
Abstract
There is an unmet clinical need for objective biomarkers to monitor disease progression and treatment response in Huntington’s disease (HD). The aim of this review is, therefore, to provide practical advice for biomarker discovery and to summarise studies on biofluid markers for HD. A PubMed search was performed to review literature with regard to candidate saliva, urine, blood and cerebrospinal fluid biomarkers for HD. Information has been organised into tables to allow a pragmatic approach to the discussion of the evidence and generation of practical recommendations for future studies. Many of the markers published converge on metabolic and inflammatory pathways, although changes in other analytes representing antioxidant and growth factor pathways have also been found. The most promising markers reflect neuronal and glial degeneration, particularly neurofilament light chain. International collaboration to standardise assays and study protocols, as well as to recruit sufficiently large cohorts, will facilitate future biomarker discovery and development.
INTRODUCTION
Huntington’s disease (HD) is an inherited neurodegenerative disorder caused by a CAG triplet repeat expansion in the gene encoding huntingtin [1]. HD is clinically characterised by a variable phenotypic expression of motor, cognitive and psychiatric symptoms [2], with typical age-at-onset in the thirties or forties [3] and a slow disease progression [4]. The age-at-onset, severity and duration of disease are very variable and depend on CAG repeat length as well as other genetic and environmental factors [3]. Although many clinical features of HD can be ascribed to the dysfunction and death of neurons, the causative mutant huntingtin protein is expressed ubiquitously and evidence is emerging of a role for non-neuronal tissues in the pathogenesis of HD [5–7].
Biomarker definition
The term biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [8]. Therefore, a biomarker can be related to the disease itself or to a treatment effect. Various biomarker categories are defined in Table 1 (based on BEST (Biomarkers, EndpointS, and other Tools) Resource [9]). An ideal disease biomarker should be closely linked to pathophysiology, reliable, accurate, sensitive, specific, reproducible, inexpensive, non-invasive and acceptable for the patient [10].
Biomarker categories and definitions
Huntington’s disease biomarkers: Unmet needs
At present there are no effective treatments in clinical practice that will prevent the disease, halt its progression or delay its onset [11–13], but as the future holds promise for disease modifying strategies [14], there is a need for reliably assessable disease progression biomarkers, as well as markers to evaluate therapeutic interventions. As a chronic progressive disorder, HD has a premanifest phase during which no motor signs are present, but the underlying pathological processes are ongoing [15, 16]. The possibility of genetic testing in HD opens up the possibility of evaluating disease-modifying treatments already in the premanifest phase. Thus, biomarkers that reflect underlying disease processes or progression are needed to monitor changes in clinically asymptomatic individuals [17]. Biomarkers providing an objective measurement for the assessment of HD severity would also be valuable in the clinical care of existing HD patients and might prove particularly useful in a clinical trial context, potentially serving as surrogate endpoints. In 2004, the U.S. Food and Drug Administration (FDA) emphasised the importance of biomarkers in improving drug development efficiency [18] and continues to promote research on potential new biomarkers to use as surrogates in future trials [19]. Since then, there has been an increase in the use of biomarkers in all phases of drug development. Impaired proteasome activity [20], transcriptional dysregulation [21], oxidative stress [22], mitochondrial and metabolic dysfunction [23], neuroinflammation [24, 25], and microglial activation [26–28] have been shown to play important roles in the pathogenesis of HD and many biomarker studies have been undertaken that reflect these processes (clinical, imaging and biofluid). Although much data has been generated, the road to clinical application is long and validation of the intended use in independent cohorts is challenging. To help identify the most promising candidate biomarkers, we reviewed “wet biomarker” literature in HD published to date. Here, we are defining a “wet biomarker” as a potential biomarker that is objectively measured in a bodily fluid. We have limited our focus to studies in blood, urine, saliva and CSF and have excluded DNA, RNA and microRNA studies. While these studies are also of great interest, they require a different set of criteria from markers discussed here and are beyond the scope of this review. We have built our discussion around a table giving an overview of wet biomarker studies to date. The supplementary table is an unbiased summary including all studies regardless of methodology and reproducibility, describing the markers evaluated, the number of subjects, disease stage and the main finding of the study. At the outset, our intention was to also include a table showing “validated” wet biomarkers that have been consistently reproduced and validated longitudinally; however, only one of the markers reviewed here met these criteria. None of the studies to date have yielded biomarkers for accurately predicting either age at onset or disease progression for HD. Since the first step towards clinical implementation of a newly discovered biomarker is independent replication, we focus on biomarkers that have been validated in at least two independent cohorts and we discuss recent data challenging earlier findings. The goal of this critical review is to highlight the most promising markers for future validation studies and to help investigators design future biomarker studies in HD.
METHODS
A literature search was performed using the Pubmed electronic database up to the 17th of February 2018 employing the following search terms: “Huntington’s disease” or “Huntington disease” and “saliva” or “urine” or “blood” or “plasma” or “serum” or “cerebrospinal fluid” or “CSF” and filtering on English language articles. The search yielded 1351 results 1 . Abstracts were screened for relevance and studies assessing biofluid markers in HD were included in the final list, with the exception of hormone function tests since they are measuring the response to an activating stimulus. In addition, we checked reference lists published in review articles and other scientific reports and thus added additional references to the final list that were considered relevant and not retrieved by the PubMed research. The final list contained 160 studies.
