
Abstract
At fifteen different genomic locations, the expansion of a CAG/CTG repeat causes a neurodegenerative or neuromuscular disease, the most common being Huntington’s disease and myotonic dystrophy type 1. These disorders are characterized by germline and somatic instability of the causative CAG/CTG repeat mutations. Repeat lengthening, or expansion, in the germline leads to an earlier age of onset or more severe symptoms in the next generation. In somatic cells, repeat expansion is thought to precipitate the rate of disease. The mechanisms underlying repeat instability are not well understood. Here we review the mammalian model systems that have been used to study CAG/CTG repeat instability, and the modifiers identified in these systems. Mouse models have demonstrated prominent roles for proteins in the mismatch repair pathway as critical drivers of CAG/CTG instability, which is also suggested by recent genome-wide association studies in humans. We draw attention to a network of connections between modifiers identified across several systems that might indicate pathway crosstalk in the context of repeat instability, and which could provide hypotheses for further validation or discovery. Overall, the data indicate that repeat dynamics might be modulated by altering the levels of DNA metabolic proteins, their regulation, their interaction with chromatin, or by direct perturbation of the repeat tract. Applying novel methodologies and technologies to this exciting area of research will be needed to gain deeper mechanistic insight that can be harnessed for therapies aimed at preventing repeat expansion or promoting repeat contraction.
Keywords
INTRODUCTION
The first discoveries in the 1990s that inherited unstable trinucleotide repeats caused human neu-rological and neuromuscular diseases stimulated considerable early interest in understanding the mec-hanisms underlying repeat instability (see [1] for a historical perspective). These so-called repeat ex-pansion disorders exhibit instability of the expanded repeat tract upon transmission to the next generation as well as instability in somatic cells [2–30]. To study mechanisms of repeat instability, a wide variety of model systems in different organisms, from E. coli to mice, have been implemented. In particular, a large body of research on repeat instability has been conducted in S. cerevisiae [31–35] and has guided many of the subsequent mammalian studies. Collectively across all organisms, many genes and candidate pathways have been identified that can play a role in altering repeat dynamics in the various model systems. Much less well understood is the relevance of most of these genes and pathways to human disease, as most have not been studied in the context of relevant cell-types in animal models or have no direct validation in patients.
Here we review the modifiers and potential mechanisms underlying the instability of CAG/CTG re-peats, gleaned primarily from mammalian cell-based and animal models, and put these observations into the context of recent human genetic data. We focus on the coding CAG repeat causing Huntington’s disease (HD) (OMIM #143100) and the non-coding CTG repeat causing myotonic dystrophy type 1 (DM1) (OMIM# 160900). These are the most common of the thirteen CAG/CTG repeat expansion disorders [22], upon which the majority of the mouse and patient cell-based models are based.
The HD CAG repeat is located within exon 1 of the HTT gene. CAG repeat lengths of 6–35 are not clearly disease-associated, whereas 36–39 CAGs and 40+ CAGs are associated with incompletely or fully penetrant disease, respectively [3, 36]. A rarer juvenile-onset form of the disease is usually associated with > ∼60 CAGs [37]. Intergenerational changes in repeat length, associated in males with instability in sperm DNA, are seen in the disease range (36+ CAGs) as well as in the high normal range (27–35 CAGs), the latter giving rise to new mutations [26, 38]. Somatic instability has been studied in individuals with disease-associated repeat lengths. The instability in somatic cells is expansion-biased, occurs in both in the brain and in peripheral tissues, but shows tissue- and cell-type specificity; forebrain regions show relatively high levels of expansion, the cerebellum exhibits low levels of expansion, and neurons have greater expansions than glia [2, 30]. Of the peripheral tissues, liver is relatively unstable [2]. Somatic expansion in postmortem cortex was found to be inversely correlated with the age of disease onset [4].
The DM1 CTG repeat is located within the 3’ untranslated region (UTR) of the DMPK gene. Individuals are classified into five categories based on the age at onset: congenital, infantile, juvenile, adult, and late-onset [39, 40]. These are largely correlated with repeat size, but there is significant overlap between the categories [41]. Repeat lengths of 5–∼37 CTGs occur in the general population, and it is thought that alleles between 16 and 30 repeats are the source of rare de novo expansions [42]. Affected individuals can harbor from ∼50 to several thousand repeats, with congenital DM1 often presenting with alleles >1,000 CTGs [43, 44], Repeats of ∼50–79 exhibit high levels of male germline expansion, whereas 80+ repeats tend to exhibit greater expansions in female transmissions [41]. Somatic instability can be de-tected in fetal tissues between 13 and 16 weeks of gestation, and in patients shows expansion-biased and tissue-dependent instability, with high levels seen in heart, skin, muscle and blood, as well as in brain tissues [41, 45].
Thus, patient data indicate that there may be differences in mechanisms of instability in HD and DM1, likely due to genomic location and repeat length. However, as revealed by studies in mice there are clearly shared underlying mechanisms. Thus, observations in DM1 models may well be relevant in HD where the CAG repeat can expand somatically to hundreds of repeats [4, 29]. The reverse is also true: the observations made in HD models are likely to be relevant to DM1, in particular for repeat lengths in the lower range. Overlapping mechanisms extend to other trinucleotide repeats, as discussed in [46]. Equally important however, are aspects of repeat in-stability and modifiers that distinguish the different disease-associated repeats that may yield important mechanistic insight.
As described in the following sections, many genes that function in DNA repair pathways have been found to modify CAG/CTG instability. In several cases (see: “Non-Canonical Mechanisms Underlying Repeat Instability?”) it appears that the encoded proteins do not in fact “repair” DNA, but rather promote mutation. For the purpose of this review, we use the term “DNA repair” to describe the process or function to which genes or pathways are traditionally assigned, rather than the functional outcome of that process in the context of expanded repeats. More specifically, and as an example, we use the term “mismatch repair” (MMR) to mean the canonical post-replicative pathway and its components [47], acknowledging that the specific functions of the proteins in the pathway may be different in the context of expanded repeats.
THE LINK WITH HUMAN GENETIC STUDIES
An ongoing challenge is to understand which of the many modifiers identified in model systems is relevant in humans. Recent genome-wide association studies (GWAS) for modifiers of onset in HD have identified genes in the MMR pathway (MSH3, MLH1, PMS2, PMS1) as modifiers of the rate of disease onset in patients [48–51]. MSH3, also modified a measure of HD progression [52]. Other DNA repair genes identified in the age-at-onset GWAS were LIG1, encoding a DNA ligase that can function in several DNA repair pathways, and FAN1, encoding FANCD2 and FANCI Associated Nuclease 1, with a role in repairing interstrand crosslinks. Several MMR pathway genes had been previously identified as critical drivers of somatic repeat expansion in mouse models (see below), thus providing strong support for somatic expansion as the underlying mechanism for disease modification in HD. Notably, the genes that have emerged in the human genetic studies to date do not obviously highlight a role for a general DNA damage response as a driver of disease onset, where key players include ATM and ATR for example [53]. Interestingly, two HD onset-modifying DNA repair genes, FAN1 and PMS2, were also associated with age at onset in the spinocerebellar ataxias (SCAs), caused by CAG expansion mutations but having distinct neuropathological profiles [54], suggesting somatic expansion as common disease onset-modifying mechanism. MMR pathway genes MSH3, MLH1 and MLH3, as well as FAN1, are also associated with repeat instability in HD or DM1 patient blood [50, 55–57].
Human genetic discovery efforts help to define the DNA repair factors and pathways that may play prominent roles in the process of somatic repeat expansion in cell-types that are relevant to the onset or progression of disease phenotypes, prioritizing targets for therapeutic intervention [58]. Larger sample sizes would be expected to reveal genetic modifiers that are rarer in the population and/or that have weaker effects. However, the absence or rarity of naturally occurring functional variation in a particular gene will preclude its detection in genetic association studies. Therefore, complementary investigation of instability in model systems allows the identification of modifier genes that may not be revealed in human genetic studies, but which may be part of the same pathway(s), better defining pathways and mechanisms and providing additional points for possible therapeutic intervention. An example is MMR pathway gene MSH2; this gene has not emerged as a human onset modifier, yet it is clearly required for somatic expansion in mouse models as well as in patient cell-based models (Table 1). This may indicate the lack of functional MSH2 variation in the human population at sufficiently high frequency to be detected in the GWAS studies to date.
Genetic modifiers of repeat instability found in mammalian systems. Excluded from this list: modifiers that act in the context of an exogenous perturbagen or sensitizer (e.g., under replication stress, or upon double-strand break or nicks induced within the repeat tract)
@No effect of MSH3 knockout on contractions in a (CTG)33-URA3 shuttle vector assay that detects contractions only [78]. *Effect only seen with simultaneous knockdown of both TOP1 and TDP1. #In male germline transmissions only; in somatic cells, effect only seen with simultaneous knockout of both Csb and Ogg1. §Depends on the orientation of the repeat tract with respect to the origin of replication. & only for intergenerational transmissions. Ξ Only in male germline transmissions. †Only during maternal germline transmissions. HttQ111, HttQ50, HttneoQ50 and R6/1 mice have a canonical human (CAG)nCAACAG repeat structure; HttQ150 mice do not have a penultimate CAACAG. Note that the mouse knock-in models are also referred to using the original gene nomenclature as HdhQ111, HdhQ50, HdhneoQ50, and HdhQ150.
NON-CANONICAL MECHANISMS UNDERLYING REPEAT INSTABILITY?
