overall neuronal population in the striatum [178, 179].
The road well-travelled: striatal and cortical instability is shared among many disease loci
Another important consideration was the fact that striatal specific expansions are not limited to HD. Notably, the striatum was revealed as the region with the largest expansions in other expanded CAG•CTG repeat mouse models of DM1 [180, 181], SCA1 [182] and DRPLA [183] (see also Wheeler and Dion, this issue [169]). Likewise in humans, high levels of somatic expansion are observed in striatal and/or cortical regions in individuals with other CAG•CTG repeat expansions including DM1 [184, 185], SCA1 [186–192], SCA2 [193, 194], MJD [187, 195–197], SCA7 [94, 198] and DRPLA [188, 199–204]. These data suggest that regional-specific CAG•CTG repeat somatic expansion in the brain is strongly driven by major tissue-specific trans-acting factors. These observations have been further borne out by a recent study that revealed very similar somatic expansion profiles of the expanded CAG repeat in both HD and SCA1 across multiple brain regions [192]. Nonetheless, it is worth noting that in SBMA overall levels of mutation length variability in somatic tissues are lower than in the other polyglutamine expansion disorders [195, 205–207], with more expansions in peripheral tissues such as cardiac and skeletal muscle, skin and prostate, than in the central nervous system. Overall, however, the broadly preserved pattern of somatic mosaicism in the CNS in the polyglutamine expansion disorders sheds some doubt on whether somatic expansions really drive the regional specificity of neurodegeneration observed in HD, and the other polyglutamine encoding CAG repeat expansion disorders more broadly.
The view from the other side: regional and cell-type specificity in HD and the spinocerebellar ataxias
In relationship to the other polyglutamine repeat expansion disorders, particular consideration deserves to be given to the contrast between the SCAs and HD. The SCAs are characterised by early cerebellar degeneration, with loss in particular of the critical cerebellar Purkinje cells. It is very notable, however, that in addition to the six different types of SCA caused by the expansion of polyglutamine encoding CAG repeats (SCA1, 2, 3, 6, 7 and 17), the SCAs caused by other simple sequence repeat expansions (SCA8, 10, 12, 36 and 37), mutations in at least 37 additional genes involved in a wide variety of different cellular pathways can also cause SCA [208]. This is in stark contrast to HD, DRPLA and SBMA, that are each caused by a single type of mutation, the CAG expansion, in only a single gene. The massive genetic heterogeneity in the SCAs reveals that cerebellar Purkinje cells must be extremely sensitive to a wide variety of cellular insults. Thus, given that HTT is highly expressed throughout the brain, with very high levels in the cerebellum, including in Purkinje cells [158, 209–212], one way of viewing the dichotomy between the SCAs and HD is to ask, why is HD not SCA49, and for that matter why DRPLA is not SCA50, and SBMA SCA51? It is possible that the default state might be that in the absence of a very pronounced striatal-specific expansion process, HTT germline expansions might elicit a late onset SCA like phenotype. Another important consideration in this regard relates to critical role that Purkinje cells play in the cerebellum, especially considering the relatively low density and absolute of numbers of Purkinje cells. Purkinje cells are estimated to comprise less than one in a thousand cells in an intact cerebellum. Thus, it remains very possible that investigations of somatic length variability in bulk DNA analyses have failed to account appropriately for this gross disparity in relative cell ratios. Additional application of laser capture microdissection to better investigate the mutation profiles of defined cell types within the cerebellum in both HD and the SCAs are clearly warranted. Indeed, laser-dissected cerebellar cells in DRPLA patients have demonstrated that the expansion lengths in granule cells are significantly smaller than in Purkinje cells and glia [202, 203]. Additionally, caution must be applied when interpreting studies of tissue from patients who died after a long disease course—most commonly the case in the published literature cited above. In end stage-disease tissue, the residual cell profile is much altered due to the loss of vulnerable neurons and the proliferation of cells such as astrocytes. Therefore, in order to more clearly define the relationship between somatic mutation length and pathological vulnerability in polyglutamine diseases, more detailed analysis of defined cell types (isolated by techniques such as laser capture microdissection or single cell sequencing) from candidate brain regions in the rare, early disease cases where cell loss is minimal are really needed.