REVIEW OF WET BIOMARKER LITERATURE IN HUNTINGTON’S DISEASE
Supplementary Table 1 is an unbiased summary of wet biomarker literature in HD, showing analytes that were tested in biofluids as potential biomarkers for HD. Supplementary Table 1 contains the following information: PMID, analyte, type of sample (e.g., CSF), number of subjects the data is based on (and whether the controls are matched), significant correlations with clinical measures (or lack of correlation), method (technical platform, time of sample collection, fed/fasted status, and whether the subjects are medication-free), first author, last author and year of publication. Out of these, 24% were tested only once and found to be significantly different in HD compared to control, and 34% were tested only once with no significant difference reported. Of the analytes tested, 22% were tested on more than one occasion and were found to be similar in HD and control cohorts (Table 9). We considered all relevant biomarker studies, and divided the biomarkers into several categories reflecting the strength of evidence; this classification was primarily based on the number of replication or validation cohorts and the number of studies that confirmed the findings (described in the “codes” tab of the supplementary Table). The markers that are most promising based on this selection criteria are subdivided into traditional functional classifications in Tables 2 to 7 and are discussed below.
Immune markers in HD compared to control
Analytes were measured in blood, unless otherwise specified. Abbreviations: 55-kDa-type soluble tumour necrosis factor receptor (sTNF-R). C-reactive protein (CRP). Interleukin-6 (IL-6). Soluble interleukin-2-receptor (sIL-2R).
Immune markers (Table 2)
Both innate and adaptive immune systems have been suggested to play a role in HD pathology [29] and a number of studies have highlighted the existence of a peripheral immune response in HD patients. An unbiased proteomics screen of HD plasma identified immune proteins that are elevated in HD compared to healthy controls [30], including proinflammatory cytokine IL-6, acute-phase protein alpha-2-macroglobulin, complement factors and a complement inhibitor clusterin (Table 2). The authors hypothesised that IL-6 induces the release of acute-phase proteins and this in turn leads to the activation of the complement cascade and modulating factors such as clusterin [30]. A follow-up study demonstrated that immune changes are apparent even during the preclinical stage of HD [24] with IL-6 increased even in premanifest subjects, presenting the possibility that inflammatory markers in plasma can be used to track HD. The findings of increased IL-6 levels in HD plasma were replicated in three independent studies [31–33], however, no change has also been reported [34–36] (Table 2).
Acute-phase proteins alpha-1 antitrypsin [37], alpha-2-macroglobulin [30, 37] and C-reactive protein (CRP) [31, 37–40] have been investigated as potential biomarkers in HD. One study found decreased levels of CRP in one HD cohort and no significant difference in another [37], whereas several other studies reported raised CRP levels in HD subjects [31, 40] (Table 2). Increased CRP level may reflect an acute-phase response due to mutant huntingtin expression in HD. However, as HD mutation carriers that use antipsychotics are prone to develop an acute phase response [39], it is possible that the CRP increase reflects the use of antipsychotics by HD subjects studied. It still remains to be confirmed whether CRP is indeed increased in HD and if yes, whether this increase tracks with disease progression.
Neopterin is synthesised by macrophages following interferon gamma stimulation and is a marker of cellular immune system activation. Several studies have consistently reported increased neopterin levels in late stages of HD [40–43]; however, no follow-up studies in early stages of the disease have been performed.
Metabolic markers (Table 3)
Metabolic markers in HD compared to control
Analytes were measured in blood, unless otherwise specified. Abbreviations: 24S-hydroxycholesterol (24OHC). 3-hydroxyanthranilic acid (3HAA). 5-hydroxytryptamine / Serotonin (5-HT). Apolipoprotein A4 (ApoA4). Phosphatidylcholine acyl-alkyl C36:0 (PC.ae.C36.0). Premanifest HD (P).
It is reported that there is a negative energy balance in HD [44] and many of the peripheral manifestations of HD, such as weight loss [45] and muscle wasting are indicative of metabolic alterations. This is reflected in the fact that many of the analytes tested as potential biomarkers for HD are metabolites (Table 3). Advances in metabolomics provide the opportunity to exploit this catabolic phenotype to discover novel biomarkers for HD. An untargeted metabolomic study using gas chromatography and mass spectrometry to analyse serum from control subjects, premanifest and early stage HD found metabolic alterations associated with a catabolic phenotype [46], thus confirming longstanding reports that catabolic changes in amino acid metabolism occur before onset of symptoms [47]. Levels of branched chain amino acids (valine, leucine and isoleucine) have been consistently reported to be correlated with weight loss, disease progression and abnormal triplet repeat expansion [36, 46–55] (Table 3). However, there are also a number of studies that report no change in branched chain amino acids [38, 56–58].
Many studies have investigated markers of lipid metabolism, in particular, cholesterol metabolism in HD. Most studies have shown no alteration in peripheral levels of total cholesterol, HDL-cholesterol and LDL-cholesterol [57, 59–64], with the exception of Wang et al. [34] that showed a reduction of all three analytes in premanifest as well as manifest HD subjects. The major brain cholesterol metabolite 24(S) hydroxycholesterol (24OHC) reduction has consistently been observed in plasma of individuals with HD [59, 66] and was paralleled by the reduction of caudate volume, suggesting that the reduction of 24OHC may reflect progressive neuronal loss [59] (Table 3). Additionally, HD patients’ plasma also showed reduced levels of cholesterol precursors lanosterol and lathosterol, and of the bile acid precursor 27-hydroxycholesterol [65]. Future studies need to follow subjects longitudinally to examine the rate of change of metabolic markers as the disease progresses.
