
Abstract
Background:
Currently, no evidence exists on the expression of apoptosis (CASP3), autophagy (BECN1), and mitophagy (BNIP3) genes in the CA3 area after ischemia with long-term survival.
Objective:
The goal of the paper was to study changes in above genes expression in CA3 area after ischemia in the period of 6–24 months.
Methods:
In this study, using quantitative RT-PCR, we present the expression of genes associated with neuronal death in a rat ischemic model of Alzheimer’s disease.
Results:
First time, we demonstrated overexpression of the CASP3 gene in CA3 area after ischemia with survival ranging from 0.5 to 2 years. Overexpression of the CASP3 gene was accompanied by a decrease in the activity level of the BECN1 and BNIP3 genes over a period of 0.5 year. Then, during 1-2 years, BNIP3 gene expression increased significantly and coincided with an increase in CASP3 gene expression. However, BECN1 gene expression was variable, increased significantly at 1 and 2 years and was below control values 1.5 years post-ischemia.
Conclusions:
Our observations suggest that ischemia with long-term survival induces neuronal death in CA3 through activation of caspase 3 in cooperation with the pro-apoptotic gene BNIP3. This study also suggests that the BNIP3 gene regulates caspase-independent pyramidal neuronal death post-ischemia. Thus, caspase-dependent and -independent death of neuronal cells occur post-ischemia in the CA3 area. Our data suggest new role of the BNIP3 gene in the regulation of post-ischemic neuronal death in CA3. This suggests the involvement of the BNIP3 together with the CASP3 in the CA3 in neuronal death post-ischemia.
Keywords
INTRODUCTION
For several years, intensive experimental research on the responsibility of ischemic brain episodes in the development of Alzheimer’s disease (AD) has shown changes characteristic of AD, i.e., progressive hippocampal atrophy, increased amyloid production, and alterations of the tau protein [1–10]. Clinical studies after ischemia have revealed the deposition of diffuse and senile amyloid plaques and the formation of neurofibrillary tangles characteristic of AD [11–15]. These observations clearly indicate that after ischemia, changes in folding proteins occur with the development of dementia with an AD phenotype [16]. Based on these data, some research groups have been proposing an ischemic etiology of AD for several years [2, 17–23].
Interestingly, neuronal mechanisms called autophagy, which maintain order and control protein quality in the cell, have been shown to be insufficient and/or defective in the brain of AD patients [24–27]. Autophagy is a key system for clearing the cell cytoplasm, especially of abnormal conformation or aggregated proteins and worn-out organelles through degradation [28]. The phenomenon of autophagy is inextricably linked to the development of pathologically folded proteins and cell death in the brain. Studies of apoptosis (CASP3), autophagy (BACE1), and mitophagy (BNIP3) genes in CA3 area in an ischemic model of AD with survival up to 30 days showed dysregulation of their activity similar to alterations in the brain in AD (Table 1) [29]. Survival of up to 30 days in this model appears to be consistent with the preclinical stage of AD, which takes 10–20 years to progress in humans [30]. In this study, we set out to evaluate the behavior of the above genes during 2 years survival post-ischemia, which likely corresponds to the symptomatic stage of AD in humans. The work is a continuation of research on an ischemic model of AD with survival of 0.5, 1, 1.5, and 2 years and focuses on the quantitative evaluation by RT-PCR of genes associated with neuronal death, such as CASP3, BECN1, and BNIP3 in the CA3 area.
Changes in BACE1, BNIP3, and CASP3 genes expression post-ischemia of the hippocampal CA3 area with survival up to 30 days
Expression: ↓-lowered; ↑-increased. Genes: autophagy (BECN1); mitophagy (BNIP3); apoptosis (CASP3).
