
Abstract
Amyloid protein-β (Aβ) concentrations are increased in the brain in both early onset and late onset Alzheimer’s disease (AD). In early onset AD, cerebral Aβ production is increased and its clearance is decreased, while increased Aβ burden in late onset AD is due to impaired clearance. Aβ has been the focus of AD therapeutics since development of the amyloid hypothesis, but efforts to slow AD progression by lowering brain Aβ failed until phase 3 trials with the monoclonal antibodies lecanemab and donanemab. In addition to promoting phagocytic clearance of Aβ, antibodies lower cerebral Aβ by efflux of Aβ-antibody complexes across the capillary endothelia, dissolving Aβ aggregates, and a “peripheral sink” mechanism. Although the blood-brain barrier is the main route by which soluble Aβ leaves the brain (facilitated by low-density lipoprotein receptor-related protein-1 and ATP-binding cassette sub-family B member 1), Aβ can also be removed via the blood-cerebrospinal fluid barrier, glymphatic drainage, and intramural periarterial drainage. This review discusses experimental approaches to increase cerebral Aβ efflux via these mechanisms, clinical applications of these approaches, and findings in clinical trials with these approaches in patients with AD or mild cognitive impairment. Based on negative findings in clinical trials with previous approaches targeting monomeric Aβ, increasing the cerebral efflux of soluble Aβ is unlikely to slow AD progression if used as monotherapy. But if used as an adjunct to treatment with lecanemab or donanemab, this approach might allow greater slowing of AD progression than treatment with either antibody alone.
Keywords
The hallmark neuropathological findings in Alzheimer’s disease (AD) are amyloid-β protein (Aβ), containing senile plaques (SPs), and tau protein, containing neurofibrillary tangles. The amyloid hypothesis [1, 2] suggested that deposition of insoluble Aβ as SPs initiated AD-type neurodegeneration, with tau pathology and neuronal loss developing downstream. Following reports of weak associations between SP counts and cognitive impairment in AD patients [3–5] and the finding that Aβ oligomers rather than fibrillar Aβ may be the most neurotoxic Aβ conformation [6, 7], the hypothesis was revised to suggest that Aβ oligomers may initiate AD pathology [8]. The amyloid hypothesis led to multiple approaches which attempted to slow AD’s clinical progression by lowering brain Aβ levels [9–13], most recently via administration of monoclonal anti-Aβ antibodies [14–16]. These approaches failed until clinical trials with the monoclonal anti-Aβ antibodies lecanemab [17, 18] and donanemab [19, 20]. A third monoclonal antibody, aducanumab, reduced brain levels of insoluble (PET-detectable) Aβ in a phase 1b trial [21] but its two phase 3 trials produced conflicting results regarding its ability to slow AD progression [22].
In phase 3 trials, lecanemab slowed AD’s clinical progression by 27% [18], while donanemab slowed disease progression by 35% for subjects with low/medium cerebral tau levels and by 22% when subjects with high tau levels were included in the analysis [20]. Although these effects were statistically significant (p < 0.001), questions were raised as to whether they were clinically meaningful [23–26]. The findings in the lecanemab and donanemab trials are encouraging, but additional approaches may be needed to further slow disease progression. More effective targeting of Aβ oligomers may be required; the effects of the antibodies on brain Aβ oligomer levels in AD subjects in the clinical trials are unknown because the PET radioligands currently used to detect cerebral Aβ bind only to insoluble Aβ [27]. Another option would be targeting of downstream neuropathological processes in addition to Aβ [28–30]. A third approach would be to combine the administration of anti-Aβ monoclonal antibodies with interventions to increase the efflux of soluble Aβ from the brain. In late-onset AD (LOAD), which accounts for 90–95% of AD cases [31], the increase in cerebral Aβ has been attributed to impaired removal of Aβ [32]. Aβ is cleared from the brain by enzymatic degradation, including the endosomal-lysosomal system, the ubiquitin-proteasome system, and autophagy [33], and soluble Aβ can leave the brain via the blood-brain barrier (BBB) [34], the blood-cerebrospinal fluid (CSF) barrier (BCSFB) [35], glymphatic (paravascular) drainage [36], and intramural periarterial (IPAD) drainage, also referred to as perivascular drainage [37–41]) (Table 1). This review discusses experimental approaches for increasing cerebral Aβ efflux, clinical applications of these approaches, and the effects of these approaches in clinical trials in which they have been evaluated with subjects with AD or mild cognitive impairment (MCI). Although most studies of cerebral Aβ efflux have used either monomeric Aβ or Aβ species whose levels of aggregation were unclear [42], a study in C57BL/6 mice comparing clearance of monomeric and low molecular weight oligomeric Aβ40 from brain interstitial fluid (ISF) to CSF suggested that efflux of these Aβ conformations from the brain may be via similar routes [42].
Mechanisms of clearance of soluble Aβ from the brain
ABCB1, ATP-binding cassette sub-family B member 1; AD, Alzheimer’s disease; AQP4, aquaporin-4; BBB, blood-brain barrier; BCSFB, blood-cerebrospinal fluid barrier; CAA, cerebral amyloid angiopathy; IPAD, intramural periarterial drainage; LOAD, late onset Alzheimer’s disease; LRP1, low density lipoprotein receptor-related protein 1.
BBB CLEARANCE OF Aβ
The BBB (Fig. 1) consists of capillary endothelial cells separated by tight junctions, adherens junctions, and gap junctions [43]. Because of its tight junctions the BBB is less permeable than peripheral blood vessels to solutes [44–46]. Adherens junctions are organized similarly to tight junctions and influence paracellular permeability to solutes [43, 47]. The BBB is supported by astrocytes, pericytes, and extracellular matrix components [48–50]. Cellular elements of the BBB are in contact with the basement membrane (produced by endothelial cells and pericytes), which encloses the pericytes and attaches astrocytic endfeet processes [51]. The basement membrane undergoes age-related alterations [52] including microvascular fibrosis [53], changes in proportions of collagen IV, agrin, and laminin [54], and lipid accumulation [51]. The percentage of Aβ cleared by the BBB was estimated by Qosa et al. to be 62% [55] and by Goulay et al. to be 85% [56], although Roberts et al. suggested that direct transport of Aβ across the human BBB may account for only 25% of its clearance from the brain [57]. Distinguishing between Aβ’s clearance via the BBB and its clearance by other pathways may be difficult because of technical limitations [58]. In AD, BBB clearance of Aβ is decreased by approximately 30% [59]. Impaired BBB clearance of Aβ is thought to play a role in the pathogenesis of cerebral amyloid angiopathy (CAA) as well as AD [60].