The underlying mechanisms by which DNA repair proteins modulate repeat instability remain to be clearly defined. However, it is conceivable that these proteins could act in non-canonical ways or in non-canonical pathways. DNA repair pathways are traditionally defined based on the repair of a specific lesion, such as a mismatch or a single-strand break. However, the substrate(s) or lesion(s) at repeat tracts may be highly unusual, and the classical definition of DNA repair pathways may break down. Therefore, it is plausible that DNA repair proteins have roles promoting or protecting against repeat instability that differ from those usually described during the repair of a specific lesion. Most notably, the same MMR proteins that normally act to suppress genomic instability by repairing post-replicative errors, promote CAG/CTG instability in non-dividing cells. This cou-nterintuitive finding suggests that repeat tracts might engage components of the MMR machinery in unusual ways, and/or could implicate other pro-mut-agenic processes in which MMR factors play roles [59, 60]. There is also considerable cross-talk bet-ween proteins in DNA repair pathways (see non-exhaustive examples in these reviews: [61–65]). Thus, DNA repair proteins may work together in unexpected ways to modulate repeat dynamics. For example, FAN1 is not a component of the classically-defined MMR pathway, yet Fan1 interacts genetically with Mlh1 to control somatic CAG instability in mice [66].
Although it remains helpful to broadly classify modifiers of repeat instability by the pathways in which they were first described, as categorized below and illustrated in Fig. 1, we also present genetic mod-ifiers identified in the various mammalian model systems in the context of known associations between them, in a manner that is agnostic to their classically defined functions (Fig. 2). This allows a better appreciation of the extent to which many of the modifiers (Table 1), regardless of the system(s) in which they were identified or their specific role in modifying repeat instability, are connected as part of a network. Importantly however, the vast majority of these genes have not been validated as modifiers of repeat instability in mouse models, and/or their relevance to patients is unknown (see color coding in Fig. 2). The connections in this network may therefore provide testable hypotheses for future discovery of genes and pathways that might be relevant to repeat instability in disease.
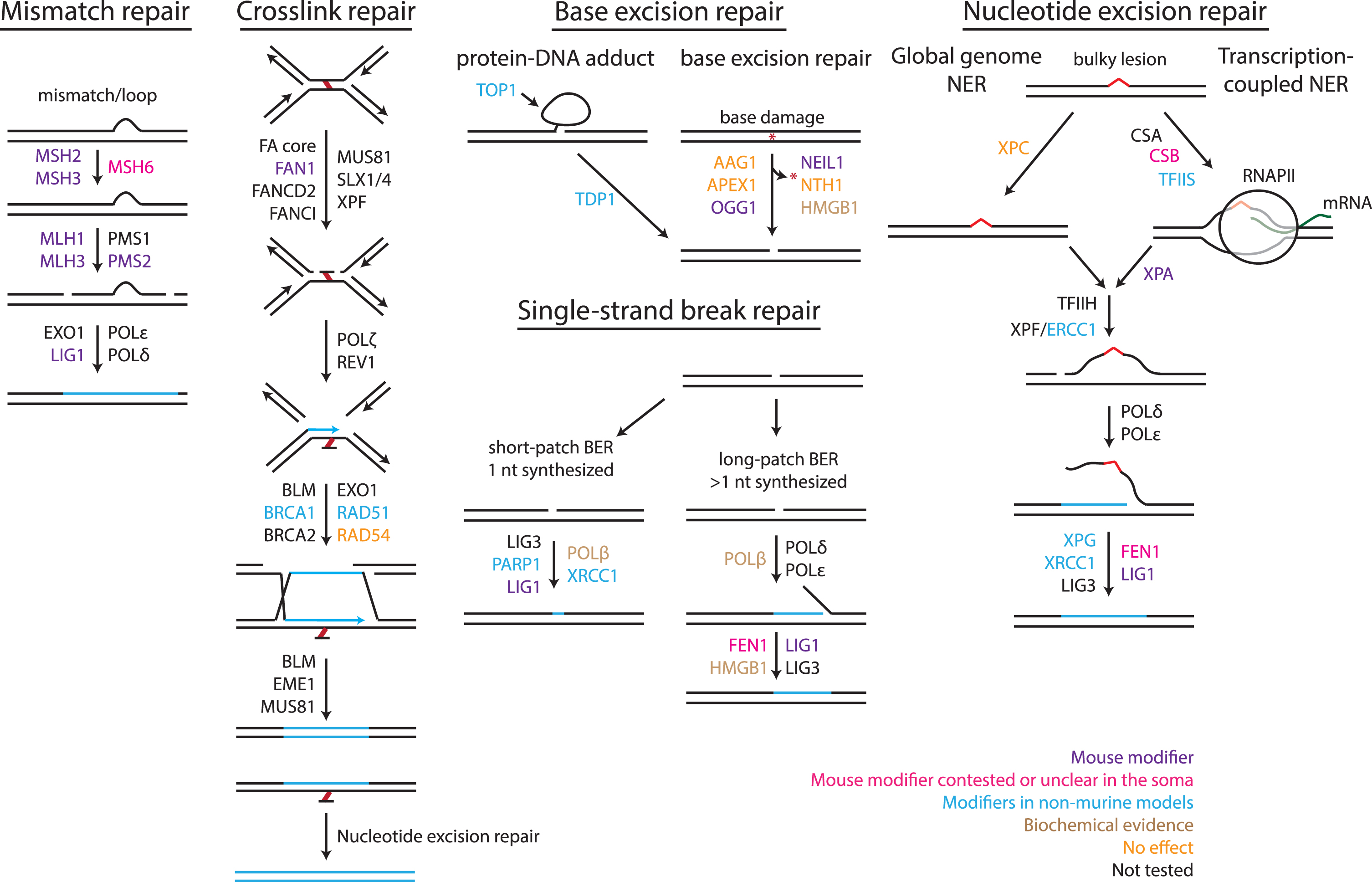
Classically defined DNA repair pathways implicated in repeat instability. Outline of four different repair pathways, indicating a non-exhaustive list of proteins within those pathways, either based on in vivo evidence or from reconstituted systems. Modifiers of repeat instability are color-coded as follow: purple: modifier of repeat instability in murine models; pink: modifier with contested or unclear somatic instability data; blue: modifiers in non-murine models; brown: modifier in biochemical assays; orange: no effect when tested in a mammalian system; black: not tested for an effect on repeat instability. See Table 1 for direction of effect of modifiers in mammalian systems. Figure update from [109] with permission.
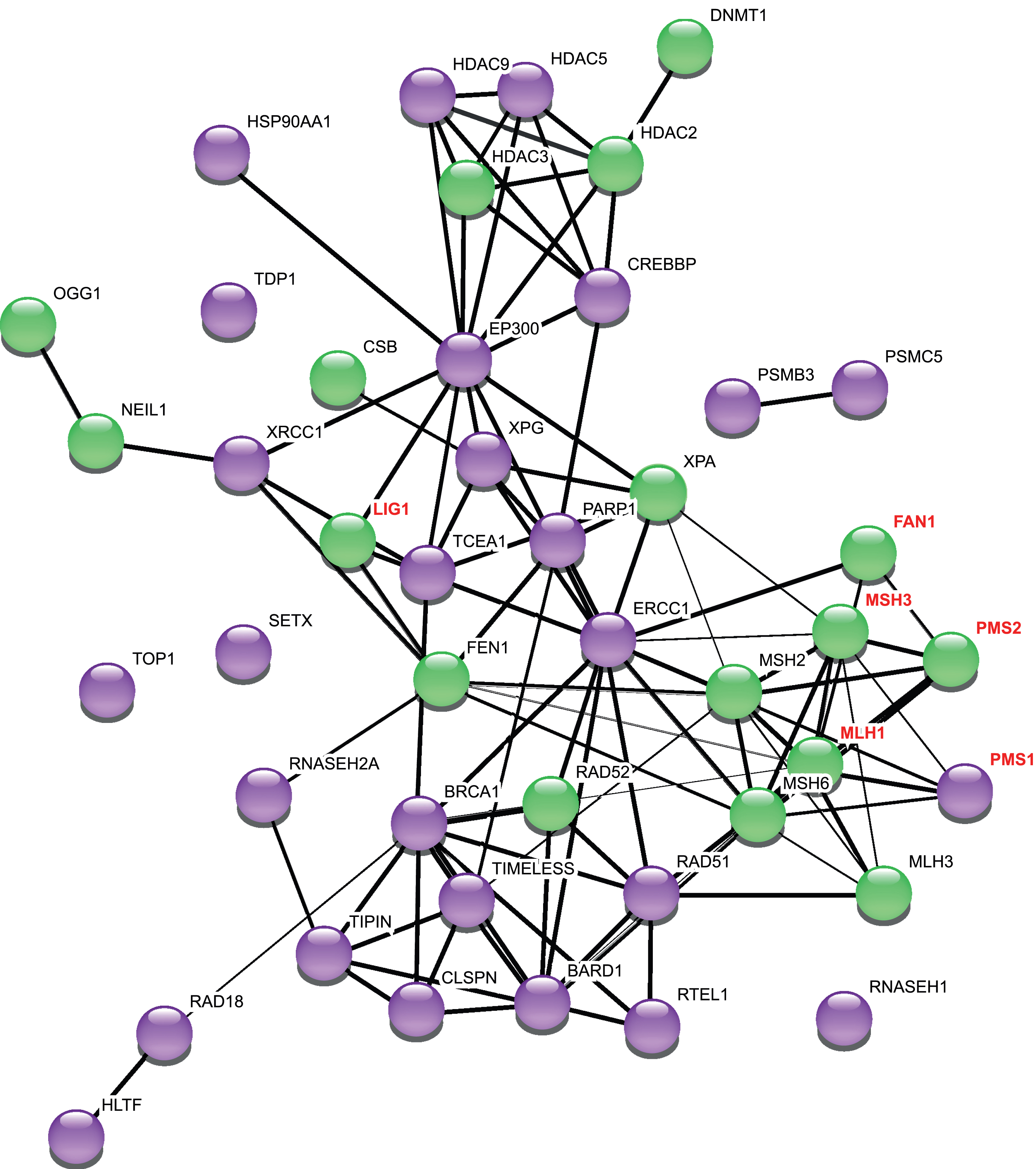
Network of repeat instability modifiers. This network was generated using StringDB v.11. The thickness of the edges refers to the confidence score (CS). 1 pt edges represent a CS of 0.7, 2 pt edges have a CS of 0.8, and 3 pt edges have a CS of 0.9 or greater. The String output categorizes interactions as “binding” (direct or indirect), “catalysis” and “reaction”: for clarity we have not included this in the Figure, but refer the reader to the StringDB v.11 for this information [126]. Purple nodes are genetic modifiers in at least one mammalian system for studying repeat instability. Green nodes are modifiers of repeat instability in murine models of CAG/CTG diseases. Names of genes additionally identified as modifiers of HD age at onset or progression [48–50, 52] are highlighted in red font. PMS1 promotes repeat expansion in a mouse cell model with an expanded CGG/CCG repeat [127].