MATERIALS AND METHODS
Wistar rats (n = 30) body weighing 120–150 g, 2 months old, were exposed to 10-min brain ischemic episode [1, 2] with survival post-ischemia 0.5, 1, 1.5, and 2 years [31]. Wistar rats (n = 30), about the same conditions after sham surgery, served as control group with survival 0.5, 1, 1.5, and 2 years. Ischemia and control rats were housed in the animal house under a 12-h light-dark cycle. All studies were done within the day. The rats were maintained in pairs per cage in a room temperature of 24±2°C, with 55±5% humidity. All animals had free access to commercial laboratory chow and tap water ad libitum. Isoflurane (2%) with oxygen was used as anesthesia [31]. Anesthesia was discontinued before the induction of brain ischemia. Brain ischemia was induced by cardiac arrest [1, 2]. A hook made of an L-shaped steel needle was introduced into the chest by the right parasternal line and the third intercostal space. Next the hook was gently moved towards the spine. Subsequently, the hook was gently tilted 20° towards the tail and this meant that the hook in this position was under the heart vessels. The hook was next pulled to the sternum, which led to closure of the heart vessel bundle through the sternum. In order to prevent chest movements and ensure closure of the heart vessels, external pressure was applied to the sternum with the index and middle fingers, which resulted in complete hemostasis and cardiac arrest. After 3.5 min, the hook was removed from the chest and the rats remained in this state for next 6.5 min until resuscitation starts [1, 2]. After ischemia resuscitation started of artificial ventilation and external heart massage until cardiac function returned and breathing occurred [1, 31]. During this time, air was administered using a respirator through a polyethylene tube inserted into the trachea. The heart massage frequency range was from 150 to 240/min [1, 31].
All studies were handled in accordance with the NIH recommendations for the Care and Use of experimental animals and the Directive of the Council of the European Community. In addition, the Local Ethics Committee for Animal Experiments (Nr 64/2010) approved all used experimental procedures.
Before collecting samples from the CA3 region, rat’s brains were perfused with cold 0.9% NaCl. Next the brains were removed from the skulls and transferred on ice-cooled Petri dishes. Samples with a volume of about 1 mm3 have been taken from the CA3 ischemic and control brains from both sides and placed in RNAlater solution (Life Technologies, USA) [31, 32]. Next cellular RNA was isolated. RNA quantity and quality was performed using a Nano Drop 2000 spectrophotometer (Thermo Scientific, USA) [31, 32]. RNA was stored in 80% ethanol at –20°C [31, 32]. Finally, 1μg of RNA was reverse transcribed into cDNA [31, 32]. Veriti Dx (Applied Biosystems, USA) was used for cDNA synthesis using the manufacturer’s SDS software [31, 32]. The cDNA obtained was amplified by real-time gene expression analysis (qPCR) using the manufacturer’s SDS software [31, 32]. The tested genes were assessed in relation to the control Rpl13a gene, and the relative amount (RQ) of the examined genes were presented as ΔCT, and the final values were showed as RQ = 2-ΔΔCT [31, 32]. The results were showed after logarithmic adaptation of the RQ (LogRQ) [31, 32]. LogRQ = 0 means that the expression of the examined genes post-ischemia did not change. LogRQ < \.0 meant reduced genes expression post-ischemia, and LogRQ > 0 indicated raised genes expression post-ischemia compared to the control.
Statistica v. 12 was used to statistically evaluate the data using the non-parametric Kruskal–Wallis test with the “z” test for multiple analyses of differences between groups. Results in figures are presented as means±SD. p≤0.05 was considered statistically significant changes in expression of presented genes.
RESULTS
BECN1 gene changes up to 2 years after ischemia
Six and 18 months after ischemia, BECN1 gene in CA3 was below control values. Six months after ischemia, the lowest value was −0.502-fold change and the maximum −0.237-fold change and the median was −0.296-fold change. Eighteen months after ischemia, the lowest value was −0.340-fold change and the maximum −0.220-fold change and the median was −0,272-fold change. Whereas, 12- and 24-months following ischemia BECN1 gene was above control values. The lowest value was 0.972-fold change and the maximum 2.296-fold change and the median was 2.122-fold change after 12 months of survival. However, 24 months after ischemia, BECN1 gene was above control values with the lowest value 0.626-fold change and the maximum 1.224-fold change and the median was 0.905-fold change (Fig. 1). Figure 1 shows statistically significant changes in BECN1 gene changes. Significant changes in BECN1 gene were seen between 0.5 and 1 year (z = 4.319, p = 0.000094); and 0.5 and 2 years (z = 2.854, p = 0.025884); and 1 and 1.5 year (z = 3.234, p = 0.007316) after ischemia (Fig. 1). No significant alterations were noted between 6 and 18 months and 12 and 24 months and 18 and 24 months (Fig. 1).