Endocrine markers (Table 4)
Endocrine markers in HD compared to control
Analytes were measured in blood, unless otherwise specified. Abbreviations: Insulin-like growth factor 1 (IGF-1). Growth hormone (GH). Premanifest HD (P). Early HD (E). Transthyretin (prealbumin).
In the tuberal nucleus of the lateral hypothalamus of HD patients there is progressive neuronal death that could have an impact on the function of most of the pituitary axes [67–72]. Consequently, numerous neuroendocrine studies have been carried out in HD, although sometimes with conflicting results (Table 4).
Melatonin
One neuroendocrine marker that shows great promise is melatonin (Table 4). Three studies that analysed 24-hour profiles of melatonin secretion have reported melatonin alterations in HD subjects [73–75]. One study found a delay in evening rise of melatonin in HD [74], whereas another found reduced melatonin levels and reported that the evening rise of melatonin was significantly more temporally spread in both premanifest and stage II/III HD subjects [75]. A study measuring melatonin at a single time point (in the morning after an overnight fast) in advanced HD patients found no difference compared with controls [43]. In the three studies measuring 24-hour levels of melatonin, morning melatonin levels were similar in controls and HD subjects [73–75], highlighting the fact that single measures cannot reflect the dynamic range of melatonin over 24 hours. Thus, when it comes to measuring levels of markers that have a circadian rhythmicity it may be necessary to examine the 24-hour profile in the first instance to see if the circadian rhythm is disrupted and to determine at what time of day the marker is most informative. As such, studies measuring hormones at only one time-point may not provide enough information.
Cortisol
Abnormal hypothalamic-pituitary-adrenal (HPA) axis function has been reported in HD mouse models and in HD patients [76–81]. HPA axis hyperactivity has been reported in early disease stage, primarily in the morning and early afternoon period [76] and elevated salivary cortisol awakening response (CAR) was found in premanifest HD mutation carriers compared to diagnosed HD individuals [82, 83], but no significant difference was found in overall parameters of HPA axis activity between HD mutation carriers and controls (Table 4). Several other studies also reported no significant difference in cortisol levels between mutation carriers and controls [84–86]; however, in two of these studies morning cortisol levels were higher in premanifest HD subjects compared to symptomatic HD subjects [84, 86]. It is difficult to directly compare studies that examined HPA axis functioning in HD because various biofluids were used and this can lead to different results. Also the studies were inconsistently adjusted for potential confounders such as smoking status, alcohol consumption, BMI, use of psychotropic medication and presence of depression. In addition, some studies recruited HD family members as the control group [82, 84], and it may be that this control group also has raised cortisol levels compared to the general population [82]. Also, due to circadian rhythmicity in secretion, it may be necessary to examine the 24-hour profile of cortisol in order to fully understand possible alterations in the secretion profile. Taken together these studies seem to indicate that there may be a hyperactivity of the HPA axis in the premanifest stage, as suggested by increased CAR and morning cortisol levels [76, 86]. Studies have also shown that approximately 30% of people with depression have increased cortisol secretion [87–90] and there are some indications that HPA axis alterations may contribute to depressive symptoms during early stages of the HD [82, 86]. Only one of these studies was longitudinal [82], thus more longitudinal studies are needed to elucidate whether there is indeed disturbed HPA axis functioning that varies with disease stage and what influence this has on depressive symptoms.
In the future it may be beneficial to also measure cortisol levels in hair because this offers a longer-term view of systemic cortisol exposure than biofluids such as saliva, urine or blood; a centimetre of hair shows the average cortisol over a month [91].
Growth hormone (GH)
Morning fasting GH levels were reported to be higher in stage I/II HD compared to controls suggesting early dysfunction of the somatotropic axis, and this increased somatotropic activity was associated with disease severity [81]. However, studies analysing GH concentration in HD have yielded conflicting results with three studies reporting an increase in HD patients compared to controls [81, 93] and five studies reporting no significant difference [84, 93–97] (Table 4).
Similarly to cortisol, GH and the tightly linked hormone, IGF-1 (insulin growth factor 1), have multi-organ effects and play major roles in whole body metabolism. It may be necessary to study multiple pathways and endocrine markers in association, to find patterns of changes that track with HD progression.
Oxidative stress markers (Table 5)
Evidence from studies in both humans and animal models suggests the involvement of energy metabolism dysfunction and oxidative stress in the pathogenesis of HD [98, 99]. It is thought that impairment in the electron transport chain and mitochondrial dysfunction are the major mechanisms involved in increased reactive oxygen species (ROS) production in HD [100, 101]. It has been reported that HD individuals have an increased level of oxidative stress markers (Table 5) accompanied by a reduction in antioxidant systems compared to healthy subjects [102, 103]. Several studies have reported enhanced lipid peroxidation in individuals with HD [40, 102–106] (Table 5). Chen et al. [103] detected a correlation between lipid peroxidation products in plasma and degree of severity in patients with HD, while Klepac et al. [105] reported an increase in plasma lipid peroxidation accompanied by reduced glutathione content (Table 5). An increase in Cu/Zn-superoxide dismutase (Cu/Zn-SOD), an important antioxidant enzyme, has also been reported in HD [62, 64] (Table 5). In addition, alterations in the kinetics of the kynurenine pathway (a major route of tryptophan catabolism) have been reported in patients with HD [40, 107] (Table 5), although some studies find no alterations [43, 108].