Nevertheless, it is also important to recognise that the question as to whether somatic expansion drives the regional specificity of neuropathology between the disorders, is entirely separable from the question as to whether somatic expansion drives onset and progression in any one disorder. A striatal-specific expansion process may not delineate HD from the SCAs, but it is unlikely to be helping in HD, and cerebellar expansions in the SCAs, even if they occur at an overall much lower rate than in the striatum, may still be critical since cerebellar Purkinje cells are acutely sensitive to perturbation.
The drivers of instability: somatic expansion is cell-division independent and mismatch repair dependent
Shortly after the first expansion mutations were identified, attention quickly turned to what molecular mechanisms were driving instability. Not unreasonably, it was widely assumed that expansions were most likely to be mediated by DNA replication slippage [154–156]. This concept appeared to be supported by observations that polymerase slippage in vitro could generate products with altered numbers of repeats—these data arising from experiments that dated back to the 1960s when repeating oligonucleotide tracts were being synthesised as part of efforts to crack the genetic code [213–215]. This concept was further reinforced by the observation of similar slippage products that arose during PCR of non-disease associated simple sequence microsatellite repeats [216, 217]. This concept was further reinforced when, as presaged by studies in microbes [218], in 1993 and 1994 it was shown that mutations in [post-replicative] DNA mismatch repair genes were associated with genome-wide microsatellite instability in the tumours of individuals with Lynch syndrome (a hereditary predisposition toward non-polyposis colon cancer) [219–223], the mechanism by which variation in non-disease associated microsatellites arose appeared to be firmly established as DNA replication slippage and mismatch repair avoidance. It seemed a very reasonable assumption that the expansion of disease-associated loci would be similarly mediated by DNA polymerase replication slippage errors that simply overwhelmed the DNA mismatch repair machinery.
Additionally, the fact that for most of the CAG•CTG repeat expansions disorders, the repeats were relatively stable in the female germline, and more unstable and prone to large expansions during male transmission, appeared to fit nicely to the greater number of premeiotic mitoses in spermatogenesis relative to oogenesis. Indeed, detailed single sperm analysis of male germline dynamics in HD did at least appear to partially support such a mitotic model [224]. Moreover, a premeiotic origin for at least some of the HTT CAG mutations in the male germline was directly established using laser capture microdissection of testicular cells [225]. However, these experiments also suggested that meiotic events were important too [225]. A primarily premeiotic replication dependent mechanism for male germline mutations would also predict a strong age effect. In comparing HTT CAG repeat length distributions in sperm between men, an age effect was not detected [224]. Likewise, there was no significant difference in HTT CAG repeat length distributions in two sperm samples from the same man obtained two years apart [224]. Cross-sectional and longitudinal small pool PCR analysis of sperm DNA variation in DM1 males has similarly failed to find evidence for an age effect [146, 227]. In addition to the absence of an obvious age effect in the male germline, these studies in HD [224] and DM1 [146, 227], and similar studies in SBMA [228, 229] and SCA7 [230], have revealed substantive differences between the dynamics of expanded repeats in the male germline and those observed in somatic tissues, including: a much greater frequency of germline mutations than somatic mutations (at least in blood, and for DM1 males inheriting <80 CTG repeats); and, despite a bias toward expansions, a greater frequency of contractions in the male germline, including reversions into the non-disease associated range. Although male germline instability is observed in many expanded CAG•CTG repeat mouse models (e.g., [164–167, 231]; see also Wheeler and Dion, this issue [169]), the very frequent and very large expansions observed in the male germline in humans have not yet been faithfully mirrored in mice, especially when one considers the relatively large allele sizes with which the majority of the mouse models have been generated. It thus remains unclear to what extent the mechanism of expansion is shared between the germline and soma, and what mediates the obvious differences.