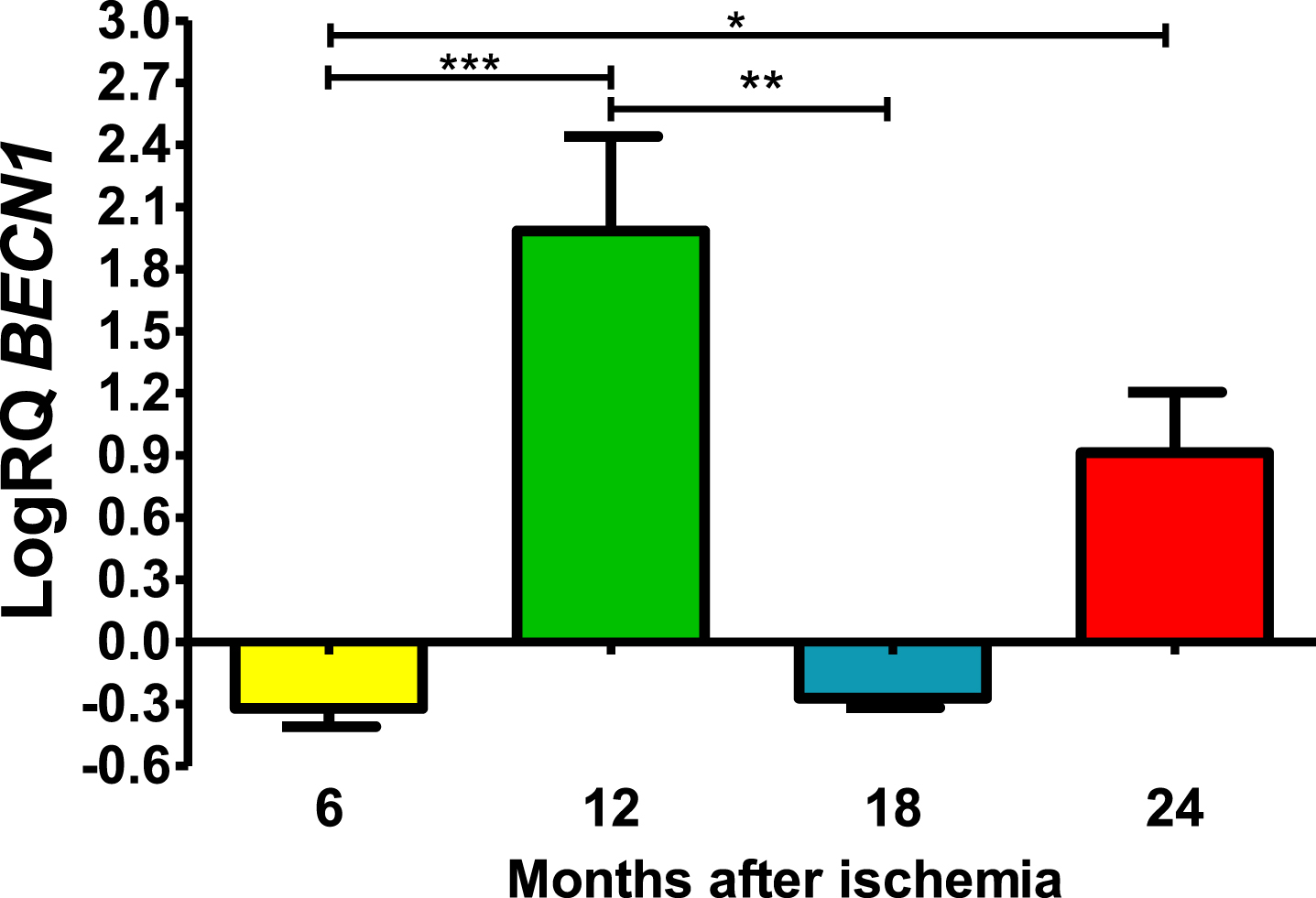
The mean BECN1 gene expression levels in the CA3 area of the hippocampus in rats with 6 (n = 10), 12 (n = 7), 18 (n = 6), and 24 (n = 6) months after 10-min brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test, followed by the “z” test. *p≤0.05, **p≤0.01, ***p≤0.001.