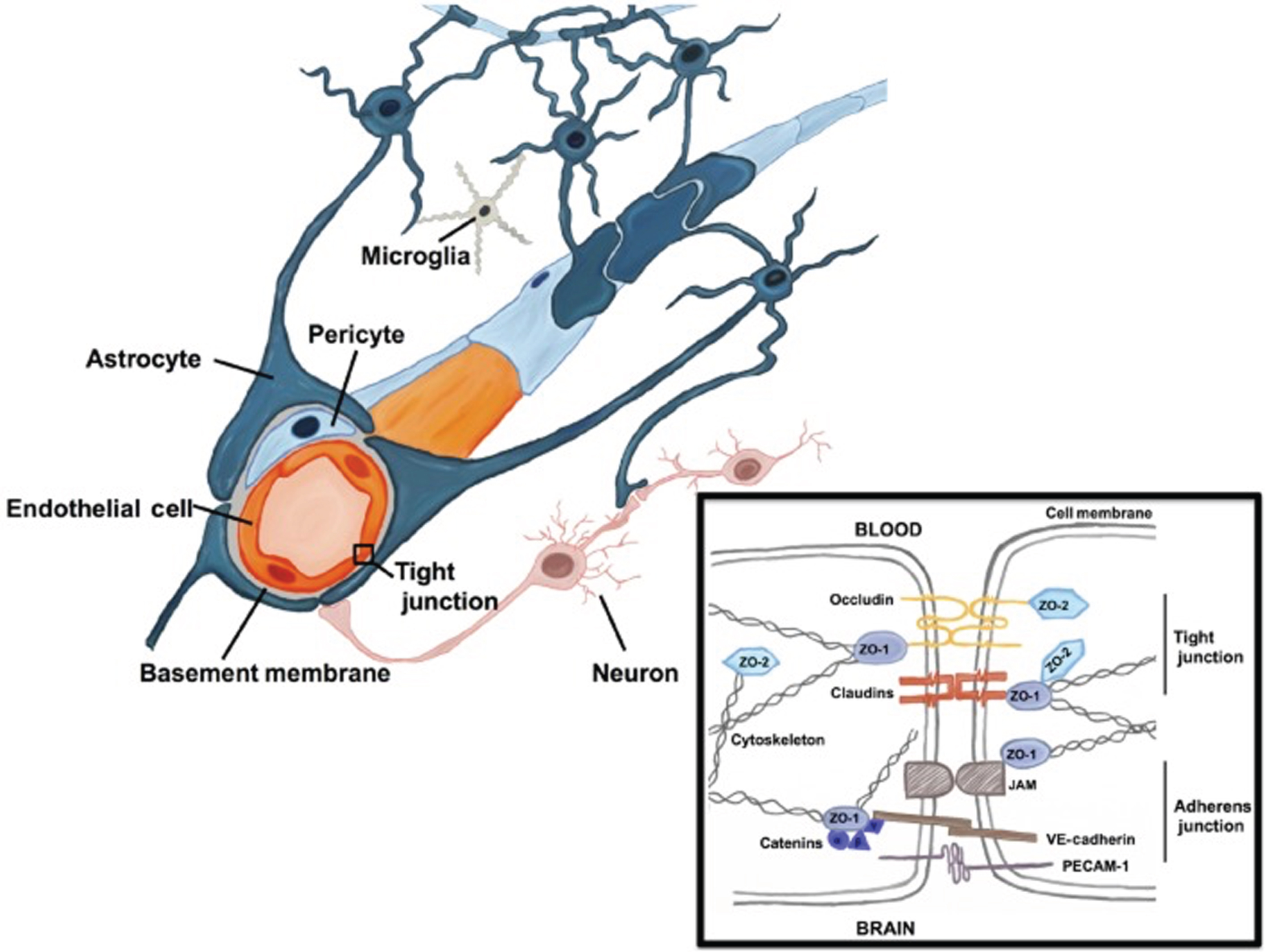
The blood-brain barrier (BBB). The BBB consists of capillary endothelial cells surrounded by extracellular matrices formed by cellular basement membrane (shared with pericytes) and astrocytic endfeet. Tight junctions, adherens junctions, and gap junctions are present between the endothelial cells. The tight junctions limit passage of solutes between the cerebral microvasculature and brain parenchyma. Junctional proteins in tight junctions and adherens junctions are shown in the black box. Aβ can cross the BBB via transcytosis (membrane-bound carrier-mediated transport, facilitated by LRP1 and ABCB1) as well as via paracellular transport. (From: Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol. 2014 Mar;171(5):1210-30 [British Pharmacological Society; publisher: Wiley-Blackwell, Hoboken, NJ, USA]). Permission to use obtained via RightsLink.
In addition to transcytosis (membrane-bound carrier-mediated transport) of Aβ across the BBB, paracellular transport of monomeric Aβ across the BBB through the BBB’s tight junctions has been described. Interestingly, Aβ was found to transiently downregulate the tight junction-associated proteins claudin-5 and occludin in the Tg2576 transgenic mouse model of AD, thus increasing its clearance across the BBB via this mechanism [61].
The length and conformation of Aβ influence its BBB clearance. Aβ42 is cleared via the BBB more slowly than Aβ40 [62] and BBB clearance of aggregated Aβ has been suggested to be less effective than clearance of monomeric Aβ [63]. (In the study by McIntee et al. discussed above [42] in which clearance of monomeric and low molecular weight oligomeric Aβ40 from ISF to CSF was examined, efflux was faster for monomers than for oligomers). The effects of oligomeric and fibrillar Aβ on cerebral microvascular endothelial cells may differ, because Aβ oligomers induce apoptosis of these cells while exposure of these cells to fibrillar Aβ increases BBB permeability but does not induce apoptosis [64]. Normal BBB functioning in the hippocampus may be required to maintain normal cognition, based on a study which found that hippocampal BBB damage was associated with early cognitive decline in human subjects irrespective of the presence or absence of AD-type pathology [65].