MAMMALIAN MODELS USED TO STUDY CAG INSTABILITY
The principal types of cell or animal-based mammalian model systems and assays that have been developed to investigate CAG/CTG instability, are summarized in Table 1 and in Fig. 3. Knock-in and transgenic mice have been generated with unstable repeats, typically containing more than 100 units [110]. Repeat instability is analyzed in these models by PCR, using either small pool-PCR with Southern blot detection, or using fragment analysis on a DNA sequencer [13, 87]. These models exhibit repeat length-and time-dependent, expansion-biased somatic instability that is tissue or cell type-specific [87, 111–117]. In the HD models, high levels of expansion are seen in the striatum, in particular in the medium-spiny striatal neurons (MSNs), and in the liver, attributable to hepatocytes [87, 117]. The cerebellum exhibits relative stability [87, 116]. Overall, there is very good correlation with tissue-specific instability patterns observed in both adult and juvenile onset HD [2]. DM1 mouse models can exhibit relatively high instability in kidney, liver, skeletal muscle and brain, including the striatum, with relatively low instability in the spleen, heart and cerebellum [112, 118]. There appears to be more variation in the tissue-specific instability patterns between different DM1 lines than apparent across HD mouse models, and in general, extensive comparisons with humans have been limited by the lack of availability of human tissues. Both HD and DM1 models exhibit intergenerational repeat length changes that recapitulate some, but not all of the features of intergenerational instability in patients. A paternal expansion bias is seen in transmissions in HD and DM1 models [112, 119] with maternal expansions in DM1 depending on the model [112, 119]. However, absolute mutation frequencies are much lower than those seen in patients, requiring very long CAG lengths relative to those in humans, to achieve frequencies of intergenerational changes and/or large jumps in repeat size that are seen in patients [112, 121]. Thus, parallels in many of the features of repeat instability in patients and in mice indicate that the mice models are likely to afford insight into mechanisms of instability that are relevant to disease in patients. Other advantages of mouse models are that they allow the dissection of modifiers that might act in a tissue/cell type-specific manner, including distinguishing mechanisms that act in the germline and in the soma. They also permit the analysis of naturally occurring strain-specific variation that is associated with different levels of instability [90, 122]. Moreover, comparisons between models that differ in repeat length and genomic contexts can provide information on the role of cis elements that might contribute to repeat instability [111, 123–125]. While yielding rich in vivo insights into potential mechanisms of repeat instability, the throughput in mice is low due to the need for extensive breeding to test genetic modifiers, and the time (several weeks to months) for somatic repeat expansions to accumulate. This is especially challenging in models with shorter inherited repeat lengths, e.g., within the adult-onset range of CAG lengths for HD (∼40–50); although these models would better model the typical time-course of somatic expansion seen in most HD patients, the relatively slow rate of somatic expansion and the limited extent of expansion within the lifespan of a mouse [66] may make some modifier studies impractical. Finally, the ability to query a particular genetic modifier will depend on germ cell or mouse viability when the gene is knocked out or mutated.
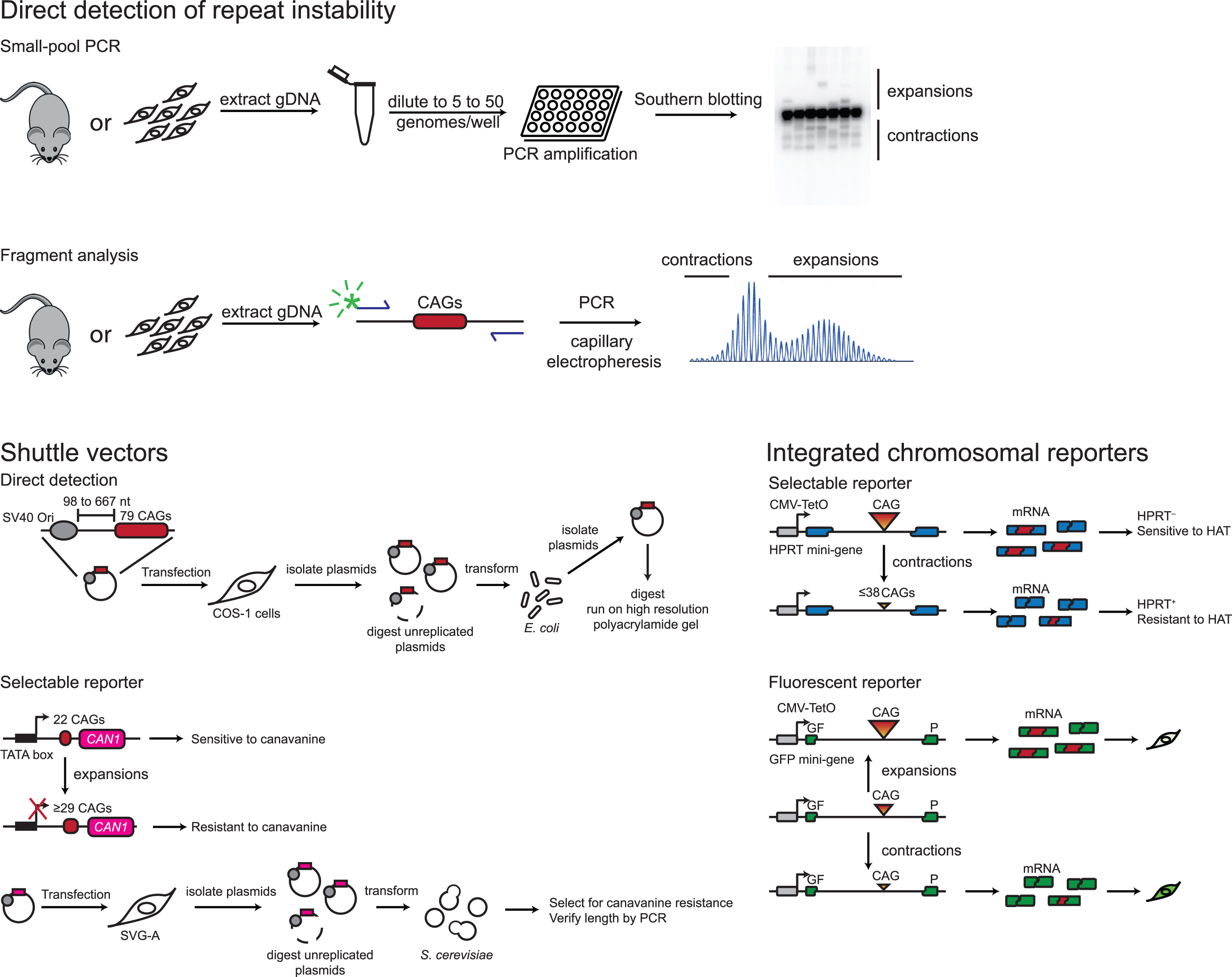
Common assays for measuring repeat instability. Direct detection of repeat length changes is used to analyze instability in mouse models, patient-derived cells and cell-based models harboring repeat tracts at ectopic loci. Small-pool PCR is the current gold standard as it reduces bias against amplification of longer alleles, it is quantifiable, and can detect both expansions and contractions. It is prone to carry-over contamination and is time-consuming. Recently published methods address those concerns [144, 145]. Fragment analysis, e.g., using GeneMapper [111] is quicker and easier than the small-pool PCR, is not as sensitive to rare events but is also quantitative [87]. Methods for studying repeat instability are discussed in detail in [146]. A shuttle vector assay in which repeat length changes are detected directly has been used for mapping cis-acting elements, including origin of replication distance and direction as well as DNA methylation [129, 147]. It has not been used in conjunction with genetic perturbation. Shuttle vector assays using selectable reporters were first developed as a unidirectional contraction assay [148, 149] and later as an expansion-only assay [75, 128]. Two versions of the latter exist, one using the S. cerevisiaeCAN1 gene, the other the URA3 gene. The CAN1 reporter assay has been extensively used for genetic studies (see Table 1). Integrated chromosomal reporters have the advantage of being ultra-sensitive, but limited in the types of events they can detect. The first ones relied on APRT and HPRT function. An expansion assay was developed but was found to have impractically low frequencies of expansions [150]. The expression can be controlled with a doxycycline-inducible promoter [67]. HAT: medium containing hypoxanthine aminopterin and thymidine. An integrated chromosomal reporter based on GFP fluorescence can detect both expansion and contractions, but has not yet been used to uncover genetic modifiers [130, 133].