A clear prediction of the DNA replication slippage model for expansion would be that tissues with higher levels of cell turnover would show higher levels of somatic mosaicism. This does not appear to be borne out in either humans with, or animals models of, the CAG•CTG repeat expansion disorders, with somatic expansions accumulating in post-mitotic tissues such as skeletal muscle and brain (e.g., [94, 193–204]; see also Wheeler and Dion, this issue [169]). Another clear prediction of the DNA replication slippage model is that loss of function mutations in the post-replicative DNA mismatch repair pathway should increase the frequency of expansions. This prediction was turned on its head in 1999, when Manley et al. demonstrated that the complete reverse was true and that the obligate mammalian MutS homologue Msh2 was absolutely required to generate somatic expansions [232] (see Iyer and Pluciennik, this issue, for more details on the DNA mismatch repair pathway [233]). These insights were extended when it was shown that Msh3, but not Msh6, was also essential for the somatic expansion of CAG•CTG repeat expansions, directly implicating the MSH2/MSH3 MutSBeta complex [234]. One potential explanation for the requirement for MSH2 and MSH3 could have been that MutSBeta stabilises [232, 235] the slipped strand [236] and/or hairpin [237] DNA structures that are the presumed length change intermediates in the expansion pathway. However, involvement of various downstream MutL homologues in CAG•CTG repeat expansion [238, 239], and the requirement for MSH2 ATPase activity [240], suggest instead that expansions may be mediated by an actual mismatch repair reaction of small slipped strand loop-outs in which the loop is preferentially incorporated [232, 240]. In addition to candidate gene studies using knock-out mismatch repair gene null alleles, it is notable that naturally occurring mouse strain-specific differences in CAG•CTG repeat somatic expansion profiles could be detected [234, 241] and some of these associated with naturally occurring variants in Mlh1 and Msh3 [239, 242], presaging the identification of similarly acting human variants (see below). Cell division-independent inappropriate DNA mismatch repair has thus come to the fore as a likely mechanism of expansion of CAG•CTG repeats [238]. Along with other in vitro experiments, these animal model studies were critical in establishing the key players in the expansion pathway (for more details, see Wheeler and Dion, and Iyer and Pluciennik, in this issue [169, 233]). However, in most cases, these studies did not lead directly to insights into the accrual of somatic expansions in mediating pathology (although see below). Nonetheless, the identification of the key players in the expansion pathway, would later prove critical in providing an explanation for the results of the genome-wide associations studies for modifiers of age at onset in HD, and for providing suitable targets for candidate gene studies (see below).
The journey to pathology: genetically suppressing somatic expansion in HD mice slows the accumulation of pathological hallmarks of disease
The critical dependence of somatic expansion on functional Msh2, Msh3 and Mlh1 genes has been used to demonstrate that genetically slowing the rate of somatic expansions can also slow the rate of accumulation of pathological markers of HD such as polyglutamine aggregates [239, 244]. One of the primary reasons that more definitive data directly linking somatic expansion and disease pathology in HD models has been difficult to generate relates to the length of the CAG repeat in mice necessary to generate sufficient levels of somatic expansion and/or a disease phenotype during the lifetime of a mouse, or the even shorter length of a typical project grant. Simply put, mice with small germline CAG expansions in the range typically observed in humans do not develop an overt HD phenotype during their lifetime [245, 246]. HD mice with larger germline expansions typically beyond the length that is observed even in most juvenile HD cases (>80 repeats), can display robust HD phenotypes in a matter of months (e.g., [163, 168]). In such cases, the repeat is likely already well beyond any cell-dependent toxic threshold and pathology can proceed in the absence of somatic expansion. Of course, that is not to say, as evidenced above [239, 244], that somatic expansions may not exacerbate the phenotype in HD mice already inheriting large expansions. Indeed, such an effect may explain the greater frequency of HTT aggregates observed in a minimally CAA interrupted yeast artificial chromosome HD mouse model (presumed to be at least partially somatically unstable), relative to a bacterial artificial chromosome HD mouse model with a somatically stable highly interrupted polyglutamine encoding CAG/CAA repeat tract [247, 248]. Furthermore, detailed studies in mouse models with germline alleles that are small enough not to cause an overt early onset phenotype, but that have the capacity to somatically expand, are clearly warranted. Nonetheless, it remains unclear if short-lived mice can be effectively used to determine the absolute requirement for the somatic expansions that typically accumulate over 30 to 40 years in HD patients inheriting alleles of 40 to 50 CAG repeats.