Oxidative stress markers in HD compared to control
Analytes were measured in blood, unless otherwise specified. Abbreviation: 3-Hydroxyanthranilic acid (3HAA). Cu/Zn-superoxide dismutase (Cu/Zn-SOD). Reduced glutathione (GSH)/ L-γ-glutamyl-L-cysteinylglycine.
Axonal and glial degeneration markers (Table 6)
HD is associated with a variety of pathological changes affecting both glial and neuronal brain tissue and these changes may be mirrored in the release of proteins into the CSF, and to lesser extent into the blood. Markers that reflect CNS pathology are needed for disease-modifying agents that target the CNS specifically. Currently, the most promising marker of HD onset and progression is neurofilament light protein (NF-L), a subunit of neurofilaments that make up the neuronal cytoskeleton (Table 6). Four studies have consistently found raised NF-L in CSF of individuals with HD [109–112] and several studies have found CSF NF-L to correlate with clinical measures [109, 111–114]. A recent, longitudinal study has found raised plasma NF-L in HD individuals, a finding confirmed in a separate, cross-sectional cohort [112] (Table 6). This is a very important finding because plasma NF-L levels reflect CNS pathology and may be a useful marker for trials involving CNS-delivery of disease-modifying agents. Importantly, both CSF and plasma NF-L levels have been found to correlate with disease onset in HD [112, 113] and brain atrophy [115]. A neuronal damage marker, NSE, is also increased in serum of HD patients [62, 64] (Table 6).
Neuropeptide, glial and axonal degeneration markers in HD compared to control
Abbreviations: γ-aminobutyric acid (GABA). Chitinase-3-like-protein 1 (YKL-40). Nerve growth factor (NGF). Neurofilament light subunit (NF-L). Neuron-specific enolase (NSE).
Several other markers of axonal and glial degeneration have been reported to be increased in HD individuals’ CSF including myelin basic protein (MBP) [111], total tau (T-tau) [110, 116], and chitinase-3-like-protein 1 (CHI3L1) / YKL-40 [110, 117]. T-tau and YKL-40 have also been found to correlate with clinical measures in some, but not all, of the studies [110, 118].
These studies suggest that the profile of glial-related inflammatory CSF biomarkers (such as YKL-40, GFAP, MCP-1, sCD14) and cytoskeletal and myelin markers of neurodegeneration (such as NF-L, T-Tau, P-Tau181, MBP, NSE, UCHL1), and their relation to disease severity should be further investigated.
CSF mutant huntingtin (Table 7)
Unsurprisingly, mutant huntingtin (mtHTT) has been found to be absent in healthy controls and increased in manifest HD CSF compared to premanifest HD CSF [114, 119] (Table 7). It is promising to note that CSF mtHTT levels have shown associations with clinical phenotype [114, 119].
Mutant huntingtin in HD compared to control
Abbreviations: Mutant huntingtin (mtHTT).
CRITICAL EVALUATION OF WET BIOMARKER LITERATURE IN HUNTINGTON’S DISEASE AND FUTURE DIRECTIONS
Why are studies not reproducible?
When conducting a biomarker study, it is important to consider the future purpose of the biomarker (its context-of-use) [120, 121] (see Fig. 1). For instance, if the purpose of the biomarker is to track disease progression, it should alter with advancing disease. Therefore, it is important to note that although many of the analytes in Supplementary Table 1 are not useful markers for HD in terms of disease progression, that does not mean that they would not be useful as, for example, pharmacodynamic markers.
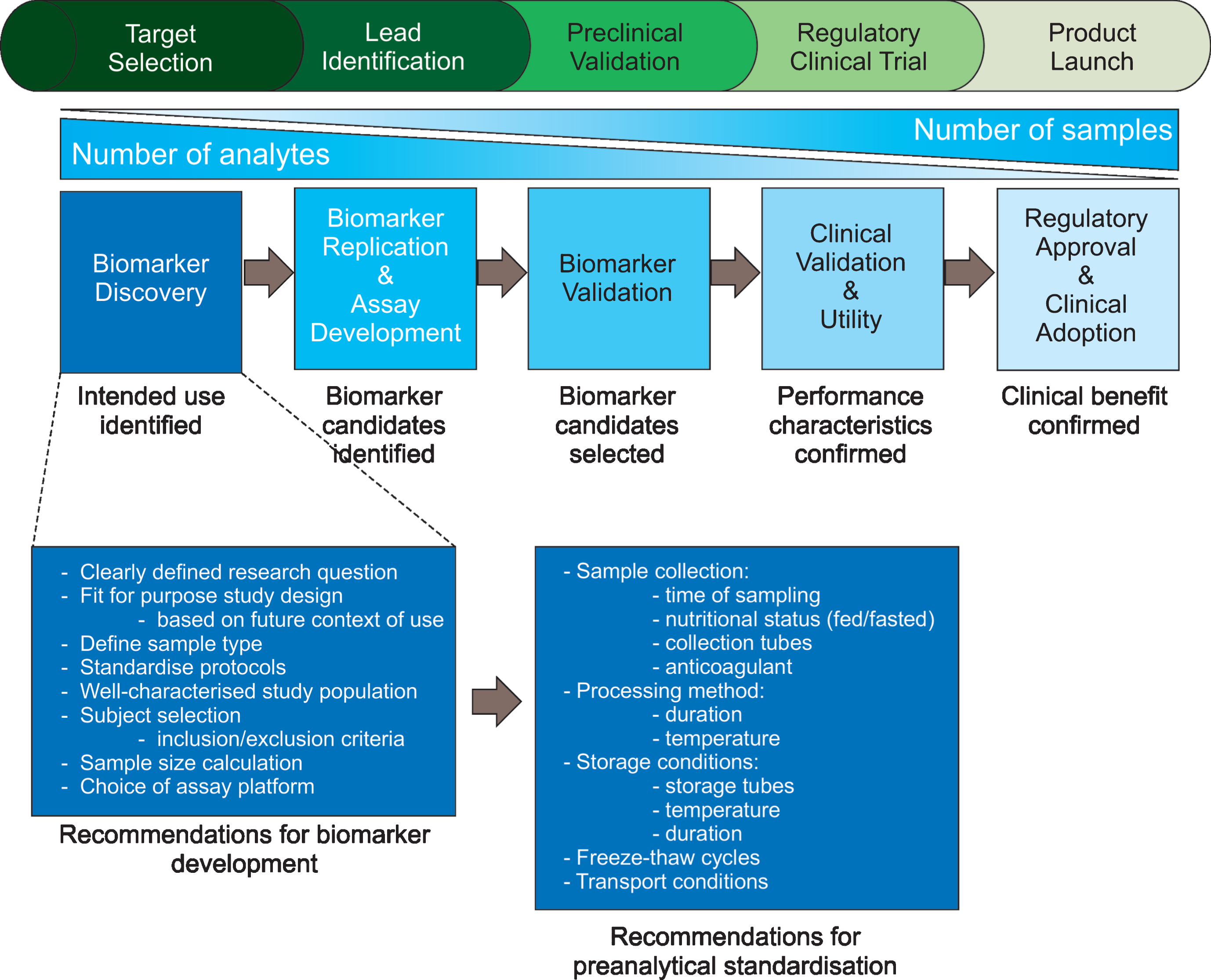
Phases of biomarker development and recommendations for biomarker discovery studies. Biomarker development from target selection and biomarker discovery to the product launch of a clinical assay that has been validated in large cohorts is a time-consuming, expensive process. It is, therefore, important that makers put forward for clinical validation are based on solid evidence. Here, we suggest factors to consider during biomarker study design to ensure meaningful data are produced. Adapted from [273].