At the other end of the spectrum are systems in cultured human and primate cells, either utilizing plasmid-based mammalian-E. coli/S. cerevisiae shuttle vectors, or chromosomally-integrated reporters (Fig. 3). In the shuttle vector systems, CAG/CTG-containing plasmids are first introduced into mam-malian cells and, following the experimental perturbation, are transformed into E. coli or S. cerevisiae for read-outs of instability. In one system, which has been used extensively to test genetic modifiers repeat length changes can be determined in S. cer-evisiae based upon a CAG length-dependent resistance to 5-fluorooritic acid (5-FOA) [128] or to canavanine [75]. In another system, repeat length changes are measured by digesting plasmids isolated from single E. coli colonies and run on high-resolu-tion polyacrylamide gels [129]. Shuttle vector sys-tems have the major advantage of requiring only a few days. Integrated chromosomal reporters have been developed based on a CAG length-dependent sensitive hypoxanthine-guanine phosphoribosyltransferase (HPRT) or adenine phosphoribosyltransferase (APRT) activity, which can be selected in the app-ropriate culture media, or based on CAG length-dependent GFP fluorescence. These chromosomal reporter systems require a longer time for read-outs of instability compared to shuttle vectors but offer a higher throughput [67–73, 130–135]. Notably, the largest small molecule screen conducted so far was performed using a selectable chromosomal reporter system and included 880 compounds [72]. The use of selectable markers, in the context of either the shuttle vector or integrated reporter assays allows for the sensitive detection of low frequency instability events, and the ease of combining both of these systems with knockdown, and more recently knockouts, has greatly facilitated the dissection of DNA repair pathways that are involved in CAG/CTG instability in cultured mammalian cells [69, 74–76]. The disadvantages of these systems are that they work in immortalized and rapidly dividing cells, they probe the instability of repeats outside of their endogenous genomic loci, they detect rare events, and are only sensitive to specific types of repeat length changes that can be selected for in the respective assays. For example, the chromosomal reporter systems based on a (CAG)95 repeat in an intron of the HPRT gene or the APRT gene are only sensitive to large repeat contractions that bring the repeat size below a threshold of ∼38 repeats, which is required to restore HPRT or APRT function [132]. Conversely, the human astrocyte cell line (SVG-A)/S. cerevisiae shuttle vector system is only sensitive to expansion of a (CTG)22 tract to 29 or more repeats or more, which alters transcription initiation and blocks expression of the CAN1 or URA3 reporter [75, 128]. Therefore, these systems are unlikely to recapitulate fully what occurs in disease-relevant cell types. Use of a green fluorescent protein (GFP) reporter mitigates one of these issues by enabling the simultaneous read-out of both expansions and contractions while still providing an assay system that is compatible with high throughput screening [130, 134].
Other mammalian cell-based systems are those in which repeats are detected directly by PCR, allowing monitoring of both expansions and contractions that occur at relatively high frequencies. These include cell lines stably transfected with plasmids harboring expanded repeats at ectopic loci [79–82], those derived from mouse models [87, 136] or from patient-derived cells including lymphoblastoid cell lines and fibroblasts [80, 137–139], embryonic stem cells [140, 141], and induced pluripotent stem cells (iPSCs) [82–84]. Pluripotent cells have the major advantage of having the potential to differentiate into many different cell types. In practice, differentiation protocols can often yield a variable fraction of the intended cell type. Modeling repeat instability in patient cells has the advantage of providing direct insight into human-relevant modifiers of instability of repeats in their appropriate genomic contexts. However, these—and all patient-derived cell models—are slow, typically requiring long-term culturing of weeks to months to observe measurable instability. Dividing cells may also introduce selection or clonal artefacts that need to be considered [136, 142]. Differentiating stem cells into specific cell types found to exhibit high levels of instability in mice and patients, e.g., MSNs, may increase the rate of repeat expansion in cultured cells. However, improved differentiation protocols are needed to increase the purity of the desired cell type. Human organoids have not yet been used for the study of repeat instability, but have the potential to model complex tissue environments in the context of human mutations [143].
Despite the disadvantages inherent to all these systems and approaches, each has yielded important insights into modifiers of CAG/CTG instability, contributing to our understanding of potential underlying mechanisms. Many factors, particularly MMR genes, modulate instability across multiple systems, indicating the value of initial high-throughput systems as screening tools to generate candidates for testing in mice or human cells (Table 1).
MISMATCH REPAIR FACTORS
The MMR pathway is best characterized for its role in correcting DNA mismatches generated during DNA replication but has additional functions in DNA recombination and in DNA damage sig-naling (reviewed in [47, 152]). In mammals, MSH2-MSH6 dimers (MutSα) primarily recognize base-base mismatches, whereas MSH2-MSH3 dimers (MutSβ) primarily recognize insertion-del-etion loops. During the process of MMR, DNA recognition is followed by binding of MLH1-PMS2 (MutLα) that mediates the recruitment of downst-ream effector proteins to excise and repair the lesion. Two additional MutL dimers have been described: MLH1-MLH3 (MutLγ) plays a role in meiosis, whereas the role of MLH1-PMS1 (MutLβ) is yet unclear. Mutations in MSH2, MSH6, MLH1 and PMS2 underlie the cancer prone Lynch syndrome in which biallelic mutations result in elevated instability of microsatellite repeat tracts [153]. This observation prompted an early interest in the role of MMR factors in the instability of disease-associated trinucleotide repeats.
A role for MMR pathway genes in CAG instability in an animal model of disease was first demonstrated by Manley et al., in which genetic knockout of Msh2 eliminated somatic HTT CAG expansion in R6/1 exon 1 HD transgenic mice [97]. Constitutive, or striatal MSN-specific, Msh2 knockout in HttQ111 HD knock-in mice abrogated CAG expansion in the striatum [86, 89], demonstrating that MSNs harbored the most highly expanded alleles and that a process dependent on a MMR gene driving CAG expansion was active in post-mitotic neurons. Knockout of Msh2 also suppressed male gametic expansion [98] and inherited repeat length changes in paternal transmissions [86, 88]. The absence of Msh2 promoted repeat contraction in paternal transmissions [86, 88], indicating a distinct role for MSH2 in protecting against CAG contractions. Similarly, in a DM1 transgenic mouse model harboring a long (>300 CTG) repeat tract (DM300-328), Msh2 knockout suppressed expansions and enhanced contractions in both the soma and the germline [106].
Genetic knockout of Msh3 in multiple knock-in and transgenic models implicates MutSβ as the major driver of somatic CAG/CTG expansion [88, 105]. In the germline, Msh3 knockout had a moderate impact relative to that of the Msh2 knockout in HttQ111 mice, but strongly suppressed germline expansions in DM300-328 mice [88, 105]. Msh3 knockout also promoted contractions in DM300-328 mice, indicating a contraction-suppressor role for MutSβ [105]. In a cell-free SV40 replication assay MutSβ also protected against contractions [154], with the suggestion that a contraction-suppressor role may be relevant to dividing cells in vivo. The contrasts between the HttQ111 and DM300-328 models suggest potential differences in MMR-related mechanisms in the soma and germline that might depend on the disease gene context and/or CAG/CTG repeat length. Notably, in both HD and DM1 models, heterozygous Msh3 knockout was sufficient to reduce CAG/CTG expansion, indicating that MSH3 levels are rate limiting in the expansion mechanism [88, 105]. Consistent with this, Msh3 expression levels correlated with CAG expansion in mouse strains harboring naturally occurring Msh3 variants [94] and in a human SVG-A astrocyte cell-based model [78]. The extent to which MSH3 levels contribute to CAG expansion that differs between tissues or between cell types [30, 156] is not well understood and warrants further investigation.
The role of Msh6 is much less clear, with variable effects in mice depending on the model. In HttQ111 mice, Msh6 knockout had no impact on striatal ex-pansions, whereas heterozygous knockout promoted contractions in the male germline [88]. In R6/1 mice, Msh6 knockout slightly reduced expansion in a tissue-specific manner [93]. In DM1 models, Msh6 knockout had either no impact [105] or pro-moted expansion [118] in somatic cells, and suppressed expansions and promoted contractions in the female germline [105]. Some of these effects may be attributable to altered levels of the MutSβ complex as a consequence of reduced MSH6 protein. However, this remains to be tested, and direct roles of MutSα are also possible. Overall the data indicate that although Msh6 is not a key driver of somatic CAG/CTG expansion, it may modulate various aspects of repeat instability with the extent of its involvement perhaps depending in part on the relative levels of MSH6 and MSH3 in different cell types [157]. Cell-based systems, including reporter-based assays that exclusively detect either expansion or contraction events, have reinforced CAG/CTG instability-promoting roles of MSH2 and MSH3 observed in mice, whilst showing minimal impact of MSH6 [67, 83].