The passage of the extremes: the frequency of large cortical expansions is associated with variation in age at onset in HD
A key prediction of a critical role for somatic expansion in HD pathology would be that individual-specific variation in the rate of somatic expansion would be reflected in individual differences in disease severity. To this end, the first human data demonstrating such a link were generated in 2009 [249]. Specifically, using small pool PCR analysis of cortical DNA from a cohort of HD individuals with extreme early or extreme late onset of symptoms relative to the age at onset predicted by the number of CAG repeats inherited, Swami et al. were able to demonstrate that the fraction of large somatic expansions, as quantified by the skewness of the repeat length distribution, was inversely associated with residual variation in age at onset, i.e., individuals with more large somatic expansions had an earlier age at onset than expected. These data, along with additional data defining the repeat length dependence of somatic expansions in buccal cell DNA [250], demonstrated that the somatic expansion phenotype in humans is modifiable by factors other than repeat length, tissue and age. Through this period, animal model and human data implicating somatic expansion slowly accumulated, but even as late as 2018 somatic expansion was not widely viewed as a therapeutic target in HD (e.g., [251]).
THE RETURN OF THE REPEAT
The reckoning: genome-wide association studies of variation in HD age at onset reveal DNA repair gene variants
A major limitation of the scientific process is the inherent biases and preconceptions that are inevitably brought into play in the design of an experiment. One of the beauties of genome-wide association studies (GWAS) are that they are completely unbiased, at least in terms of the having to make no predictions about the genes in which variants may modify the phenotype of interest. It was thus with great excitement that the HD community eagerly awaited the results of GWAS of modifiers of residual age at onset in HD. As detailed in the accompanying manuscript by Hong et al. [252], the results of the first GWAS to collate a large enough cohort of HD participants to achieve genome-wide significance levels was published by the GeM-HD consortium in 2015, and revealed genome-wide significant associations in two regions: the FAN1 gene; and the RRM2B gene [253]. The FAN1 gene encodes the Fanconi anaemia FANC1/FANCD2-associated endonuclease 1 DNA repair gene. Although at the time FAN1 was not known to be involved in the repeat expansion pathway, it is now clear that the levels of FAN1 are important in mediating somatic expansion in cells [254] and animal models [255, 256] (see also Deshmukh et al. [257] and Zhao et al. [258], this issue). RRM2B encodes ribonucleotide reductase regulatory TP53 inducible subunit M2B and its role in modifying HD onset currently remains unknown. A third region encompassing the MLH1 DNA mismatch repair gene almost reached genome-wide significance in the 2015 GWAS [253], and was subsequently replicated in an independent cohort [259]. MLH1 has previously been shown in animal models to be essential for somatic expansions of the HD repeat [239]. Pathway analysis of the 2015 GWAS results also revealed that polymorphisms in DNA repair genes were overrepresented in variants associated with age at onset in HD, including specifically DNA mismatch repair. These data thus strongly supported the contention that DNA mismatch repair processes mediate differences in HD age at onset not accounted for by inherited CAG length. Given the prior association of the mismatch repair proteins in the somatic expansion process, it seemed logical to assume that these polymorphisms mediate a role in HD pathology via a more direct role in the somatic expansion pathway [253].
The land of light: the same variants in MSH3 are associated with disease severity in HD and DM1
Providing an amazing example of the utility of careful longitudinal clinical characterisation of the disease phenotype, in 2017 Hensmann Moss et al. were able to demonstrate that a combined multi-phenotype CAG and age-adjusted disease progression score was able to reveal a genome-wide significant association with variants in the MSH3 DNA mismatch repair gene in only just over two hundred HD participants in the TRACK-HD cohort [260]. These associations were further strengthened when it was revealed that some of the same variants in and around a polyproline/alanine encoding polymorphic 9 bp repeat in MSH3 exon 1 were also associated with both somatic expansions rates in blood DNA, and residual variation in disease severity, in both HD and DM1 [261]. It is a bizarre coincidence that MSH3 contains its own variable repeat, but the fact that MSH3 is absolutely required for repeat expansion in animal models, suggests that the association between MSH3 variants with variation in disease severity is not a coincidence, and that the causative MSH3 variants modifying disease severity are acting directly through their effects on somatic expansion.