BNIP3 gene changes up to 2 years after ischemia
Six months post-ischemia, BNIP3 in CA3 was under control. Six months after ischemia, the lowest value was −0.245-fold change and the maximum −0.124-fold change and the median was −0.167-fold change. But 12–24 months, gene manifestation was above control. Twelve months post-ischemia, gene expression was maximal with the lowest value 0.625-fold change and the maximum 2.309-fold change, the median being 2.192-fold change. During eighteen months of survival, the lowest value was 0.133-fold change and the maximum 0.338-fold change and the median was 0.243-fold change. Whereas, 24 months after ischemia BNIP3 gene expression was still above control values, the lowest value was 0.873-fold change and the maximum 1.288-fold change and the median was 1.139-fold (Fig. 2). Figure 2 shows the mean values and significant changes in BNIP3 gene changes. Significant levels in BNIP3 gene changes were seen between 0.5 and 1 year (z = 4.498, p = 0.000041) and 0.5 and 2 years (z = 3.377, p = 0.004406 after injury (Fig. 2). No significant changes were noted between 6 and 18 months and 12 and 18 months and 18 and 24 months (Fig. 2).
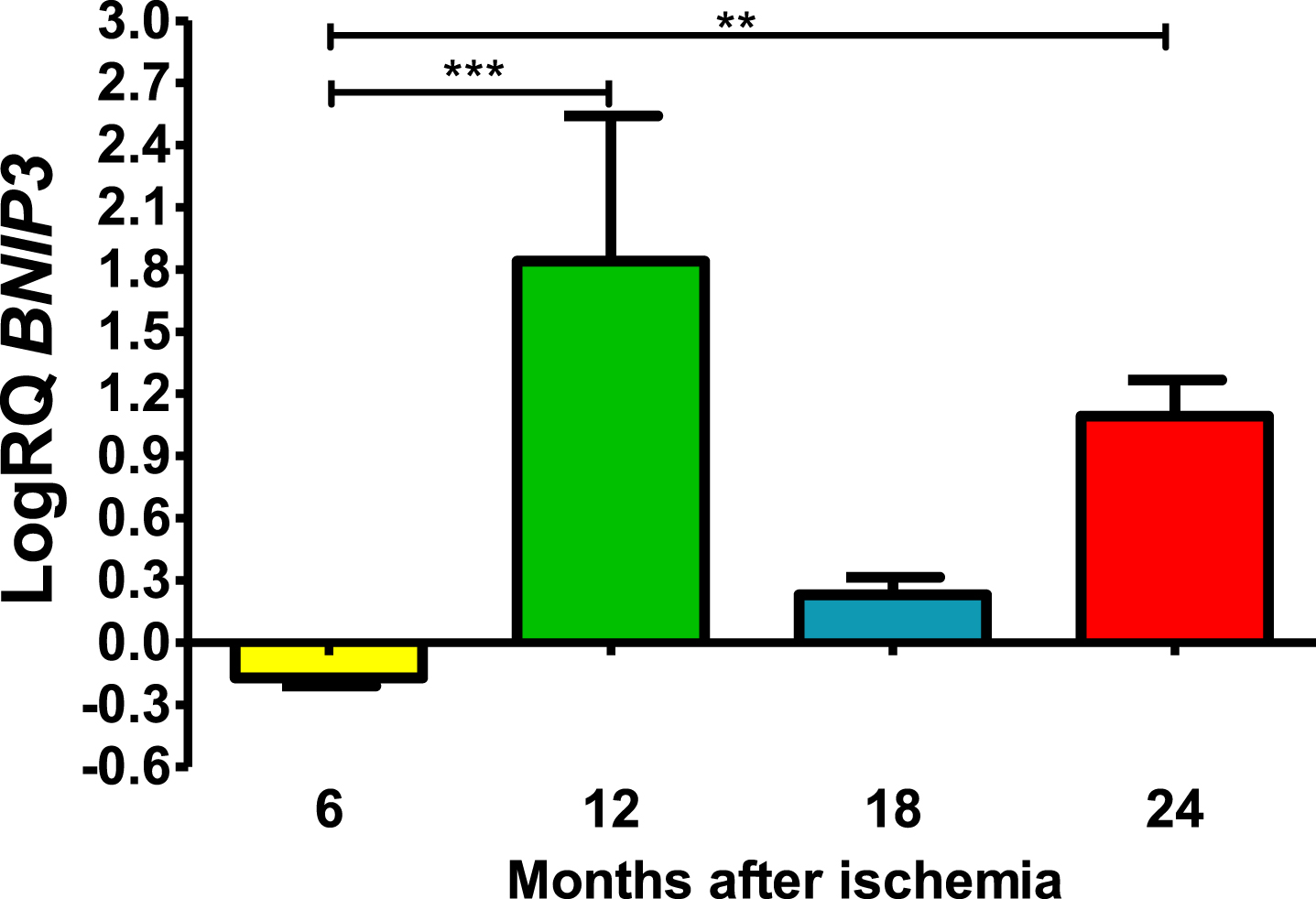
The mean BNIP3 gene expression levels in the CA3 area of the hippocampus in rats with 6 (n = 8), 12 (n = 8), 18 (n = 6), and 24 (n = 6) months after 10-min brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test, followed by the “z” test. **p≤0.01, ***p≤0.001.
CASP3 gene expression up to 2 years after ischemia
In all post-ischemic observation periods, CASP3 gene in the CA3 was above control (Fig. 3). Six months after ischemia, the lowest value was 0.408-fold change and the maximum 1.207-fold change and the median was 0.915-fold change. Twelve months after ischemia, maximal expression was noted with the lowest value of 1.721-fold change and the maximum 2.162-fold change and the median was 1.946-fold change. However, 18 and 24 months after ischemia, CASP3 gene was reduced but still remained above control. Eighteen months after ischemia, the lowest value was 0.188-fold change and the maximum 0.365-fold change and the median was 0.294-fold change. However, 24 months after ischemia, CASP3 gene expression increased again, with a lowest value of 0.797-fold change and a maximum value of 1.093-fold change and a median value of 0.918-fold change (Fig. 3). Figure 3 shows the mean values and significant changes in CASP3 gene changes. Significant levels in CASP3 gene were seen between 0.5 and 1 year (z = 2.797, p = 0.030927); and 1 and 1.5 years (z = 4.838, p = 0.000008); and 1 and 2 years (z = 2.655, p = 0.047554) after injury (Fig. 3). No significant changes were noted between 6 and 18, 6 and 24 and 18- and 24-months post-ischemia (Fig. 3).