In addition to Msh2 and Msh3, both Mlh1 and Mlh3 are absolutely required for somatic CAG expansion in HttQ111 mice [90], implicating MutLγ in this process. Pms2 knockout partially reduced expansions in DM1 transgenic mice, also implicating MutLα [104]. Naturally occurring strain-specific Mlh1 variation was associated with CAG expansion, an effect that could in part be explained by altered MLH1 expression [90]. Both MutLα and MutLγ possess endonuclease activities that, in reconstituted systems, can result in the retention of CAG or CTG loop-outs, representing expansion events [158, 159]. MutLα was also capable of eliminating such loop-outs in this system, representing potential contractions [158]. MutLγ endonuclease activity has been implicated in the expansion of GAA repeats [160]. Note that in cell-based systems, modifier roles of MutL genes were not found [79] or were inconsistent with observations in mice [69]. Importantly, the critical requirement for MutL genes for somatic expansion in mouse models supports the idea that active DNA repair is required to drive CAG/CTG expansion, rather than subversion of a normal MMR process (see below). This is also supported by data in mice indicating that the ATPase activity of MSH2, which is essential for the recruitment of MutL proteins, is required for repeat instability [95]. Similarly, an ATPase mutant in MSH3 behaved like a knockout in a human cell system for repeat expansion [78]. Downstream MMR factors (exonucleases, polymerases, ligases) that are involved in repeat expansion have not been delineated. The identification of LIG1 as a human onset modifier [50], and the knowledge from reconstituted systems that this DNA ligase can function in MMR [161], suggests that it may be part of the MMR mechanism that drives somatic expansion. In mice, Lig1 heterozygosity leads to more expansions and fewer contractions exclusively in the female germline of DM1 transgenic mice [107], and LIG1 expression levels were found to correlate with repeat instability in a cell-free replication assay [162]. Further studies on the role of LIG1 in somatic expansion are of interest.
To gain insight into the mechanism by which MMR proteins promote expansion, several biochemical studies have also been conducted to understand the nature of the repeat substrate that can be bound by MutSβ and the consequences of repeat binding. Both short (1–3 repeat) loop-outs and longer CAG repeat-containing hairpin structures can bind MutSβ [93, 163–166]. Some, but not all, of these studies indicated that CAG hairpins can alter properties of MutSβ binding or activity [93, 166]. These observations have provided some controversy as to whether MutSβ binding to repeats inhibits the nor-mal process of MMR. In cell-free assays that mea-sure the repair of repeat-containing substrates, the repair of short loop-outs was found to be dependent on MutSβ and MutLα [163, 167] and hairpin repair was stimulated by MutSβ [164]. Although these assays do not provide a direct readout of instability per se, these findings support the idea that MutSβ binding to repeat structures stimulates, rather than inhibits, a repair process that ultimately results in expansion, consistent with genetic data in mice. Although CAG/CTG slipped-strand structures have been identified in patient tissues [168], and enrichment of MMR proteins can be observed close to CAG/CTG repeat tracts using chromatin immunoprecipitation [76, 169], the nature of the substrate(s) bound by MutSβin vivo remains unknown. Further studies are needed to tie biochemical observations to repeat instability outcomes.
FAN1: A PROTEIN INVOLVED IN INTERSTRAND CROSS LINK REPAIR
FAN1 is required for the repair of interstrand DNA crosslinks (ICLs), possessing both 5′–>3′ exonuclease activity and a structure-specific endonuclease [170–174]. A potential role in CAG repeat instability was first indicated by human GWAS that identified FAN1 as a modifier of HD onset [49]. Subsequently, it was shown that FAN1 knockout in a human U2OS cell line model containing HTT exon 1 with 118 CAGs, or FAN1 knockdown in HD patient iPSCs with ∼109 CAGs or ∼70 CAGs enhanced HTT CAG expansion [82, 84]. Knockout of Fan1 also enhanced somatic CAG expansion in the striatum and other tissues of HttQ111 mice, in a manner that is dependent on Mlh1 [66]. Significantly, Fan1 knockout also promoted expansion of a 48-CAG repeat tract in HD knock-in mice, indicating that FAN1 normally acts to suppress the expansion of CAG repeats that fall into both the adult-onset and juvenile-onset inherited length ranges of the human disease [66]. Notably, FAN1 physically interacts with MLH1, MLH3, PMS1 and PMS2 [170, 175]. The mechanism by which FAN1 normally suppresses CAG expansion, whether physical interaction(s) between FAN1 and MMR proteins are required in the process of CAG repeat instability and whether additional ICL proteins play a role in repeat instability are currently unknown.
BASE EXCISION REPAIR AND SINGLE STRAND BREAK REPAIR
Base excision repair (BER) is initiated by the detection and excision of damaged bases in the DNA [176]. This is followed by the formation of an apurinic/apyrimidinic (AP) site that is cleaved by APEX1, which leaves a single base pair gap. DNA polymerase beta (POLβ) fills in the missing base pair for a short-patch BER followed by a ligation event or coordinates a long-patch synthesis that involves strand displacement and FEN1 activity in addition to DNA ligases (I and III). Single-strand break repair (SSBR) is often referred to as a sub pathway of BER [177–180] as it uses some of the same proteins, e.g., XRCC1, PARP1, and LIG1 (Fig. 1).
In the context of repeat instability, a chemical screen identified molecules targeting several enzymes in the SSBR pathway including topoisomerase 1, TDP1, and POLβ, to stimulate repeat contractions in a selectable assay [72]. Knockdown of TDP, TOP1 as well as other SSBR genes, including XRCC1 and PARP1, confirmed that SSBR protects against contractions [72]. It is unknown whether these factors influence expansions as well. In a different system with 800 CTG repeats inserted at an ectopic site, individual knockdown of TDP1 or TOP1, had no effect on repeat instability, though their simultaneous knockdown dramatically increased repeat contraction [79]. SSBR proteins have not been investigated beyond these two systems, but downstream factors in the SSBR pathway are shared with other pathways and several are involved in repeat instability (Fig. 1), notably XRCC1 and LIG1.
Extensive studies have been carried out to understand the role played by the repair of oxidized bases in repeat instability. Knockout of Ogg1, encoding a glycosylase that excises oxidized guanines from DNA, reduced somatic expansion in ∼70% of R6/1 mice [100], as well as in HttQ150 knock-in mice [85]. Kovtun et al. proposed a model [100] whereby 8-oxoguanine (8-oxoG) excision from within the repeat tract by OGG1 triggers error-prone repair, involving FEN1 and long-patch BER. This process then leads to expansion either by DNA polymerase slippage or because the secondary structure formed within the displaced flap prevents FEN1 action and is ligated in rather than digested [181]. It has been further suggested that the stoichiometry of FEN1 and POLβ and coordination between these proteins is important in determining the efficiency of gap-filling synthesis that would be expected to promote expansion [182–184]. Recent studies indicate that interaction between POLβ and MutSβ [185, 186] also promotes gap-filling synthesis, suggesting cross-talk between BER and MMR pathways [185]. However, a role for FEN1 that is predicted by this model, and previously shown in S. cerevisiae to protect against CAG/CTG instability, including expansions [187–189], has been more difficult to test directly in mice due to the lethality of the homozygous knockouts. Heterozygous Fen1 knockout had no impact on somatic CAG expansion in R6/1 mice [99] or in DM1 knock-in mice [103]. There is some evidence that Fen1 promotes expansions and suppresses contractions in the male germline [99] and suppresses contractions during DNA replication in a human cell-based model [81] but neither OGG1 nor FEN1 knockdown modified contraction frequency in a transcription-dependent chromosomal reporter assay [67, 68]. The role of POLβ in CAG/CTG expansion in mice is unknown, though it was found to modify FMR1 CGG instability in a mouse model of Fragile X-related disorders [190].
The idea that DNA repair triggered by oxidized bases leads to repeat instability is also supported by the observation that knockout of Neil1, encoding another glycosylase with a preference for oxidized pyrimidines and minimal activity towards 8-oxoG, reduced somatic expansion in R6/1 mice [92]. Oxidized bases or AP sites have also been implicated in CAG/CTG repeat instability in cell-free systems [191–196], supporting a role for oxidative damage triggering repair within a repeat tract. Interestingly, guanines are more susceptible to oxidation when in the loop of a hairpin, yet are inefficiently repaired [191, 196], further supported by the CAG length-dependent accumulation of oxidative lesions within the CAG repeat tract in HD knock-in mice [183]. These data appear to present somewhat of a paradox inasmuch as they imply the lack of repair at repeat tracts, rather than the active repair of oxidized lesions. However, a model has been proposed in which oxidative lesions that arise transiently in susceptible hairpin structures may be inefficiently repaired and thus incorporated into the repeat [196]. It has also been suggested that oxidized bases themselves may be incorporated [192].
A key tenet of the models involving recognition of oxidized bases is a “feed forward” toxic oxidation cycle whereby the accrual of oxidative lesions with aging and disease pathogenesis progressively increases the susceptibility of the repeat tract to error-prone repair [100], thus accelerating the rate of expansion. As the brain is highly susceptible to oxidative stress, and as there is evidence for the further accumulation of 8-oxoG in brains of R6/1 and R6/2 HD mice [100, 192] as well as in HD post-mortem caudate [197], this model provides a potential link between repeat instability and cumulative damage as part of ageing and neurodegeneration. However, additional observations do not support a clear link between DNA oxidation and CAG instability; for example, oxidative lesions within the HTT CAG repeat tract itself were not age-dependent, nor did they correlate with tissue-specific instability in HD mice [183]. In support of this, the extent to which disease pathogenesis itself, including the dysregulation of DNA repair processes that occurs in HD [198–200], contributes to somatic CAG expansion is unclear. Rather, there is evidence to support the idea that the primary driver(s) of somatic CAG expansion are independent of disease manifestation. Indeed, mouse models for clinically distinct diseases show similar tissue-specific patterns of instability [87, 201] and genetic and bioinformatic analyses in HttQ111 mice indicated that the susceptibility of a tissue to CAG expansion was independent of the ongoing pathogenic process [87]. Taken together, therefore, data provide support that the repair of oxidative DNA lesions within the repeat tract can play a role in CAG instability but that they may not be necessary to drive expansion.