This review of published HD biomarker studies to date reveals that very few wet biomarkers have been consistently and reliably reproduced; there are many conflicting studies that could be due to a myriad of variability factors. In biomarker discovery studies, great attention needs to be paid to several aspects of study design, sample collection, sample measurement and data analysis [122–125]. Factors to consider during biomarker discovery are outlined in Fig. 1. Below we discuss potential sources of variability in biomarker studies and suggest considerations for further developments and harmonisation of standard operating procedures (SOPs).
Biological variation
Within-subject (intra-individual) biological variation is the inherent change in analyte results around a homeostatic setting point within an individual over time [126, 127]. Individual homeostatic setting points are different and this difference between individuals is termed between-subject (inter-individual) biological variation [126, 127]. Some analytes have predictable biological rhythms that may cycle daily (e.g. cortisol, GH) [128], monthly (e.g., luteinizing hormone (LH), follicle-stimulating hormone (FSH), progesterone) [129, 130] or seasonally (e.g., vitamin D, cholesterol) [131–133]. With advancing age blood levels of some hormones increase (e.g., cortisol [134, 135], FSH [136], LH [137], noradrenaline [138]); decrease (e.g., melatonin [139], aldosterone [140, 141], GH [142], oestrogen in women [143], prolactin in women [144]); or change only slightly (e.g., adrenaline [145], insulin [146], thyroid hormone T4 [147]). In addition, some hormones are metabolised more slowly with age [147].
Some analytes can even be affected by activity levels; for instance, exercise prior to sample collection can result in higher cytokine and myokine levels in plasma [148]. Many HD individuals require medication for their symptoms and it is often difficult to get a medication-free cohort. Medication can influence analyte levels, e.g., [149, 150], and thus needs to be taken into consideration when interpreting results.
A large biological variability requires studies with larger subject cohorts. Lessons learned from cardiovascular biomarker research illustrate the importance of effect size; the prognostic effect was shown to be significantly stronger in datasets from observational studies than in datasets from randomised controlled trials [151]. As can be observed from Supplementary Table 1, many HD biomarkers studies to date have used less than 20 subjects per group, making it likely that the studies are underpowered.
Biological variation due to factors such as age, gender, diet, ethnicity, smoking, alcohol consumption, or medication may, at least in part, explain the contradicting results seen in different studies [152]. For example, for haemoglobin, the main sources of between-subject variation is clinical indication, but following that gender, age and nationality greatly influence variation [153], whereas CRP is not only affected by age, but also, to a large extent, gender [154, 155]. Confounding factors may be controlled for by selecting the most appropriate time of day, month or year for sample collection or may be taken into consideration in the interpretation of results. A study examining classification of control subjects from Alzheimer’s disease patients showed that removal of causes of variability such as age and gender can significantly improve classification accuracy [156]. Therefore, it is paramount to have a well-characterised cohort and a balanced design that minimises the number of uncontrolled covariates such as age and sex. If they are not matched, confounding factors should at the very least be controlled for in the statistical analyses [157, 158].