Low-density lipoprotein receptor-related protein 1 (LRP1) and ATP-binding cassette sub-family B member 1 (ABCB1, also known as P-glycoprotein 1) have been suggested to be key transporters in BBB clearance of cerebral Aβ [66, 67]. (However, Ito et al. [68] concluded that LRP1 was not important for clearance of Aβ40 across the mouse BBB). LRP1 is expressed on the abluminal surface of the BBB while ABCB1 is expressed on its luminal surface. The expression of these transporters on the BBB decreases during normal aging [69–71] and AD [72–75]. LRP1 and ABCB1 have been suggested to be functionally related via phosphatidylinositol-binding clathrin assembly protein (PICALM) [66], therefore in AD, PICALM downregulation by Aβ may contribute to the decreased BBB clearance of Aβ [76, 77]. Oxidation of LRP1 may also play a role in this decrease [78]. Because LRP1 binds other ligands including apolipoprotein E (apoE) and amyloid-β protein precursor (AβPP) [79] in addition to Aβ, therapeutic interventions to increase LRP1’s expression in AD could have unintended (and possibly deleterious) consequences [80] unless this upregulation is specific for LRP1’s binding to Aβ, as in the site-mutagenesis approach suggested by Sagare et al. [81].
ApoE also plays a role, possibly a negative one, in clearing Aβ across the BBB. This may be true to a greater extent for apoE4 than for apoE2 and apoE3 [82–85]. ApoE competes with Aβ for binding to LRP1 and for LRP1-mediated transport across the BBB [58], and apoE-Aβ complexes are cleared from the brain more slowly than unbound Aβ [86]. Of relevance is that increasing apoE’s lipidation increases its efficiency of binding to Aβ, and this decreases the ability of Aβ to aggregate [87]. The ABC transporter ABCA1 regulates lipidation of cerebral apoE [87, 88]. Another apolipoprotein, apolipoprotein J (apoJ, also known as clusterin), also influences Aβ’s BBB clearance by binding to it [89] and mediating its clearance via low-density lipoprotein-related protein 2 (LRP2) [62, 90]. LRP2 does not bind to Aβ but it does bind to Aβ-apoJ complexes [91]. Aβ’s binding to apoJ increases its BBB clearance [62, 92], and viral vector-mediated increased expression of apoJ in astrocytes led to decreased cortical and hippocampal Aβ levels in the APP/PS1 transgenic mouse model of AD [93]. ApoJ’s role in the pathogenesis of AD is unclear; although it increases BBB clearance of Aβ and limits Aβ aggregation and neurotoxicity [91, 95], genome-wide association studies have found some single nucleotide polymorphisms of the CLU (clusterin) gene (which encodes for apoJ) to be associated with increased risk for AD [96, 97]. ApoJ concentrations are increased in AD hippocampus [98] possibly as a compensatory response to the increased levels of Aβ.
Approaches which have been used to improve BBB clearance of Aβ in experimental systems are shown in Table 2. Therapeutic increasing of Wnt/β-catenin signaling, which plays a role in maintaining BBB function [99, 100], has also been suggested as an approach [101, 102] but no specific activators of this mechanism are known to cross the BBB [102]. Wnt/β-catenin signaling is reduced in AD brain [103].
Experimental approaches for increasing BBB clearance of Aβ
ADAM10, A Disintegrin And Metalloproteinase Domain containing protein 10; LDLR, Low-density lipoprotein receptor; MMP-9, matrix metalloproteinase-9; Nedd4, Neuronal precursor cell-expressed developmentally downregulated 4; PICALM, phosphatidylinositol binding clathrin assembly protein); PCSK9, proprotein convertase subtilisin/kexin type 9). aADAM10 inhibition improved BBB-mediated clearance of Aβ in an in vitro BBB model by reducing proteolytic shedding of LRP1 at the endothelial cell surface [104]. bLowering of systemic amylin concentration by treatment with amylin-specific antisense microRNAs reversed amylin-induced lowering of LRP1 expression in an in vitro model of the BBB [105]. cAdministration of Annexin A1 restored normal BBB functioning in mice lacking Annexin A1 [106], and also in 5xFAD and Tau-P301 L mice [107]. dThe antioxidant astaxanthin was suggested as a possible therapy for increasing BBB clearance of Aβ because it increased in vitro expression of ABCA1, ABCG1, and LRP1 [108]. eIn an in vitro BBB model exposed to fibrillar Aβ42, catalpol increased the expression of LRP1, ABCB1, and tight junction proteins, decreased the expression of MMP-2, MMP-9, and the receptor for advanced glycation end products (RAGE), and enhanced the efflux of soluble Aβ [109]. fTreatment of mice with the PET imaging agent copper diacetyl bis(4-methyl-3-thiosemicarbazone) increased the expression of P-gp (ABCB1) in brain microvasculature [110]. gThe dihydropyridine L-type calcium channel blockers nilvadipine and nitrendipine lowered brain Aβ in Tg PS1/APPsw mice and increased Aβ clearance in an in vitro BBB model [111]. hIntraperitoneal administration of 1α,25-dihydroxyvitamin D3 increased 24-h clearance of 125I-labeled human Aβ40 from mouse brain [112]. iTransplantation of endothelial progenitor cells into the hippocampus of APP/PS1 mice upregulated tight junction proteins and promoted Aβ clearance [113]. jIn TgCRND8 mice, late running (five months of wheel running, started four months after disease onset) increased BBB clearance of Aβ [114]. kInhibiting the serine protease kallikrein-8 in a mouse transgenic model of AD increased BBB clearance of Aβ [115]. lIncreasing the expression of the apoE receptor LDLR (by developing LDLR transgenic mice) increased BBB-mediated clearance of 125I-Aβ. This was suggested to be mediated in part by LRP1 [116]. mInfusion of mesenchymal stem cells to spontaneously hypertensive rats resulted in remodeling of microvasculature, in part by activation of transforming growth factor-β and angiopoietin 1 signaling pathways [117]. nThe matrix metalloproteinase 9 (MMP9) inhibitor SB-3CT prevented lipoprotein receptor shedding in Aβ42-treated human brain microvascular endothelial cells, and increased Aβ clearance via the BBB in C57BL/6 mice expressing human apoE4 [118]. oPirenzepine, a selective muscarinic acetylcholine receptor inhibitor, increased BBB clearance of Aβ in AβPPPS1, hAβPPSL, and AβPP/PS1 mice [119]. pNEDD4-1 is a ubiquitin E3 ligase that ubiquitinates P-gp (ABCB1); siRNA-mediated knockdown of Nedd4 expression in CHO-APP cells increased their P-gp expression and their secretion of Aβ [120]. qAdministration of olive leaf extract to 5xFAD mice for three months improved BBB integrity and increased cerebral Aβ clearance [121]. rPICALM was found to regulate BBB transcytosis and clearance of Aβ by regulating, in endothelial cells, PICALM/clathrin-dependent internalization of Aβ bound to LRP1 and guiding of Aβ to the endosomal regulators Rab5 and Rab11. Increasing of PICALM expression in AD brain endothelial cells (by adenoviral-mediated transfer of PICALM) increased transcytosis of Aβ [77]. Treatment of PICALM-deficient 5XFAD mice with the anti-malaria drug artesunate increased cerebral capillary PICALM expression and prevented brain Aβ accumulation [122]. sPCSK9 binds to low-density lipoprotein receptor proteins, promoting their lysosomal degradation. Treatment of 5xFAD mice with anti-PCSK9 antibodies reduced cerebral Aβ [123]. tSomatostatin prevented Aβ-induced BBB permeability in human CMEC/D3 (human temporal lobe microvessel) cells through its effects (upregulation) on LRP1 and tight junction proteins [124]. uα-tocopherol upregulated LRP1 and ABCB1 in an in vitr o model of the BBB and in 5XFAD mice, reducing cortical and hippocampal Aβ by 75% and 59% respectively [125].