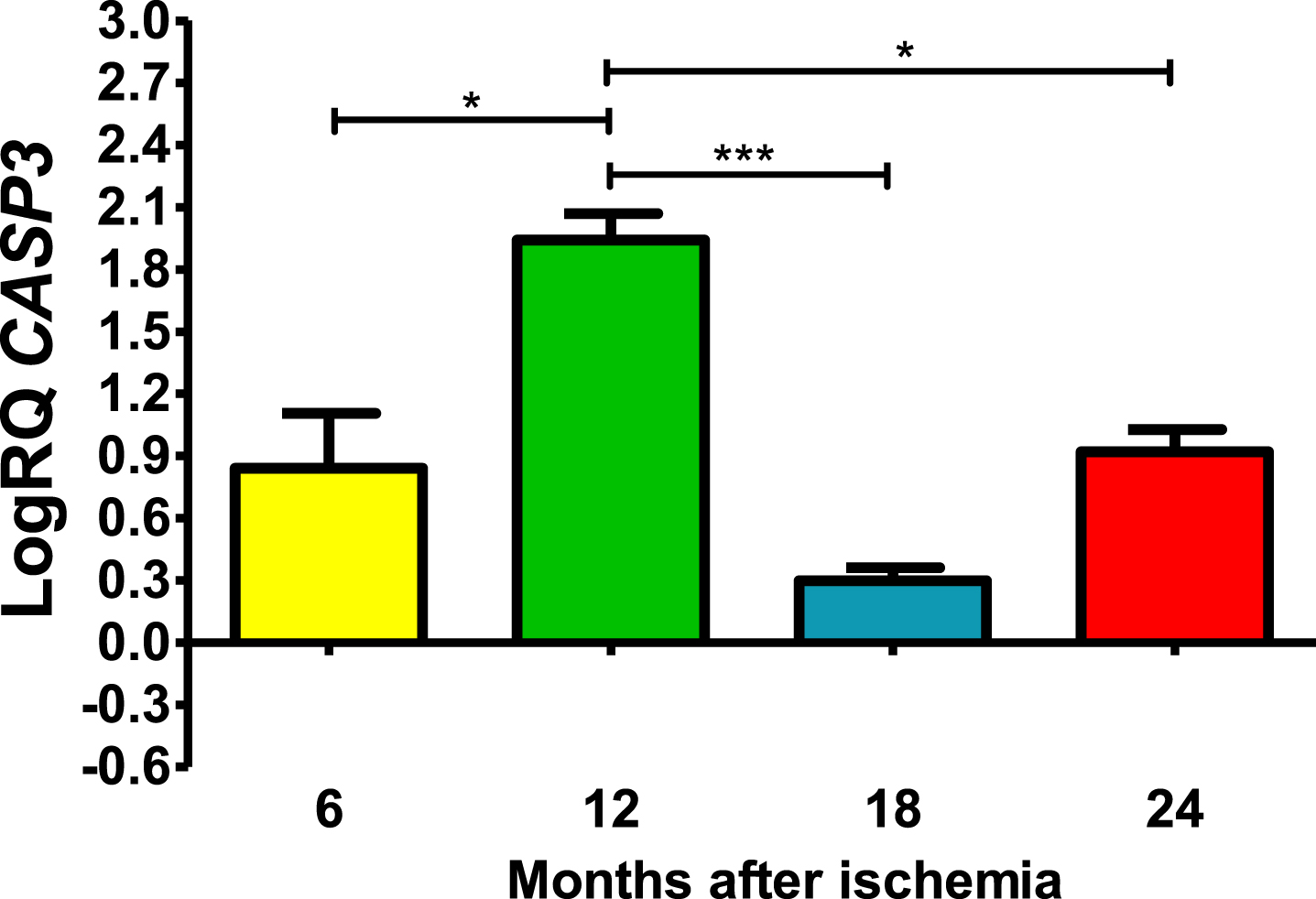
The mean CASP3 gene expression levels in the CA3 area of the hippocampus in rats with 6 (n = 8), 12 (n = 8), 18 (n = 6), and 24 (n = 8) months after 10-min brain ischemia. Marked SD, standard deviation. Kreskas-Wallis test, followed by the “z” test. *p≤0.05, ***p≤0.001.
DISCUSSION
In this study, we deliver for the first-time data of a significant increase in CASP3 gene expression in the CA3 subfield from 0.5 to 2 years following ischemic brain injury. Overexpression of the CASP3 gene go together with 6-month reduction in the activity levels of both BECN1 and BNIP3 genes. Then, over a 12–24-month period, BNIP3 gene expression was significantly increased and coincided with an increase in the expression of the apoptotic CASP3 gene. However, BECN1 gene expression was variable, i.e., it was elevated significantly after 1 and 2 years and was under control 18 months after ischemia. It has been proposed that ischemic injury induces classic apoptotic neuronal cells death throughout the hippocampus [6, 33], but our data indicate that the process is more complex and that this mechanism of pyramidal neuronal cells death in CA3 is less severe and extended in time as a result of protective overexpression of the BECN1 gene.