The pure repeat: CAG repeat number, not encoded polyglutamine length best predicts HD severity
The polymorphic HTT CAG that expands in HD is succeeded by an additional CAACAG cassette that also encodes glutamine such that the total length of the encoded polyglutamine tract equals the number of CAG repeats plus two in a typical HTT allele [61]. It has been known for many years that a subset of atypical HTT alleles can differ in this regard with some alleles containing a duplication of the CAACAG cassette, and some alleles lacking this cassette completely (see Hong et al. [252], this issue, and Ciosi et al. [262]). These variants can give rise to CAG sizing errors as estimated using fragment length analysis, and recent high-throughput DNA sequencing analyses by Ciosi et al. have revealed that failure to take these sizing errors into account can yield highly atypical genotype to phenotype associations in HD [262]. Similar effects were also reported by Wright et al. [263] and in the latest results from the GeM HD Consortium GWAS for modifiers of age at onset in HD [264] (see Hong et al., this issue [252]). An early observation after the disease-causing mutations were first identified, was that in several disorders non-disease associated alleles were interrupted with stabilising variant repeats, whilst genetically unstable disease-causing expansions were pure (e.g., [186, 265–268]). In the DM1, the vast majority of non-disease associated alleles are pure CTG, as are most disease-causing expansions. However, a subset of approximately 5% of DM1 disease-causing expansions are interrupted by primarily CCG variant repeats [269, 270]. In addition to being genetically more stable in both the germline and soma, such alleles are typically associated with delayed onset and/or milder DM1 symptoms, thus linking variant repeat interruptions with increased somatic stability, and further linking somatic instability with disease onset [150, 269–274]. Thus, the most logical explanation for the greater predictive value of the number pure CAG repeats, rather than total glutamine number encoded, is that it is pure CAG number that drives somatic expansion and ultimately disease onset/progression. Indeed, pure CAG length accurately predicts the relative ratio of somatic expansions observed in the blood DNA of HD individuals [262], effects that appear to be mirrored in the male germline [275]. However, it should be noted that whilst correcting for pure CAG more accurately predicts age at onset and disease progression than does the number of glutamines encoded, individuals lacking the CAACAG cassette on their mutant chromosome still tend to have an earlier age at onset than expected. Likewise, individuals with the CAACAG cassette duplication on their mutant chromosome do not have a worse disease course, despite the fact they inherit alleles expressing two additional supposedly toxic glutamine codons relative to individuals with a typical expanded allele [262–264]. The reasons for these residual effects remain unknown, but could include additional effects on somatic expansion in the brain not detectable in blood DNA, or some other effect on HTT transcription [276], RNA stability and/or translation efficiency for instance [252, 278]. Alternatively, it is possible that the ultimate pathogenic moiety in HD is not the canonical polyglutamine containing HTT protein, but some directly toxic effect of the HTT CAG RNA [279], an alternative truncated transcript [280] or a repeat associated non-ATG translation product [281].
More of the same: GWAS reveals even more DNA mismatch repair gene variants associated with variation in HD age at onset
In 2019 the GeM HD Consortium revealed the results of the latest GWAS for modifiers of age at onset in HD incorporating just over 9,000 participants [252, 264]. In addition to further confirming associations with FAN1, MLH1 and MSH3, these data elevated variants in the PMS2 and PMS1 DNA mismatch repair genes, in addition to the LIG1 DNA ligase gene, also required to complete a DNA mismatch repair reaction, to genome-wide significance. These data further highlight the critical role that DNA repair gene variants have in mediating symptomatic variation in HD, most likely through their action on somatic expansion [264].