BCSFB CLEARANCE OF Aβ
The term BCSFB refers to the barrier posed by the tight junctions between choroid epithelial cells to passage of solutes between CSF and blood [126]. Although the choroid plexus (Fig. 2) is a major component of the BCSFB, the BCSFB also includes the arachnoid membrane and circumventricular organs [127]. The choroid plexus lines the lateral ventricles, third ventricle, and fourth ventricle, and is the main source of CSF [128, 129], secreting about 80% of CSF [130]. Its functions include transport of glucose and other nutrients from blood to CSF, reabsorption and elimination of brain metabolic waste products from CSF, synthesis of proteins including transthyretin (TTR), cytokines, growth factors, and neurotrophic factors, regulation of trafficking of immune cells into the brain, and maintenance of CNS homeostasis by modulation of solute exchange between CSF and brain parenchyma [129, 131–134]. Choroid plexus blood flow has been estimated to be 5-fold [135] or 10-fold [136] higher than in the cerebral vasculature. The choroid plexus includes an epithelial cell monolayer whose microvilli extend into the ventricles and whose basolateral side rests on basal lamina. These cells surround and enclose stroma containing peripheral blood capillaries [137, 138]. Dendritic cells, fibroblasts, macrophages, and lymphocytes are also present in the stroma [139, 140]. The surface area of the choroid plexus epithelial cell microvilli is approximately half of the surface area of the BBB [141]. Stromal capillaries in the BCSFB are fenestrated, unlike BBB capillaries, allowing free movement of molecules up to 800 kDa between stroma and peripheral blood [135]. However, tight junctions, adherens junctions, and desmosomes are present between the apical borders of the epithelial cells [127, 133]. The Aβ in CSF is thought to originate primarily from ISF draining into the ventricles [142]. Active transfer of solutes between CSF and peripheral blood occurs in both directions across the choroid plexus [143–145] although for Aβ, movement from CSF to peripheral blood predominates [145]. Permeability of Aβ at the choroid plexus is approximately 10-fold greater than its permeability in either direction of the BBB [146].
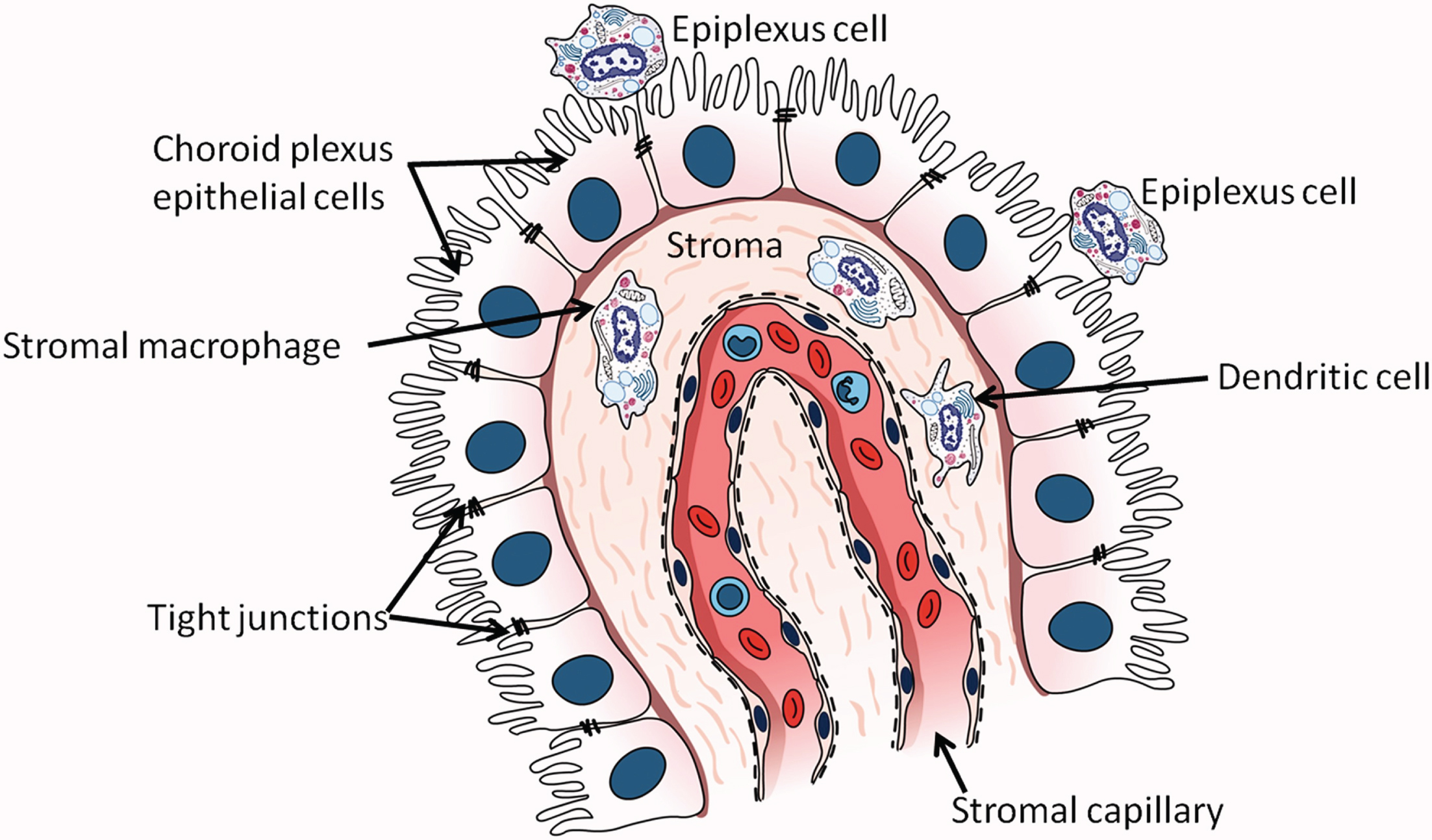
The choroid plexus in the lateral ventricle. Epithelial cells of the choroid plexus rest on a basement membrane and contain microvilli projecting into the lumen of the lateral ventricle. Tight junctions are present between cells near their apical surfaces. (From: Kaur C, Rathnasamy G, Ling EA. The Choroid Plexus in Healthy and Diseased Brain. J Neuropathol Exp Neurol 2016;75(3):198–213; copyright Oxford University Press. Reproduced with permission of Oxford University Press).