NUCLEOTIDE EXCISION REPAIR FACTORS AND TRANSCRIPTION-BASED MECHANISMS
Nucleotide excision repair (NER) removes bulky DNA lesions, including thymine dimers and 6-4 photoproducts [202]. The initial steps of NER differ depending on how the lesion is detected. In cycling cells, the global genome repair branch of NER (GG-NER) detects lesions and leads to the recruitment of TFIIH. By contrast, lesions in transcribed regions will activate the transcription-coupled branch of NER (TC-NER) by causing the RNA polymerase to stall, CSB and CSA to be recruited, followed by TFIIH. The pathways merge with XPA binding to the lesion, followed by helicases and nucleases that remove 24–32 nucleotides around the lesion, leaving a DNA gap, which is filled by a DNA polymerase and ligated.
TC-NER was first found to be involved in repeat instability in immortalized human HT1080 cells that harbored a selectable reporter system to monitor exclusively large contractions [67, 68]. Knockdown of CSA, CSB, XPA, ERCC1, and XPG all reduced the frequency of large contractions in this system. In a Drosophila model of CAG/CTG intergenerational instability, a null allele in the XPG ortholog mus201 suppressed expansions and contractions [203]. It is worth pointing out that CAG instability in Drosophila models has not been shown to be dependent on MMR genes, raising the possibility that some of the mechanisms of instability in Drosophila may not be shared with mammalian systems. Subsequently, using a mouse model for spinocerebellar ataxia type 1 (SCA1), knockout of Xpa was shown to suppress somatic CAG expansion [108]. Surprisingly, Xpa knockout had a tissue-specific effect whereby, of the tissues analyzed, only the neuronal tissues had stabilized repeats [108]. In contrast to Xpa, knocking out Xpc, which is specifically involved in GG-NER, did not impact either somatic or intergenerational instability in HttQ111 mice [88]. This is consistent with the lack of impact of XPC knockdown in a selectable contraction-based reporter assay [67]. CSB was also implicated in repeat instability in R6/1 mice, but only had an effect on intergenerational instability when OGG1 was also simultaneously knocked out [101], again suggesting possible cross-talk between DNA repair pathways.
A role of TC-NER is in line with observations that transcription enhances CAG/CTG instability [67, 204]. It is also notable that many NER prote-ins are part of the core transcription machinery [205]. In transgenic R6 mouse models with distinct sites of transgene integration it was suggested that transcription is necessary for repeat instability [111]. However, transcription does not necessarily account for differences in instability of a particular repeat in different chromatin environments [121, 124]. Further, steady state mRNA level does not correlate with tissue-specific instability across tissues [87, 125], indicating a potentially more complex interaction between transcriptional state and instability. It was suggested that transcription elongation may better determine somatic expansion than transcription initiation in HD mouse models [182]. In support of this, transcription elongator factor TFIIS (TCEA2) promoted instability in cell-based assays [68, 79], and downregulation of BRCA1/BARD1, a ubiquitin E3 ligase that regulates transcriptional elongation, suppressed instability [68]. Instability is also enhanced by bi-directional transcription through repeat tracts in cell-based models [71, 204]. As disease-associated repeats are transcribed in both sense and antisense directions (reviewed in [206]), this also provides a plausible mechanism by which transcription could modulate instability at endogenous repeat loci in patient cells or mouse models, though this remains to be tested.
Transcription through the repeat tract can also lead to R-loops, stable RNA-DNA hybrids that form by annealing of the newly synthesized RNA transcript to the DNA template. They are favored by G- rich RNA and by secondary structure-forming sequences in the displaced single stranded DNA, such as CAG/CTG repeats [32, 207–209]. R-loops are thought to be removed by either NER or dissolved by the action of RNase H enzymes. Indeed, the latter have been implicated in large contractions in human cells, in which knockdown of RNAseH1 or RNaseH2A stimulated instability [71]. Moreover, in a cell-free system, human cell extracts from HeLa and SH-SY5Y cells could process R-loops, promoting repeat instability, an effect that was partially suppressed by treatment with RNase H [207, 208]. In HT1080 cells harboring 800 CTG repeats, knockdown of SETX (Senataxin), a putative RNA/DNA helicase with a function in resolving R-loops, stimulated repeat contractions [79]. Interestingly, R-loop mediated CAG/CTG repeat instability in S. cerevisiae was found to be dependent both on cytosine deamination and BER, and on MutLγ nuclease activity [210]. These studies suggest that there may be multiple pathways by which R-loops at CAG/CTG tracts can be processed.
Although not thoroughly tested, transcription-based mechanisms are attractive in the context of post-mitotic neurons as transcription-associated re-pair of DNA lesions remains active in the absence of replication [211]. Potential intersection with R-loop biology, TC-NER, and other DNA repair pathways is an interesting area of further investigation.
ROLES OF ADDITIONAL DNA REPAIR OR REPLICATION FACTORS
In contrast to the involvement of MMR, BER, and NER-related mechanisms in various aspects of CAG/CTG instability, there has been little support for a prominent role of double strand break (DSB) repair mechanisms in repeat instability in mammalian cells. Although RAD51, involved in homologous recombination (HR), protected against large contractions [73], there was no impact of knocking out HR genes Rad54 and Rad52 on CTG somatic expansion in a DM1 mouse model [106]. Rad52, but not Rad54 knockout did increase the size of intergenerational expansions in this model, potentially implicating single-strand annealing (SSA) mechanism of DSB repair, in which RAD52 is also involved. Non-homologous end-joining (NHEJ) is the most commonly used DSB repair pathway in mammals. This has not been studied in depth in the context of repeat instability; however, knockout of NHEJ gene DNAPKcs had no impact on somatic or intergenerational instability in DM1 transgenic mice [106]. CTIP and MRE11, genes involved in the sensing and signaling of DSB repair, did not modify CAG expansion in a human selectable assay [76]. BRCA1, which enhanced contractions in a selectable assay [68], has diverse roles that include transcription, coordinating repair of DBSs, HR, as well as ICL repair [212]. Given the multifunctional nature of BRCA1, it is plausible that in the unusual context of expanded repeats, this protein might also intersect with other DNA repair mechanisms.
Several modifier genes have been identified that influence CAG/CTG instability in the context of DNA replication. Replication-based mechanisms may be more important in rapidly dividing cells, including the germline and during early embryonic development. In human cells, the RTEL1 helicase blocked repeat expansions [74]. It is proposed to act via its hairpin unwinding activity, together with HLTF, a nucleosome remodeling factor and RAD18, an E3 ubiquitin ligase, both of which suppress expansions [74]. RTEL1 appears to be a functional homolog of the Srs2 helicase, originally found in S. cerevisiae to inhibit repeat expansion [213], and for which there are no sequence homologs in metazoans. It was proposed that RTEL1/HLTF/RAD18 acts to prevent repeat expansions during post-replication repair mechanisms that allow replication forks to progress through lesions on damaged templates [74]. In a HeLa cell-based model containing an ectopic integrated cassette that includes the c-myc replication origin and CAG/CTG repeats, as well as in DM1 fibroblasts, knockdown of one of CLSPN, TIMELESS, or TIPIN, which all play a role in replication stress and fork stabilization, resulted in a substantial increase in repeat contractions [80]. Whether any of the factors that appear to sense or resolve CAG/CTG-mediated replication stress play a role in modulating somatic or intergenerational repeat instability in mouse models is unknown.
CIS ELEMENTS, CHROMATIN, AND POST TRANSLATIONAL MODIFICATIONS IN REPEAT INSTABILITY
Some instability modifiers provide insight into ways in which DNA metabolic processes that influence repeat instability might be regulated. In addition to factors that act in trans, there is evidence that the instability of CAG/CTG repeats can be modulated in cis by DNA elements other than repeat length, repeat sequence and purity (reviewed in [206, 214]). For example, the expansion propensity of different disease-associated CAG/CTG repeats correlates with the GC-content of the flanking DNA [123, 216]. Interestingly, there is an association between ancestral HTT haplotype and the mean length of the unexpanded CAG repeat, leading to the speculation that certain HTT haplotypes may be predisposed to CAG instability due to cis-acting elements [217]. In mouse models, the genomic contexts of transgenes or knock-in alleles can modulate instability [111, 123–125]; notably, repeats flanked by genomic DNA are more unstable than those within cDNA transgenes [123, 124]. Insights into the precise nature of the cis-factors and the mechanisms by which they modulate repeat instability are currently very limited. Chromatin context regulates DNA repair, transcription and replication [218–220], and chromatin marks, including trimethylation of histone H3 at Lys36 (H3K36me3), and chromatin-remodeling factors have recently been shown to regulate MMR (reviewed in [221]), indicating possible routes for modification of CAG instability in cis. Replication fork dynamics, which can influence instability, are also influenced by cis-factors [222].