Study design: Considering preanalytical variables
Preanalytical variables such as time of day that the sample is obtained, anticoagulant choice, feeding state and acute stress of the patient, and method and duration of sample storage can influence reported analyte levels and should be documented [159, 160]. Below we discuss how pre-analytical variables contribute to variation in biomarker studies.
Patient selection: Disease stage
The major potential criticism of the value of many of the markers we discuss here for HD is that they are non-specific or variable from day to day or that they could be epiphenomena caused by a general illness state. Certainly nutritional, metabolic and infective pathology are likely to contribute to marker variability with advancing HD. Studies performed to date are often not comparable due to heterogeneity in patient cohorts in terms of disease stage; some studies focus on specific disease stages, e.g., [161], whereas others group all the manifest HD subjects from early to late stage, e.g., [81]. Analyte alterations that are seen before the onset of overt clinical features should be pursued as there may be a direct relationship to pathogenesis with the potential to inform the search for disease markers [17].
Time of sampling
Secretion of some analytes such as cytokines [162] and homocysteine [163] as well as hormones, such as ACTH, cortisol and growth hormone, shows circadian rhythmicity [164, 165]. Time of sampling might, therefore, affect results, especially if the expected alterations are subtle. Also, for some markers, it is possible that no difference is observed if only one timepoint is evaluated, but it might be the pattern of secretion that is altered. For instance, in premanifest HD subjects, there is no significant difference in mean levels of melatonin over 24 hours but there is a disruption in circadian rhythmicity [75]. Therefore, the ideal time of sampling needs to be determined for each marker and thereafter, the time of day that samples are collected should be standardised to allow direct comparisons between studies [166].
Fed or fasted state
Levels of hormones such as growth hormone [167], insulin [168] and leptin [169] can be influenced by the feeding status and thus, levels may vary depending on whether the subject had just had a meal or if the samples were taken after an overnight fast. Also, feeding status can affect cytokine production and action [170, 171]. Circulating CRP and IL-6 are elevated following a high-fat meal, while TNF-alpha levels decrease [172]. In addition, the supplementation of particular antioxidants such as glutathione, vitamins E and C can attenuate the feeding-induced rise in plasma cytokines [173, 174]. To avoid nutritional interference, studies should firstly determine whether nutritional status affects the analyte in question and secondly, need to be consistent in whether fed or fasted samples are used [175].
Blood collection tubes and specimen processing
For many blood biomarkers, either serum or plasma can be used [176, 177], however, for some biomarkers it is important to use a specific matrix and anticoagulant [178]. For example, studies demonstrate that plasma is intrinsically more stable than serum and therefore, more useful for protein analyses in biomarker studies [179, 180].
There is a wide selection of blood collection tubes available, and analyte levels can vary according to the type of collection tube used [181–186]. Blood collection devices and insufficient filling of blood collection tubes are another source of preanalytical error [187]. Care needs to be taken to ensure that the collection tubes are appropriate for analytes to be tested and that the type of collection tube is consistent throughout the study, especially if it is a multi-centre study.
It is also vital to standardise the pre-processing duration because the length of time that the samples are kept at room temperature after collection and before processing can alter analyte levels [188–190].
Sample storage and analyte stability
Both in vivo and in vitro analyte stability needs to be considered in biomarker studies because data obtained from stored specimens of an unstable biomarker may not produce any meaningful results [191]. Some potentially useful markers have very short half-lives and cannot reliably be measured and many proteins are unstable in serum even at –80°C [189, 192]. It is, therefore, important that the storage temperature of samples is appropriate for the analyte tested [193]. Thus, biomarker studies should standardise and document the temperature and the duration of sample storage [194].
Splitting samples into multiple aliquots is recommended for analytes that have a freeze-thaw instability, e.g., cytokines [195]. In addition, it is important that the type of storage tube is appropriate [196] and that tubes are filled sufficiently to avoid oxidation.
Multi-centre studies
As highlighted above, differences in sample preparation can influence absolute analyte levels. Therefore, to ensure that preanalytical variability is kept to a minimum in multi-centre studies, SOPs for collection, processing, storage and transport of samples should be created, formalised, and strictly adhered to [194, 198]. The importance of standardising pre-analytical variables was clearly demonstrated in a study of BDNF in serum and plasma [189] where the authors found many issues concerning BDNF detection that make reliable measurement difficult. The length of time between blood collection and plasma preparation affected BDNF levels, possibly due to BDNF release from platelets [199], suggesting that more robust and standardised procedures are required for plasma preparation. Also, BDNF was unstable in serum prepared following standardised protocols and stored at –80°C, but was stable in plasma. The authors emphasise that intra-group variability can be a major problem, especially because BDNF levels are affected by a number of individual factors such as exercise, stress, age, weight and gender [200–203] and preanalytical variables, including: fasting or fed condition, types of blood collection tubes (EDTA or heparin-treated) [204], duration and conditions of sample storage [192].