The BCSFB undergoes age-related structural and functional changes. Structural changes including loosening of tight junctions, thickening and flattening of epithelial cell basal membrane, deposition of fibrillar inclusions (Biondi bodies) and lipofuscin, and stromal thickening and calcification [139, 148]. Functional changes include decreases in protein synthesis, ion transport, and clearance of CSF proteins [129, 149–152]. BCSFB capillary permeability also decreases during normal aging [153, 154]. Conflicting results have been published as to whether CSF production is reduced with normal aging [150, 155].
The choroid plexus has an extensive capacity for taking up Aβ from CSF [145, 156]. Aβ transporters LRP1, ABCB1, LRP2, and RAGE are expressed on BCSFB epithelium [35, 157]. A study of age-related changes in the expression of Aβ transporters on the rat choroid plexus found that LRP1 and ABCB1 increased, while LRP2 decreased and RAGE was unchanged [35]. LRP1 and ABCB1 are thought to function at the BCSFB as Aβ efflux transporters, while LRP2 mediates movement of Aβ in a bi-directional manner from both CSF and blood into the choroid plexus [35]. The role of RAGE with regard to Aβ transport at the BCSFB is unclear [35]; the authors of an in vitro study of Aβ uptake from artificial CSF by the rat choroid plexus concluded that RAGE did not appear to be involved in Aβ uptake into the choroid plexus [145]. The choroid plexus can also produce Aβ and can degrade it due to the presence of insulin degrading enzyme, neprilysin, and endothelin-converting enzyme-1 in choroidal epithelial cells [146, 156].
The age-related structural changes in the BCSFB described above are exacerbated in AD [158, 159]. Binding of immunoglobulins and C1q, the first component of the classical complement activation pathway, to choroid epithelial basement membrane has also been reported in AD brain [160]. Functional changes in the BCSFB which occur in AD include increases in pro-inflammatory gene expression [161] and in the expression of genes encoding for interferons, mammalian target of rapamycin (mTOR) signaling, peroxisome proliferator-activated receptors (PPARs), and acute phase proteins [162], decreased expression of genes encoding for ion transporters, tight junction proteins, and proteins involved in mitochondrial ATP synthesis, and reduced mitochondrial enzyme activity [163, 164]. Decreased BCSFB expression of vascular endothelial growth factor (VEGF) was reported in the APP/PS1 transgenic mouse model of AD [165]. Impaired functioning of the choroid plexus was suggested to be an early pathogenic event in LOAD, possibly preceding deposition of cerebral Aβ [132]. The relationship between BCSFB dysfunction and AD progression is unclear, with some studies concluding that changes in permeability of the BCSFB barrier did not correlate with AD severity [166, 167]. Aβ is deposited in the AD choroid plexus, perhaps due in part to its decreased degradation by choroid epithelial cells [141]. The presence of Aβ in the AD choroid plexus is associated with increased local levels of reactive oxygen species [164] which may induce apoptotic cell death [164, 168]. Aβ42 oligomers increase BCSFB permeability by activating MMPs [169].
CSF production decreases in AD [170, 171]. CSF turnover is also reduced by about 50% [172], which may contribute to the development of AD due to slower clearance of cerebral waste products [127]. The expression of Aβ transporters on the choroid plexus may be altered in AD although not necessarily decreased. A transcriptomic study found increased expression of LRP1, perhaps in compensation for reduced clearance of Aβ from cerebral capillaries [162], and a similar finding was reported in 3xTg-AD mice [142]. However, no differences were found for CSF LRP1 concentrations between AD patients and age-matched control subjects [173] although LRP2 concentrations were reported to be reduced in AD choroid plexus [174] and CSF [175]. No reports were found of changes in ABCB1 or RAGE levels on the BCSFB or in CSF in AD, although in 3xTg-AD mice immunoreactivity for RAGE on the choroid plexus was increased while TTR expression was decreased [142].
TTR requires further mention with regard to its expression and function on the AD choroid plexus. It is produced in the CNS primarily by the choroid plexus [176] while in the periphery it is produced mainly by the liver. In peripheral blood it binds to thyroxine and vitamin A [177, 178]. TTR expression is regulated by 17-beta-estradiol [179]. The level of TTR production by the choroid plexus reflects the health status of the BCSFB [143]. TTR was reported to be the major Aβ-binding protein in CSF [180]. Its possible neuroprotective role in AD was reviewed by Saponaro et al. [176] and Gião et al. [181]; TTR’s binding to Aβ prevents Aβ aggregation and inhibits Aβ-mediated neurotoxicity [182–186] and it may facilitate Aβ efflux from the brain via the BBB by interacting with LRP1 [180]. TTR may also participate in neuronal regeneration [187] and angiogenesis [188]. One study reported decreased TTR levels in AD CSF [189] but a later study found no evidence for altered choroid plexus production of TTR in AD [190]. TTR tetramers have been suggested to be unstable in AD, which could reduce their ability to bind Aβ [181].