Highly expanded trinucleotide repeats tend to be associated with epigenetic marks of heterochromatin such as DNA methylation and methylation of histone H3 at Lys9 (H3K9) (reviewed in [206]). Greater levels of DNA methylation at the DMPK CTG repeat locus were also found in tissues exhibiting greater somatic expansion [223]. Interestingly, disease-associated repeat loci were shown to localize to topology-associated domains (TAD) and subTAD chromatin boundaries [224]. CGG repeat expansion at the FMR1 locus disrupted this boundary [224]. However, in a different study, 4C sequencing of the HTT and DMPK loci did not identify any changes in chromatin structure in the presence of expanded repeats [225]. The extent to which local epigenetic changes associated with expanded repeats might themselves contribute to repeat instability is unknown. In model systems global demethylation destabilized repeats [147, 226], and Dnmt1 deficiency promoted germline CAG instability in a mouse model of SCA1 [70]. In these cases, instability modification may be due to local DNA methylation changes, to the altered expression of other genes that modulate instability in trans, or both. More direct evidence for a cis-modifier effect comes from SCA7 mouse models in which mutation of a CTCF binding site adjacent to the Atxn7 CAG repeat promoted instability in specific tissues, notably in the kidney [124]. A similarly high level of expansion in a kidney was observed in a mouse with a wild type, but methylated, CTCF binding site. These results suggested a model in which CTCF protects against repeat expansion in a methylation-dependent manner [124].
Other proteins with chromatin-modifying activities also influence repeat instability. In a selectable assay for CAG expansions in cultured astrocytes, knockdown of histone deacetylase genes HDAC3 or HDAC5 suppressed expansions, whereas knockdown of histone acetyltransferase (HAT) gene CREBBP, encoding CREB-binding protein (CBP) or EP300, encoding the related P300 protein, promoted expansions [75, 76]. Loss of CBP also promoted CAG expansions in the Drosophila germline [203]. These genetic data are supported by the suppression of instability upon treatment with the Class I/II HDAC inhibitor trichostatin A (TSA) [203]. Genetic knockout of either Hdac2 or Hdac3 in MSNs of HttQ111 knock-in mice moderately suppressed striatal expansions [91]. The impact of Hdac3 knockout in this model is consistent with the expansion-suppressing effect of a selective HDAC3 inhibitor [227]. Relationships between HDACs, HATs and repeat instability appear to be complex, however. For example, knockdown of HDAC9 promoted, rather than suppressed, CAG expansion in the cultured astrocyte model [76], and HDAC inhibitors promoted contractions in a sel-ectable human cell-based assay [91]. In S. cerevisiae, loss of function of different HDACs either enhanced or suppressed CAG instability [75, 228], with CAG stability being dependent on activities of both HDACs and HATs that control levels of H4 acetylation [228]. As in the case of DNA methylation, HDACs or HATs may modify repeat instability via epigenetic changes in cis and/or by altering the expression or activity of trans-acting factors. A trans-mediated effect was proposed to account for the impact of CBP loss in Drosophila [203]. In human astrocytes, epistasis experiments provided evidence that HDAC3 and MSH2 act in a common pathway to promote expansion [76]. Further molecular experiments indicated that HDAC3 knockdown did not alter the binding of MutSβ at the repeat tract, or the expression level of MSH2 or MSH3, together suggesting an alternative hypothesis that HDAC3 modulates instability by regulating MSH2 or MSH3 acetylation. Indeed, activities of both MSH2 and MLH1 can be altered by acetylation [229–231], indicating that this is a plausible mechanism for instability modification by HDACs and HATs. Recent studies show that MSH3 peptides can be deacetylated by HDAC3 and that the nuclear localization of MSH3 is regulated by a selective HDAC3 inhibitor [232]. Roles of histone modification in cis contributing to instability have not been demonstrated directly in mammalian systems; however, in S. cerevisiae a protective effect of chromatin remodeler Isw1 against CAG expansion during transcription could be attributed at least in part to a direct effect at the repeat tract as nucleosome occupancy over the CAG tract was altered in isw1 mutants [233].
CELLULAR STRESSORS IN REPEAT INSTABILITY
Cellular stresses can also induce changes in repeat instability. DNA damaging agents could work by inducing damage to the repeat tract directly (see section below), indirectly causing cellular stress, or both. For instance, Gorbunova et al. [132] treated cells with hydroxyurea, which depletes dNTP pools and slows DNA replication, ionizing radiation, which causes DNA breaks, or aphidicolin, an inhibitor of DNA polymerase α. In this early example, these treatments induced large contractions, with unknown effects on expansions. Other studies in cell-based models have found effects on CAG/CTG instability of other DNA damaging compounds that include DNA intercala-ting agents, nucleoside analogues, and hydrogen peroxide [72, 234]. These experiments can often be difficult to interpret due to effects of DNA damaging compounds on the cell cycle and potential clonal selection that could artifactually alter the distribution of CAG-containing alleles in a population without modifying repeat instability. Other types of cellular stresses that alter repeat instability include cold and heat shock [135], and pharmacological and genetic suppression of the proteasome [68, 77] or of HSP90 [73]. Cellular stressors could act indirectly by altering levels or activities of DNA repair proteins that control repeat instability and may provide a means to stimulate repeat instability to facilitate analyses in mammalian cell-based models where the natural time-course of somatic expansion is slow.
DIRECT PERTURBATION OF THE REPEAT TRACT
Alternative routes to modifying repeat instability involve direct perturbation of the repeat tract. Of the small molecule studies conducted thus far [72, 236], only one compound has been shown to engage directly the repeat tract at the DNA level. Indeed, Nakamori et al. [236], found that naphthyridine-azaquinolone (NA) can bind to CAG loops, stabilize them, and induce contractions and/or prevent expansions in human cell culture as well as in the R6/2 HD mouse model. Overall, there has been no small molecule that is non-toxic and efficiently prevents expansions or induces contractions at doses that are physiologically relevant. It will therefore be exciting to see whether NA can be turned into a viable therapeutic. Antisense oligonucleotides (ASOs) are being using in pre-clinical and clinical settings to reduce the toxic huntingtin protein or the toxic DMPK mRNA [237–239]. Interestingly, an ASO against the repeat tract itself was seen to reduce repeat instability through an unknown mechanism, though direct binding to the repeat is one possibility [238].
Directly damaging the repeat tract, by inducing a DSB within or very near it has been used to induce instability in yeast and mammalian cells [81, 240–245]. These studies have shown that homologous recombination and the DNA damage response are both involved in generating repeat instability. It is not clear that the same modifiers that affect repeat instability in the absence of a direct assault on the repeat tract, will be involved in these more artificial systems. Indeed, as pointed out above, several factors involved in DSB repair do not seem to influence repeat instability in mammalian models [106]. Nevertheless, directly damaging repeats may be harnessed for gene editing and may therefore have therapeutic relevance.
The first generation of customizable gene editing tools were Zinc Finger Nucleases (ZFNs). ZFNs consist of two FokI nuclease domains that must come together to induce a DSB at the target sequence [246]. Both halves are fused to customizable DNA binding domains, each targeting a DNA triplet. In the context of expanded repeats, they were first used in a chromosomal reporter assay that measures large contractions [245]. They led to a 14-fold increase in the number of contractions and induced rearrangements near the repeat tract. Another group, using expanded repeats inserted in the genome of HeLa cells, also found that they efficiently induced contractions [81]. Interestingly, in both studies, one of the two halves of the ZFN was shown to induce instability on its own. The first study suggested that one of the studies could not distinguish effectively between the CAG and CTG strands and thus cut both [245]. The authors of the second study rather postulated that the ZFN could cut out the secondary structures formed by the repeat tract, which led to the instability [81].
Transcription activator-like effectors (TALEs) were the basis for the second generation of programmable nucleases. They have the advantage that each DNA binding domain binds a specific base pair, making their design easier, although repetitive [247, 248]. Fusing the FokI nuclease to them as with the ZFNs allows for precise induction of a DSB in the human genome [249]. TALENs have been used against expanded CAG/CTG repeats in yeast to great effect [240, 241], but they are yet to be tried in mammalian systems.
CRISPR-Cas9 as a gene editing system was described in 2012 and has since revolutionized the way we manipulate genomes [250]. There are two major advantages of the CRISPR systems over other programmable nucleases. One is efficiency, the other is the ease of design. Indeed, Cas9, a nuclease, is guided to a sequence of choice using a single guide RNA (sgRNA) that contains a target sequence of choice [250, 251]. The most widely used Cas9, from Staphylococcus pyogenes, requires a protospacer adjacent motif (PAM) composed of three nucleotides, NGG, where N is any nucleotide, required abutting the target sequence [252]. Thus, any sequence in the genome that abuts a usable PAM can be targeted and cut. Cas9, directed to the repeat tract itself, has been used to induce DSBs within the repeat tract in the context of a chromosomal reporter [134]. This led to a modest amount of expansions and contractions in roughly equal proportion, perhaps due to the suboptimal CAG and CTG PAMs. Interestingly, inducing a DSB near, but not within, the repeat tract led to a large increase in repeat instability, including expansions, contractions, and rearrangements, in immortalized mouse myoblasts from a transgenic DM1 mouse model containing 500 CTG repeats [253] as well as in yeast [254]. Together, studies inducing DSBs within the repeat tract suggest that they cause both expansions and contractions as well as larger rearrangements near the repeat tract.
The use of the CRISPR system is ideal to test whether different types of DNA damage induced within the repeat tract could lead to changes in re-peat instability. This was tried by Cinesi et al. using a D10A mutant of the Cas9 enzyme [134]. This mutation turns Cas9 into a nickase, which cuts only one of the two strands of DNA, i.e., it introduces nicks. In this case, the same sgRNA was used, targeting the CAG/CTG repeat itself, enabling the comparison between DSBs and nicks. Surprisingly, the nickase induced predominantly contractions of the repeat tract, with up to a third of the cells having a contrac-tion after 12 days of treatment. There were no rearran-gements seen beyond those observed in the untreated population. Moreover, the effect of the nickase was repeat length-dependent with mildly expanded or short alleles not being targeted. In line with this, the number of off-target mutations at other endogenous, non-pathogenic, CAG/CTG repeat loci was below the detection level of the assay used [134]. The nickase remains to be tested in other systems, however.