The mechanistic bridge: DNA repair gene variants are associated with somatic expansion scores in HD blood DNA
The high-throughput HTT sequencing assay developed by Ciosi et al. also allows for the quantification of the relative ratio of somatic expansions in blood DNA (see Ciosi et al., this issue for comparison of approaches to quantifying somatic mosaicism in HD) [282], that after correcting for age and CAG length effects, results in an individual-specific somatic expansion score [262]. As expected, assuming that somatic expansion profiles in blood DNA at least broadly parallel those in the brain, the somatic expansion score was inversely associated with variation in age at onset and positively associated with individual-specific disease progression scores (i.e., individuals with a faster rate of somatic expansion have an earlier age at onset than expected and more rapid disease course). The somatic expansion score is also a molecular phenotype that can be used for association studies to reveal genetic modifiers of the expansion process. Indeed Ciosi et al. have used this phenotype to reveal direct associations between somatic expansion and variants in the FAN1, MLH1, MLH3 and MSH3 DNA repair genes in a candidate gene analysis [262]. As discussed, FAN1, MLH1 and MSH3 have already been implicated as modifying age at onset and disease severity in HD by GWAS analyses [253, 264]. Whilst variants in MLH3 have not yet been significantly associated with variation in HD age at onset, the latest HD GWAS data indicate a nominal association of p = 0.0001 [264]. Given the essential requirement for MLH3 in mediating somatic expansions in HD mice [239], it seems a reasonable supposition that MLH3 will reach genome-wide significance with a larger cohort. Nonetheless, the FAN1, MLH1 and MSH3 data already provide a mechanistic link between the HD age at onset modifiers, and direct modifiers of somatic expansion, that lends further credence to the model proposing somatic expansion as a key driver of disease pathology in HD.
The last debate: is somatic expansion required and does size matter?
It would now appear that the concept that somatic expansion contributes toward disease onset in HD is beyond reasonable doubt—size changes, and it clearly does matter (although see Maiuri et al., this issue, for alternative hypotheses linking DNA repair and HD [283]). More pertinent now is to consider what are the actual critical products of the somatic expansion process? Are the massive somatic expansions of hundreds or even thousands of repeats actually required? The U-shaped disease severity curve observed in the R6/2 mice where germline expansions beyond 300 CAG repeats become protective [284–286], albeit in transgenic model expressing a protein fragment, are nonetheless very intriguing. Such large expansions are at least partially hypomorphic [284–286]. However, if they were truly protective, then we might expect cells carrying such large expansions to accumulate in end-stage disease. This they clearly do not do [175]. Indeed, at any one point cells carrying such large expansions are relatively rare. However, this is exactly what might be expected if such cells have only a very short half-life and exist only transiently in such an elevated state in HD brains. It is nonetheless possible that such very large expansions may be something of a red herring. Given that transmitting an allele only one CAG repeat longer results in at least a two-year decrease in the age at onset of HD, it seems not unreasonable that even somatic gains of one or two repeats are certainly not helping and almost certainly making things worse. Some clues as to the answer to this question may come from asking an even more fundamental question as to whether somatic expansion is absolutely required to generate HD pathology. If germline expansions in the range 40 to 50 CAG repeats are inherently toxic and capable of precipitating symptoms in the absence of somatic expansion, then that would suggest that even small changes would accelerate pathology in a meaningful way. If, however, a toxicity threshold exists at some larger size, then somatic expansion may indeed be absolutely required (see Donaldson et al., this issue, for further discussion of where the toxic CAG threshold may actually lie [287]). The concept that somatic expansion is required in HD, may be supported by the observation that individuals homozygous for HD expansions (the vast majority of whom are compound heterozygotes with disease-causing expansions of two different sizes), do not appear to have an earlier age at onset than that predicted by the larger of their two alleles [288, 289]. One possible explanation for the fully dominant nature of HD onset, supported by a modelling approach, is that onset is achieved when a particular fraction of cells somatically expand the CAG repeat beyond a higher pathological threshold [290]. As somatic expansion is highly repeat length dependent, this threshold will, in the majority of cells, be achieved first by the larger of the two inherited alleles, and hence it is the larger allele that predicts disease onset [290]. Additional insight into the issue of whether somatic expansion is actually required, could also be provided by the identification of individuals with repeat stabilising interruptions in the middle of an expanded HTT CAG array. For instance, if an individual with a 45 repeat allele with a single glutamine encoding variant CAA repeat (e.g., (CAG)22CAA(CAG)22) was identified, and assuming, as expected such an allele would be somatically stable, then if they were affected it would indicate that somatic expansion is not required. If however they remain asymptomatic throughout their life, then it would indicate somatic expansion is absolutely required and the true pathological threshold at the cellular level is greater than that required to be inherited (at least for a somatically unstable pure CAG repeat). Such individuals are at the least very rare in either the HD or general population, if they exist at all. However, the availability of a high-throughput HTT sequencing assay [262], and an improved ability to genotype simple repeats from whole-genome sequencing data [291, 292] suggest that this question is closer to being answered than it ever has been.