When using samples from different clinical centres, all diagnostic groups (including controls) need to be obtained from each clinical centre to be able to investigate potential variations in biomarker levels between different clinical sites. TRACK-HD [205] is a good example of a multi-centre study where the same stringent sampling procedure has been used at different sites. It will be interesting to see if markers evaluated in this sample cohort provide potential future biomarkers for HD progression. The EURO-HD network [206] is coordinating Clinical Centres within Europe with the aim of applying standardised protocols in response to criteria identified by tissue banking specialists and encourages sample deposition in a single bank (for HD studies: BioRep s.r.l. in Milan, [207]).
Analytical variables
Assay imprecision
The biochemistry of a biomarker needs to be understood in order to target the appropriate form of the biomarker that appears in biofluids. For instance, after production many proteins undergo post translational modifications, such as phosphorylation and glycosylation [208] and the analytical specificity of antibodies to these modifications determines the specificity of the corresponding assay for the biomarker.
For an assay to be a reliable tool, reproducibility and variability are of major importance. Assay variability is affected by many different factors, including within-assay run variability (between duplicate samples), within-laboratory longitudinal variability, between-laboratory variability, and within- and between-assay kit lot variability [209]. Analytical imprecision should be assessed and taken into account when evaluating biomarker data because is important to determine if a particular result is a true result or the result of variance within the measurement procedure itself [210, 211].
For novel biomarkers there are usually no standardised or harmonised assays available and manufacturers of diagnostic tests often use different reagents. This is especially problematic in the case of immunoassays, as the performance of a test is dependent on the quality and location of the epitope that the antibody is selected against [212, 213]. As such, absolute results cannot be compared in studies that use different methods and this can lead to contrasting results. Given the lack of assay standardisation, it is important for each laboratory to ensure stability in its measurements [214] and to consider assay variability and limits of quantitation when interpreting biomarker data. Recommendations for technical validation, standardised protocols for assay evaluation and reporting templates are available [215, 216].
Importance of replication and validation
As can be seen in Supplementary Table 1, many biomarkers have been proposed for HD, however, most have been evaluated in only a few studies. For a biomarker to be recognised, there is a need for validation and independent replication [217, 218]. Validation generally refers to a replication experiment performed by the same research laboratory with a different technology or a different sample cohort, whereas an independent replication of results is usually performed by an outside laboratory (Table 8). Validation for the intended purpose, at both technical and clinical levels, is essential in biomarker studies. The best biological validation of a biomarker is via its clinical correlations and clinical relevance, showing consistent data in cross-sectional and longitudinal studies and repeatability across different clinical studies.
Validation of biomarker research for use in medicine
Adapted from [217].
The need for replication of biomarker data is best exemplified by 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of DNA damage produced when guanine is oxidised by reactive oxidative species, that was initially thought to be a promising disease severity marker for HD [reviewed by [219]]. 8-OHdG levels were reported to be elevated serum [220], plasma [102] and leukocytes [103] of HD individuals and plasma levels were found to correlate with proximity to projected clinical diagnosis in premanifest HD [221]. However, subsequent studies failed to replicate these findings [222, 223] bringing into question the validity of 8-OHdG as a marker of disease state in HD.
Comparisons can be “statistically significant” (i.e., have a p < 0.05) even if no true relationship is present. 8-OHdG has a high biological variability so it is unsurprising that 8-OHdG measurements yielded false positive results. Elevated 8-OHdG is associated with confounding factors that are common in HD patients including stress [224], depression [225], smoking and low weight [226]. 8-OHdG levels are also influenced by exposure to pollution [227] and polyphenol-rich foods such as vegetables and red wine [228, 229].
Analytes found to be similar in HD compared to controls
The study by Borowsky et al. [223] challenging previous claims that 8OHdG is a useful clinical biomarker for HD progression is a good example of a well-designed biomarker study that included good practice of carrying out blinded sample analysis, use of independent analytic methods, estimation of accuracy of analytic methods and stringent collection and storage of samples.
Blinded analysis
Interpretation of data is seldom completely objective and is, as such, vulnerable to prior convictions and conflict of interest [230]. To reduce research bias in biomarker studies, blinded sample analysis and data interpretation is recommended [231, 232].
Biomarker discovery platforms
In general, there are two principal approaches for biofluid biomarker discovery: targeted and untargeted. The targeted, “pathophysiologic” approach tests a prespecified hypothesis. It involves knowledge of the relevant human physiology and disease processes, allowing the discovery of novel biomarkers that are produced, released, or cleared during the disease process. In contrast, the untargeted approach is an unbiased approach where a range of “omics” profiling technologies are generally used to systematically screen body fluids for novel biomarkers, paving the way to panels of multiple biomarkers that reflect multiple disease mechanisms. Proteomics and metabolomics are increasingly being used for biomarker discovery in neurodegenerative diseases [reviewed by [233] and [234]], including HD [30, 46]. Since the pathophysiology of HD is heterogeneous, with multiple mechanisms involved at every clinical or pathological stage, until the mechanism of HD fully understood, we must be open to novel mechanisms, and untargeted discovery using various “omics” technologies may lead to the discovery of novel markers. An integrated, multi-omics approach combining genomics, transcriptomics, proteomics, and metabolomics technologies can simultaneously detect hundreds of molecules simultaneously, some of which can be altered in the disease state which should result in a more complete biomarker fingerprint and give insight to fundamental mechanisms of HD.
To develop accurate markers of disease progression or therapeutic response that are clinically useful, it is necessary to combine data from wet biomarker studies with experimental, clinical and imaging data using systems biology approaches [235].