The choroid plexus detects both peripheral and CNS inflammatory signals [133] and serves as a gateway for trafficking of immune cells into the CSF [191]. Tumor necrosis factor (TNF) has been suggested to be the main upstream inflammatory mediator in the AD choroid plexus, via its signaling through TNF receptor 1 (TNFR1) [126]. Systemic inflammation, a possible risk factor for AD [192–194], may increase BCSFB permeability. In the AppNL - G - F (APP knock-in) mouse model of AD, low-grade inflammation induced by intraperitoneal injections of lipopolysaccharide (LPS) reduced the immunoreactivity of tight junction proteins in choroid plexus epithelial cells [195]. An earlier study found that sepsis, induced by injection of a lethal dose of LPS, extensively damaged BCSFB permeability [196]. In vitro measurements in the study cited above involving low-grade LPS-induced peripheral inflammation [195] found that interleukin-1β, whose hippocampal concentration was increased in the APP knock-in mice after LPS administration, reduced the transport of Aβ across choroid plexus epithelial cells.
Multiple approaches have been suggested or explored in experimental systems for increasing BCSFB functioning in AD. These approaches are shown in Table 3.
Experimental approaches for increasing BCSFB functioning
CSF, cerebrospinal fluid; LRP2, low density lipoprotein-related protein 2; MMP, Matrix metalloproteinase; TNF, tumor necrosis factor; TTR, transthyretin. aCerebral implantation of choroid plexus epithelial cells in APP/PS1 mice resulted in decreases in Aβ deposition, tau hyperphosphorylation, and astrocyte immunoreactivity [197]; implantation of microencapsuled choroid plexus epithelial cells into Aβ-treated rat brain decreased apoptosis and gliosis while increasing neurogenesis [198]. bJohanson et al. [159] and Wostyn et al. [200] suggested that therapeutic strategies stimulating increased CSF secretion by the choroid plexus should be tested; for example, chronic administration of caffeine to rats increased CSF production [199]. cThe choroid plexus secretes an anti-inflammatory microRNA, miR-146a, into CSF. 5XFAD mice provided with environmental enrichment increased hippocampal expression of miR-146a, and this was associated with downregulation of nuclear factor-kappa B [201]. dRegulatory T-cells (Treg) have been suggested to cause systemic immunosuppression in AD, inhibiting the entry of immune cells (monocyte-macrophages and T cells) into the CNS. Transient depletion of Foxp3 + Tregs in 5XFAD mice increased the expression of leukocyte trafficking molecules in choroid plexus and reduced SP counts in hippocampus (dentate gyrus) and cerebral cortex [202]. eThe choroid plexus synthesizes and secretes growth factors including FGF2 and TGFβ. Pharmacological manipulation of the choroid plexus (to modify the expression of choroid epithelial cell proteins) and/or supplementation with growth factors was suggested as a therapeutic approach for AD [159]. fMice lacking gut microbiota were found to have increased BCSFB permeability, which could be reversed with gut microbiota recolonization or short-chain fatty acid (SCFA) supplementation. Treatment of AppNL - G - F mice with SCFAs lowered their brain Aβ levels [203]. gKlotho is a multifunctional protein with membrane-bound, secreted, and intracellular forms. Its functions include regulation of oxidative stress, growth factor signaling, and ion homeostasis [209]. Treatment of 10-month-old senescence-accelerated mouse prone-8 (SAMP8) mice with the herbal compound ligustilide upregulated choroid plexus expression of Klotho and decreased memory deficits, Aβ42 accumulation, and tau phosphorylation [204]. hIncreasing of LRP2 expression on choroid plexus has been suggested as an approach for increasing BCSFB clearance of cerebral Aβ [174, 175]. iIn Aβ42-treated rats, the anti-oxidant lycopene blocked activation of NF-κB p65 and toll-like receptor 4 (TLR4) on the choroid plexus, inhibiting production of pro-inflammatory cytokines [205]. Similar results were found when administration of lycopene was combined with transplantation of human amniotic epithelial cells [206]. jOligomeric Aβ42, injected into C57BL/6 mouse cerebral ventricles, increased inflammatory gene expression and reduced BCSFB integrity. The BCSFB damage was prevented by co-administration into the cerebral ventricles of a broad-spectrum MMP inhibitor, GM6001 [169]. kMouse embryonic stem cells were induced in vitro to differentiate into thymic epithelial cell precursors, then transplanted into 3XTg-AD mice. This increased the expression of leukocyte homing and trafficking molecules in the choroid plexus [207]. lTNF was found by microarray analysis to be the main inflammatory mediator upstream of the choroid plexus in AD choroid plexus specimens. TNF signaling via TNFR1 was blocked in APP/PS1 mice by crossing the mice with TNFR1-deficient mice. This lack of TNF signaling decreased the expression of the inflammatory mediators IL6 and CXCL9 in the choroid plexus. [126]. mTTR is synthesized by the choroid plexus [210]. Increasing the expression of TTR in APP23 mice by crossing them with mice overexpressing human TTR resulted in normalization of the “APP23 behavioral phenotype” (as measured with Barnes maze testing) and lower hemi-brain levels of SDS-soluble and formic acid-soluble Aβ [208].
GLYMPHATIC (PARAVASCULAR) CLEARANCE OF Aβ
In 2012, Iliff et al. described a pathway for cerebral clearance of Aβ and other solutes via the paravascular space. They referred to this pathway as “glymphatic” in reference to glial-associated lymphatic drainage [211]. In the present review the glymphatic system is considered to be separate from intramural periarterial drainage, also known as perivascular drainage (discussed in the following section), although some investigators have suggested that the two systems may be the same pathway studied under different conditions [212, 213]. (See discussion of both systems by Bacyinski et al. [214]). The paravascular space, also known as the Virchow-Robin space, surrounds the cerebral vasculature (both arteries and veins), while the perivascular space is present within the middle layers of the basement membrane of arterial smooth muscle cells. A schematic drawing of the two systems is shown in Fig. 3. The use of the terms “paravascular” and “perivascular” is confusing, and glymphatic drainage is referred to as “perivascular” in some reviews [215–219]. The glymphatic system has been the subject of other recent reviews in addition to the one by Bacyinski et al.; see, for example, Jessen et al. [220], Bakker et al. [213], Benveniste et al. [221], Plog and Nedergaard [217], and Abbott et al. [222].