One rule to bind them: trans-modifiers of somatic expansion in the other repeat expansion disorders
As somatic expansion is a common theme in many of the repeat expansion disorders, it seems not unreasonable to assume that somatic expansion might similarly drive disease onset in some of these disorders too. Indeed, it was established in 2012 that individual-specific variation in the rate of somatic expansion in blood DNA was inversely correlated with residual variation in age at onset in DM1 [148]. Moreover, individual-specific variation in somatic expansion was shown to be inherited as quantifiable trait consistent with an underlying genetic mechanism [148]. These data were further borne out with the demonstration in 2016 that using a candidate DNA mismatch repair gene study, variants in the MSH3 DNA mismatch repair gene were associated directly with variation in the individual-specific rate of somatic expansion in DM1 [151]. Similarly, preliminary evidence has been generated that variants in FAN1 and the PMS2 DNA mismatch repair gene are also associated with residual variation in age at onset of some of the other polyglutamine encoding CAG repeat expansion disorders, including SCA1 [293]. Whilst most of the data relating directly to the role of somatic expansion in disease aetiology in humans relates to the CAG•CTG repeat disorders, there is also evidence for considerable somatic instability in Friedreich ataxia (FA) [294–299]. However, the very strong bias toward net somatic expansions observed in the CAG•CTG repeat disorders, is not seen in FA where a high frequency of somatic contractions of the GAA repeat are also observed [295–299]. Nevertheless, somatic mosaicism continues throughout life, and somatic expansions accumulate in the dorsal root ganglia, one of the primary affected tissues, suggesting somatic mosaicism may play an important part in FA [298, 299]. Moreover, it appears many of the same mismatch repair proteins are implicated in somatic instability of GAA repeats in both human cells and animal models [300–306]. Mismatch repair proteins have been similarly implicated in expansion of the CGG repeat in FXS mouse models [307–312] (for more details, see Zhao et al., this issue [258]). Given the massive cross-disorder potential of therapies aimed at supressing somatic expansion (see Benn et al., this issue [313]), additional insights into the dynamics and mechanisms of somatic expansion in the other repeat expansion disorder are required. Suitably powered large-scale GWAS of modifiers of somatic expansion and disease severity in the other repeat expansion disorder should be particularly informative.
The houses of healing: somatic expansion as a therapeutic target
In addition to inherent questions of fundamental biological interest, the ultimate goal of understanding the role of somatic expansion in HD is to evaluate it as a possible therapeutic target. The data are now clear that somatic expansion at the very least exacerbates disease onset and progression, and as such somatic expansion has to be considered a very real therapeutic target worthy of further scrutiny. Suppressing somatic expansion would be expected to be therapeutically beneficial. Even more enticing is the prospect of modulating repeat instability in such a way so as to be able to elicit somatic repeat contractions. If enabled early enough, repeat contractions raise the prospect of being not just beneficial, but potentially curative. In this light, the recent data that CAG repeat somatic instability can be modified by small molecules in HD animal models and appears to be associated with reduced markers of disease is exceptionally exciting [314–317]. However, I will leave Benn et al., this issue [313], to delve deeper into this topic, and discuss some of the technical challenges involved, and leave you here with the idea that our journey has reached a significant milestone, but it has not ended. Much remains to be done to translate these findings to new treatments for HD and related disorders, but the establishment of somatic expansion as contributory to HD pathology, and the identification of potential enzymatic targets, opens up some exciting possibilities.
CONFLICT OF INTEREST
D.G.M. has been a scientific consultant and/or received honoraria or stock options from Biogen Idec, AMO Pharma, Charles River, Vertex Pharmaceuticals, Triplet Therapeutics, LoQus23, and Small Molecule RNA and has had research contracts with AMO Pharma and Vertex Pharmaceuticals.
Footnotes
ACKNOWLEDGMENTS
The author would like to thank all those whose considerable efforts have gotten us to the point where somatic expansions is more broadly recognised as an important contributory factor in HD pathology and a bona fide therapeutic target. The author would also like to apologise in advance to all those whose important contributions, particularly with regard to insights into the expansion mechanism, have not been duly acknowledged in this review. Work in the D.G.M group is supported by an award from the CHDI Foundation.