There are many articles discussing how systems biology and chemometrics can be used for biomarker discovery [236–239]. For example, Feala et al. integrated protein biomarker candidates for traumatic brain injury with publicly available canonical pathways and human protein-protein interaction networks to illustrate how to systematically generate new, testable hypotheses and identify candidate biomarkers [240]. Several tools are available that provide both pathway analysis and multi-omic integration including Open MS from KNIME [241], MetaboAnalyst [242] and XCMS online [243].
Longitudinal studies
One thing that became evident during the review work conducted was the lack of longitudinal studies. Only a small proportion of original-data studies evaluated HD biomarkers longitudinally. Most of the studies conducted so far are cross-sectional studies, which is a limitation if the aim is to examine the relationship between a change in a clinical measure and the change in a biomarker over time [244, 245]. A longitudinal design, in which the putative biomarker and clinical measure are both measured at least twice, will be useful when developing a disease progression marker. In addition, the duration needs to be long enough to observe a clinically significant change in the criterion used to draw associations with the putative biomarker. For a biomarker to be disease specific, it does not necessarily have to be different from its baseline levels in matched controls. However, levels of a disease specific marker would change with disease progression or in response to specific disease-modulating therapy while remaining constant in the controls during the same time period. Future longitudinal studies that assess the biomarker and clinical measures at several time points over a sufficient follow-up period are warranted to provide sufficient evidence of a biomarker’s potential validity [244].
It is encouraging to note that lack of longitudinal studies has already been realised by HD researchers; two large prospective, longitudinal, observational studies PREDICT-HD [246] and TRACK-HD [205] aiming to identify early HD biomarkers should address this issue.
Importance of publishing negative data (Table 9)
It is important that the biomarker research community publish all data, significant or not. Publication bias has been systematically investigated, particularly in clinical trials [247–249]. It has been demonstrated that studies reporting positive, or statistically significant, results are more likely to be published, and have higher odds of being fully reported [250]. Conversely, negative results are more likely than positive results to be published in journals with lower impact factors [251].
In this review, we have included a table for analytes unlikely to be useful markers for HD, as these markers have been assessed more than once, showing no difference in between HD and control subjects (Table 9). Of limited news value, but important knowledge to the HD biomarker field. Although these analytes do not differ between HD and control cohorts, some have been shown to correlate with specific disease measures as shown in Table 9.
Biobanks
Biomarker studies require sufficient numbers of properly annotated, well-characterised biospecimens. Good-quality biobanks that collect, store and distribute biospecimens under stringent quality control and assurance measures will be valuable for biomarker research [252]. Development of novel biomarkers is always a long process and optimal use of available resources is required. International collaboration is essential for standardisation of all aspects of biomarker studies, including biobanking procedures and study design, and to enable construction of sufficiently powered cohorts and optimally replicate findings. Lessons can be learned from other research fields. For example, the BioMS network has taken steps to standardise all aspects of biomarker research and validation to optimise biomarkers for multiple sclerosis [253]; the resulting guidelines can be utilised to benefit biomarker studies in HD [215, 254–262].
Biomarker qualification
Biomarker qualification is a mechanism that integrates the use of biomarkers into drug development programs to improve the efficiency and safety of clinical trials testing novel therapeutics [263–265]. The Food and Drug Administration (FDA) and The European Medicines Agency (EMA) have developed regulatory pathways for biomarker integration in clinical trials: the “Biomarker Qualification Program” [120] and “Qualification of Novel Methodologies for Drug Development” [266], respectively. If a biomarker lacks sufficient data to achieve full qualification, the FDA and EMA can publish a Letter of Support (LOS) [267], which provides support that the biomarker has demonstrated promise based on the level of evidence that has been formally provided to regulators.
At present, there are no biofluid biomarkers that are validated as outcome parameters for HD. Biomarker development and subsequent integration into drug development is critical to accelerating effective treatments for HD. Lessons on biomarker qualification can be learned from AD research [263] where new diagnostic criteria have been proposed that integrate pathophysiological biomarkers (imaging and/or biofluid) into all phases of the diagnostic approach to improve on the diagnostic specificity [268–272].
CONCLUSION
Our review of HD biofluid marker literature shows that no clinically validated biomarkers for HD are yet available, but there are grounds for cautious optimism. A continuous effort to find a reliable, easy to measure biomarker would improve the efficacy of coming HD clinical trials in many ways. For instance, such a biomarker could identify patients with fast and slow disease progression, thereby enabling more refined stratification and statistical analysis. Also, it would increase trial power, thereby decreasing trial duration and the number of volunteers required for the trial, and this in turn would decrease the cost of clinical trials and would expedite drug development. Finally, a reliable biomarker would provide a more objective, quantitative end point compared with the clinically based outcomes currently used in HD trials.
To clone the huntingtin gene took the combined efforts and skills of the 58 coworkers in the Huntington’s Disease Collaborative Research Group and many other researchers [1]. Similarly, finding biomarkers that will aid in the development of novel therapies for HD will most likely need a collaborative effort combining the skills of researchers from many disciplines and harmonisation of standard operating procedures will be crucial for successful biomarker discovery and validation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
Although effort was taken to include all relevant publications, limitations of the search engine may have resulted in a minority of relevant papers being overlooked.
ACKNOWLEDGMENTS
The authors would like to thank Denis Silajdžić for assistance with data mining.