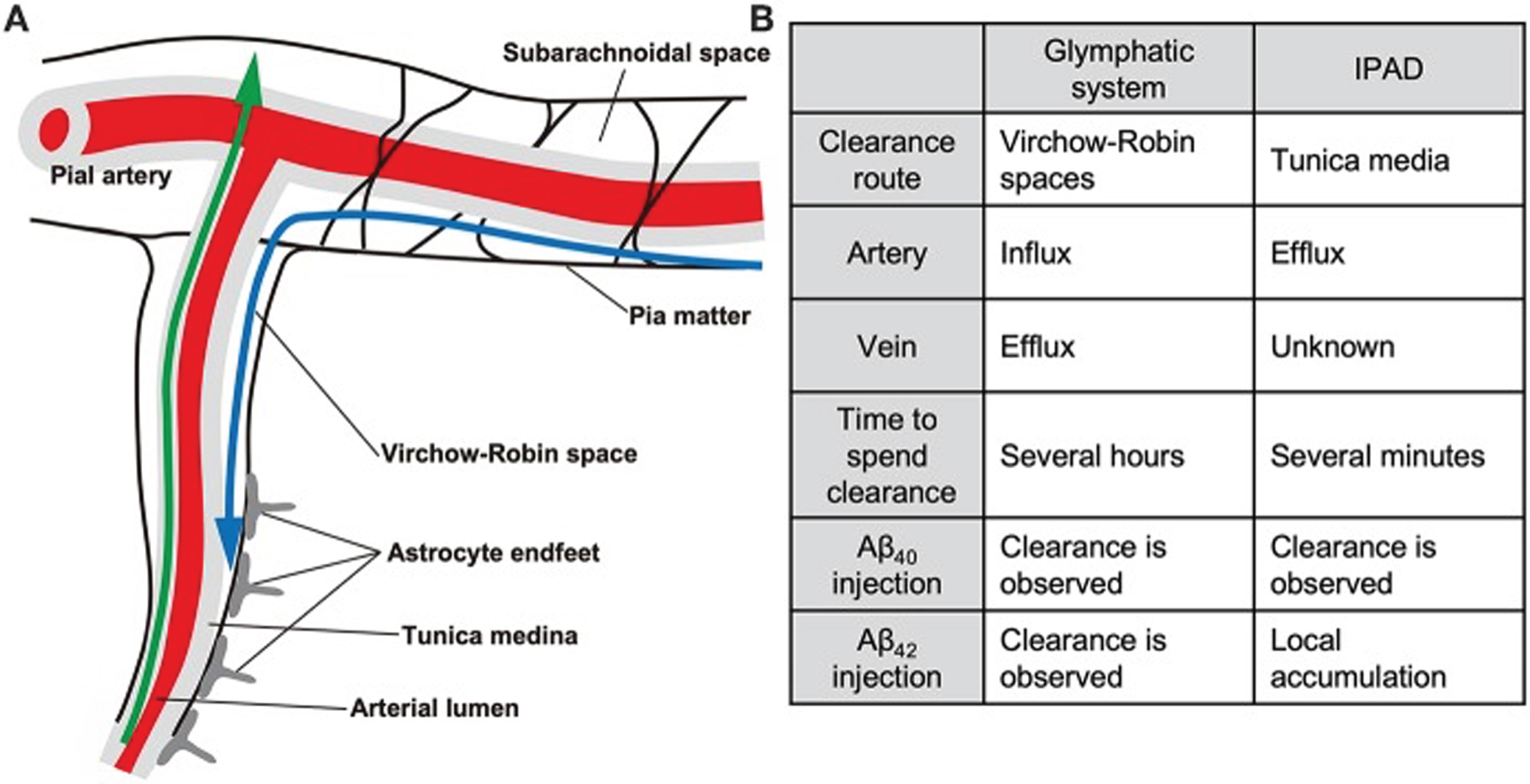
The glymphatic and intramural periarterial (IPAD) systems. In the glymphatic (paravascular) system, indicated by the blue arrow, Aβ and other solutes in interstitial fluid are cleared from the brain via the paravascular (Virchow-Robin) space, which surrounds both cerebral arteries and veins. This is facilitated by the water channel protein aquaporin-4 which is present in the astrocytic endfoot processes lining the cerebral microvasculature. CSF in the glymphatic system enters the brain through para-arterial spaces, mixes with interstitial fluid, then leaves the brain via paravenous spaces. In the IPAD system, indicated by the green arrow, solutes (including Aβ) and interstitial fluid exit the brain via basement membranes of capillaries and arteries. Drainage of solutes via IPAD is in the opposite direction to that of cerebral blood flow. The pattern of drainage of solutes via IPAD is similar to the pattern of deposition of Aβ found in CAA, i.e., along cerebral arteries and capillaries. (From: Saito S, Yamamoto Y, Ihara M. Development of a Multicomponent Intervention to Prevent Alzheimer’s Disease. Front Neurol. 2019;10 : 490 [publisher: Frontiers Media S.A].) This figure is copyrighted by Drs. Saito, Yamamoto, and Ihara, who permitted its reproduction.