At 12 and 24 months after ischemia, overexpression of the BECN1 was observed in the CA3, in parallel with a maximum increase in BNIP3 and CASP3 genes expression. This naturally occurring phenomenon suggests a possible role of the BECN1 gene in balancing the activity of the BNIP3 and CASP3 genes by partially protecting or slowing damage and delaying neuronal death caused by an ischemic event [34]. Some studies indicate that enhanced autophagy, mediated by the BECN1 gene, plays significant role in the metabolism of the amyloid protein precursor, and in the autophagic elimination of its product, i.e. amyloid susceptible to aggregation in neurodegenerative processes [25, 36]. Transient overexpression of the BECN1 gene in our study probably induced a protective effect on neurons via enhanced autophagy [34, 35]. Data indicate that an enhanced transient autophagic response in CA3 may transiently protect pyramidal neurons from sudden death during and after ischemia, allowing time for therapeutic interventions [33, 38].
We found that the reduced level of BNIP3 gene expression 6 months after ischemia was a continuation of identical changes occurring on days 2–30 [32]. However, in the period of 12–24 months after the ischemic episode, a significant rise in the expression of this gene was noted, and its changes were accompanied by alterations in the expression of the CASP3 gene. In connection with the above, attention should be paid to the existing relationship between the enhanced expression of the BNIP3 gene and mitochondrial neurotoxic proteins, i.e., endonuclease G and poly (ADP-ribose) polymerase-1, apoptosis-inducing factor, which participate in the processes of neuronal cell death [39–42]. Brain ischemia appears to redistribute BNIP3, endonuclease G, poly (ADP-ribose) polymerase-1 and apoptosis-inducing factor from the mitochondria to the neuronal nucleus which ultimately leads to the death of neurons [39, 42–46]. Apoptosis-inducing factor and endonuclease G have been shown to migrate from mitochondria to the neuronal nucleus at the same time after ischemia [45]. Furthermore, BNIP3 has been shown to interact with LC3 to direct injured mitochondria to autophagosomes, thereby initiating the mitophagy phenomenon in post-ischemic neuronal death [47]. Unlike other proteins, neuronal BNIP3 levels are usually low, but significantly increase after brain ischemia [48]. Furthermore, double staining for caspase 3 and BNIP3 showed that the majority of ischemic neuronal cells marked for both proteins [48, 49]. However, some neurons positive for BNIP3 staining did not stain for caspase 3 [48, 49]. Above data indicate that the ischemic injury regulated pro-apoptotic protein BNIP3 enters the nucleus and may cooperate with proteins implicated in DNA structure, transcription, or mRNA splicing after cerebral ischemia. The mechanism by which BNIP3, endonuclease G, apoptosis-inducing factor, and poly (ADP-ribose) polymerase-1 trigger pyramidal neuronal loss is not completely known, but probable involves both nuclear-dependent and -independent mechanisms. These observations indicate that the above proteins may be implicated in the death of pyramidal neurons post-ischemia, but the specific relationship between the above proteins and BNIP3 has not been definitively established. The above findings indicate the importance of these proteins in caspase-independent death of neurons following ischemia. Thus, our data indicate a new important role of the BNIP3 gene in the control of neurons survival and death in the ischemic CA3 area of the hippocampus.