CONCLUSIONS AND PERSPECTIVES
Through a variety of approaches and model systems considerable progress has been made in delineating factors that can modify the instability of CAG/CTG repeats. These include DNA repair proteins that act in pathways to enhance or suppress instability and reagents that interact directly with the repeat tract. Emerging data hint at possible mechanisms by which DNA repair pathways might be regulated, for example through interaction with cis-elements or by altering levels of DNA repair proteins or their activity, e.g., via post-translational modification. This is an interesting area of future investigation. Means by which repeat instability could be modulated are summarized in Fig. 4, indicating plausible routes for therapeutic intervention.
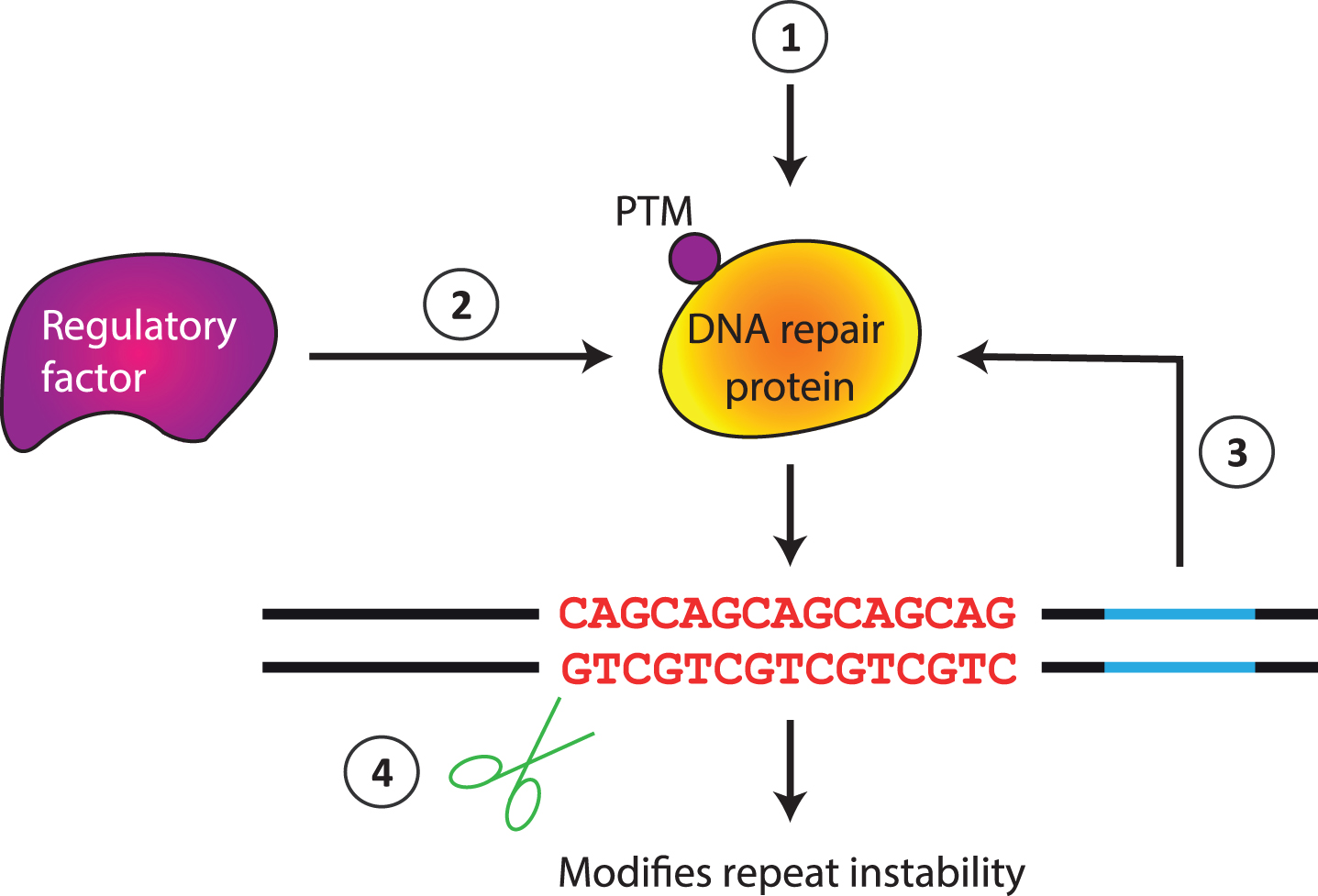
Potential mechanisms for instability modifiers. Genetic or pharmacological manipulation can directly alter the level of activity of a DNA repair protein (1). Indirect effects are also possible via a regulatory factor that controls DNA repair gene expression, stability or activity (2), e.g., altering a posttranslational modification (PTM), or via interaction with a cis-element (3). The repeat can also be modified by agents that directly interact with the repeat (4).
The mechanism(s) underlying repeat instability, however, remain poorly understood, but they are important to delineate in order to refine therapeutic approaches. For example, if DNA repair proteins act in non-canonical ways to modify repeat instability, this may provide opportunities for interventions that specifically target the repeat instability mechanism, whilst minimizing the impact on DNA repair processes more globally. Relatively few of the modifiers uncovered in simpler cell-based assays have been tested in mouse models, and their significance in human disease is unclear. Although a process dependent on MMR genes is clearly critical for somatic CAG/CTG expansion in mouse models, potential intersections with factors in other DNA repair pathways such as BER or NER are not well understood. We highlight several questions and approaches to be addressed in future work.
First, how do mechanisms of instability differ between cell types, for example between the germline and soma, or between the brain and periphery? A number of studies indicate that mechanisms may not be the same [57, 255], and although many proteins function in repeat instability across cell types, factors may be specifi-cally or preferentially involved, in a cell type-depen-dent manner. Several factors have been proposed to contribute to the tissue/cell type-specificity patterns of somatic expansion (reviewed in [109]), however the molecular underpinnings are unknown. Understanding the nature of these intrinsic cell type-specific instability drivers will also provide clues to underlying mechanisms. Differences in mechanism between post-mitotic and dividing cells are also likely, and in the germline where instability can arise at multiple stages during spermatogenesis in humans [6], processes in both dividing and non-dividing cells may be relevant. Second, although much has been learned about factors involved in somatic expansion, we know surprisingly little about repeat contraction. This is a significant question to address as promoting repeat contraction would clearly be an important therapeutic goal. Third, the majority of the genetic studies in model systems involve the knockdown or knockout of specific genes. Mechanistic insight will be enhanced by further studying the impact of functional variation, for example to test the roles of enzymatic activities of DNA repair proteins [78, 95]. Of particular significance are the functional variants discovered though human GWAS that will inform on ways in which instability can be modula-ted that, by definition, would be predicted to have an impact in patients. Fourth, what are the substrate(s) recognized by the DNA repair machinery in vivo? For example, is the substrate for MMR proteins simply an insertion loop that is recognized by the MutSβ complex in the same way this complex would recognize any other insertion loop, or does MutSβ interact with a CAG/CTG substrate in a distinct manner? Finally, there is currently a lack of connection between biochemical assays that are conducted in cell free systems and instability that occurs within cells. Developing systems to bridge this gap that would enable structure/function studies would be of value.
Novel and emerging technologies, including CRISPR-based systems, single cell analyses and next-generation sequencing will enable the development of additional platforms for modifier screening/testing and a more comprehensive knowledge of the pathways and mechanisms that control repeat dynamics. Further, as current methods provide a steady state snapshot of repeat instability, innovative methodologies, including computational modeling, will be needed to capture the dynamic nature of the unstable repeats [256, 257]. Ultimately, a greater understanding of repeat instability will provide additional therapeutic avenues in HD, DM1, and other repeat expansion disorders.
CONFLICT OF INTEREST
V.C.W. is a scientific advisory board member of Triplet Therapeutics, a company developing new the-rapeutic approaches to address triplet repeat disorders such Huntington’s disease and Myotonic Dystrophy. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. She is a scientific advisory board member of LoQus23 Therapeutics and has provided paid consulting services to Alnylam and Acadia Pharmaceuticals. V.D. is a named inventor on patent application WO2017178590A1 involving the use of the Cas9 nickase for gene editing of expanded CAG/CTG repeat disorders.
Footnotes
ACKNOWLEDGMENTS
We thank Emma L. Randall and Alysha Taylor for help with Fig. 2. Oscar Rodríguez-Lima provided the small-pool PCR figure and Ricardo Mouro Pinto provided the GeneMapper trace, both found in ![]() . Work in V.D.’s lab is supported by the Academic of Medical Sciences (AMSPR1\1014) and by the UK Dementia Research Institute, which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. V.C.W is supported by grants from the National Institutes of Health, the CHDI Foundation, Pfizer Inc. and the CCXDP, with previous support from the Huntington’s Society of Canada.
. Work in V.D.’s lab is supported by the Academic of Medical Sciences (AMSPR1\1014) and by the UK Dementia Research Institute, which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. V.C.W is supported by grants from the National Institutes of Health, the CHDI Foundation, Pfizer Inc. and the CCXDP, with previous support from the Huntington’s Society of Canada.