We found that the increased level of CASP3 gene expression 6–24 months after ischemia was a continuation of identical changes occurring in CA3 on days 7–30. In the hippocampal CA3 subfield, caspase 3 seems to be a central cause of pyramidal neuronal apoptosis. In contrast, it may explain the mechanisms of the slow spread of neuronal damage in the hippocampus itself between the CA1 and CA3 areas following ischemia with the AD phenotype. In this study, we confirmed observations of caspase 3 gene dysregulation associated with post-ischemic neuronal death in a region- or brain-structure-specific manner, as demonstrated in AD [29, 50]. Caspase can utilize substrates such as poly (ADP-ribose) polymerase-1, caspase-activated DNAse inhibitor, amyloid protein precursor, and presenilin and tau protein [49, 51–54]. Caspase 3 also cleaves a nuclear DNA repair enzyme such as poly (ADP-ribose) polymerase-1, leading to nuclear DNA injury and ultimately apoptosis [33, 38]. Moreover, caspase 3 can cleave the GGA3 protein, resulting in increased β-secretase activity [55, 56]. Importantly, the level of amyloid protein precursors is increased long-term after brain ischemia [2, 57–61] and has been presented to be a substrate for caspase 3, and caspase 3 increases the activity of β-secretase, which leads to a vicious circle [49, 56]. The above phenomenon can also have a significant impact on the expansion of neuronal alterations in the CA3 area. Some investigations have revealed that increased caspase activity significantly correlates with the level of tau protein and the development of neurofibrillary tangles [62, 63]. Furthermore, it was shown that cognitive decline was negatively correlated with rose caspase action and the level of caspase 3-truncated tau protein [54]. Thus, it is well-defined that a single ischemia can cause different type of neuronal deaths in the same neurons, and in numerous cases a single neuron can demonstrate signs of different type of neuronal cell death [48, 49].
The presented data suggest possible transient protection via the autophagy gene BECN1, which presumably slows down the damage or death of pyramidal neurons after ischemia, providing an opportunity for pharmacological intervention [34, 64]. Moreover, data suggest that brain ischemia during long-term survival induces neuronal death in CA3 through increase protein level and gene expression of CASP3 [65, 66] in cooperation with transcription of the pro-apoptotic gene BNIP3, and the process, once triggered, persists or continues to smolder, which would partially explain the long-term progression of AD from asymptomatic to fully symptomatic [30]. This study also suggests that the BNIP3 gene regulates caspase-independent death of pyramidal neurons after ischemia, possibly through effects on endonuclease G, apoptosis-inducing factor and poly (ADP-ribose) polymerase-1 in the neuronal nucleus [39, 68]. The mitochondrial BNIP3 gene is likely a signal prior to association with these proteins. Knockdown of the BNIP3 gene reduces cell death, confirming a role for this gene in activating a caspase-independent neuronal death mechanism in CA3 [46]. Thus, caspase-dependent [65, 66] and caspase-independent neuronal death may occur simultaneously in ischemic neuronal cells of the CA3 area of the hippocampus, leading to neuronal death with mixed pathophysiological features [46, 49].
We recognize that our research has strengths and weaknesses. First, the strength of our study is that rats survived 2 years after complete brain ischemia, allowing for the first time to assess the expression of selected genes associated with neuronal death. Second, this study allowed us to demonstrate that the BNIP3 and CASP3 genes are important in inducing neuronal cells death in the CA3 in long-term surviving animals despite natural transient protection from the BECN1 gene. Third, the data indicate the existence of a therapeutic window in brain ischemic pathology with long-term survival. Fourth, our investigation demonstrated four long survival periods, i.e., 0.5, 1, 1.5, and 2 years, which increased the accuracy of the assessment of alterations in the expression of selected genes and supported their participation in brain neurodegeneration after ischemia. The main limitation of our investigation for various reasons was the small number of experimental groups, which limited the availability of material for research from such structure as the CA3. Another not entirely weak point was the use of young rats for brain ischemia, when it is recognized that cerebral ischemia is preferably age-related. However, the use of old animals was unacceptable due to the experimental assumption that animals after ischemia would be observed long-term, up to 2 years. Our observations need verification in wider investigates, lasting 0.5–2 years, at proteins levels to association them to gene changes. This would allow for a precise assessment of dysregulation at the genomic and proteomic level and the impact of dysregulation on the progression of neurodegeneration from the acute phase to the chronic phase with the AD phenotype after ischemic brain injury.
AUTHOR CONTRIBUTIONS
Ryszard Pluta (Conceptualization); Anna Bogucka-Kocka (Methodology); Jacek Bogucki (Data curation); Janusz Kocki (Investigation); Stanisław J Czuczwar (Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge the support of the Medical University of Lublin, Poland (DS 721/22-SJC). Moreover, the authors would like to thank Mr. Sławomir Januszewski for technical assistance.
FUNDING
This research received no external funding.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY
The data of this study are available on request from the corresponding author.