The finding in the initial study of the glymphatic pathway by Iliff et al. [211] that cerebral clearance of radiolabeled Aβ40, after its intrastrial injection, was decreased by 55% in mice lacking the water channel protein aquaporin-4 (AQP4) compared to its clearance in wildtype mice suggested that drainage via the paravascular space may play an important role in Aβ clearance from the brain. A more recent study in C57BL/6 mice found that inhibiting AQP4 function resulted in increased accumulation of Aβ40 around cerebral vessels [223]. AQP4 is present in the astrocytic endfoot processes lining the cerebral microvasculature [224] and is thought to facilitate movement of fluid through the glymphatic system [211, 226]. Decreased AQP4 localization adjacent to cerebral vessels has been reported in AD [227]. In the glymphatic system, CSF enters the brain through para-arterial spaces [228] and after mixing with ISF [220] it leaves the brain through paravenous spaces [211, 217]. Drainage of CSF and its solutes occurs by multiple routes including cervical lymphatics, arachnoid granulations, the choroid plexus, peripheral blood, and possibly dural lymphatics [36, 229–231]. Movement of CSF from the subarachnoid space into the brain, as well as efflux of ISF from the brain, has been suggested to be driven by bulk flow rather than diffusion [217, 232–234] although this has been challenged by some investigators [218, 235–237]. The rate of fluid flow through the glymphatic system is thought to be regulated in part by pulsing of cerebral arteries, with reductions in arterial pulsatility due to cerebrovascular pathology possibly contributing to the decreased Aβ cerebral clearance in AD [36]. Vasomotion (low-frequency contractions and dilations of smooth muscle cells) has also been suggested to influence glymphatic flow [238]. Interestingly, sleep increases fluid flow through the glymphatic system by expanding the interstitial space (due to reduced noradrenergic tone), thereby reducing resistance to fluid movement [239].
In a study of elderly cognitively normal subjects, some genetic variants of AQP4 were found to modify the effects of sleep-related parameters on brain levels of Aβ [240], further supporting a role for the glymphatic system in clearance of brain Aβ. Glymphatic clearance of Aβ was decreased by 40% in old mice, with age-related lowering of perivascular AQP4 polarization and cerebral arterial pulsatility suggested to be contributing factors [241]. Reductions in glymphatic inflow (2-fold) and glymphatic clearance (1.2-fold) of intracisternally injected Aβ were found in the APP/PS1 mouse model of AD [242]. These decreases were attributed in part to disruption of glymphatic transport by Aβ oligomers, which co-localized with AQP4-expressing astrocytes and may have contributed to depolarization of AQP4 from astrocytic endfeet. Glymphatic transport of Aβ42 was less efficient than for Aβ40, possibly because of the greater propensity of Aβ42 to aggregate. Notably, reduction in glymphatic drainage of Aβ in APP/PS1 mice was detected in 3-4-month-old mice, prior to the development of extensive Aβ pathology. In another study in APP/PS1 mice, AQP4 knockout resulted in increased numbers of cortical and hippocampal plaques, as well as deposition of Aβ in cortical and leptomeningeal vessels [243]. In the tg-ArcSwe mouse model of AD, which develops perivascular as well as neuropil SPs, loss of astrocyte depolarization was found at sites of perivascular amyloid deposition [244]. Similar findings were reported in APP/PS1 mice [243]. Further, experimentally induced impairment of glymphatic drainage was found to increase Aβ deposition in APP/PS1 mice [245] and in 5xFAD and J20 mice [246]. These findings suggest that reduced glymphatic clearance of Aβ may contribute to the increased cerebral Aβ burden in AD. However, the feasibility of slowing the development of Aβ pathology by therapeutic increasing of glymphatic functioning has been questioned. Bakker et al. [213] noted poor correlation between the preferential deposition of Aβ along arteries and its clearance along veins, and Saito et al. [247] argued that Aβ rarely accumulates in the venous system, and that arterial Aβ accumulation is most prominent within the tunica media, not the paravascular space. van Veluw et al. [248], citing literature from studies in patients with CAA and animal models of CAA, noted that Aβ clearance is more likely to occur along arteries than veins.
Glymphatic drainage is decreased in experimental models of other CNS disorders in addition to AD including stroke [249], traumatic brain injury [250], and microinfarcts [251]. However, a review published in 2018 [217] noted that there had been no therapeutic efforts to improve glymphatic functioning in these disorders. The possibility has been suggested, with regard to idiopathic normal pressure hydrocephalus (in which glymphatic drainage is also compromised [252]), that improving sleep quality might reduce the development of this condition by improving glymphatic function [219].
Approaches to increase glymphatic drainage of Aβ have been investigated in experimental models. These approaches are shown in Table 4.
Experimental approaches for increasing glymphatic drainage of cerebral Aβ. Although the number of experimental approaches for increasing glymphatic drainage of cerebral Aβ is less than for BBB and BCSFB clearance of cerebral Aβ, two of the approaches, melatonin and omega-3 polyunsaturated fatty acids, have been evaluated in AD clinical trials and are used to treat many other conditions
AT1, angiotensin II type 1; NBP, L-3-n-butylphthalide; VEGF-C, vascular endothelial growth factor C. aTraumatic brain injury to mice was reported to decrease the polarized localization of AQP4 at endfoot processes of reactive astrocytes [264]. Deficiency in angiotensin II type 1 (AT1) receptors (induced by knockout of AT1 in C57BL/6 mice) inhibited AQP4 depolarization after traumatic brain injury, improving glymphatic drainage of Aβ [253]. bTreatment of APP/PS2 mice with 5-caffeoylquinic acid, also known as chlorogenic acid, resulted in upregulation of LRP1 and normalization of perivascular AQP4 polarization in the hippocampus, together with decreased hippocampal Aβ deposition [254]. cA study in SAMP8 mice subjected to electroacupuncture for 8 weeks suggested that this treatment might improve AQP4 polarity and glymphatic drainage [255]. dWheel running improved glymphatic clearance, including astrocytic AQP4 expression and polarization, in aged mice [256]. Similar findings were reported in 3-month-old but not 7-month-old APP/PS1 mice [257]. eAdministration of melatonin to Tg2576 mice increased drainage of cerebral Aβ into cervical and axillary lymph nodes. This was associated with decreased brain levels of oligomeric Aβ40 [258]. The melatonin-induced increase in lymphatic drainage of Aβ was suggested to be due to enhanced glymphatic clearance [265]. fNBP (L-3-n-Butylphthalide), an extract from Chinese celery, improved glymphatic clearance of cerebral Aβ in wild-type mice by increasing cerebral pulsation. Administration of NBP to APP/PS1 mice reduced Aβ deposition and parenchymal Aβ levels [260]. An earlier study with 3xTg-AD mice found that treatment with NBP decreased diffuse but not fibrillar plaques [259]. gCerebral clearance of Aβ in fat-1 transgenic mice was increased by treatment with omega-3 polyunsaturated fatty acids. This effect was abolished in AQP4-knockout mice. Omega-3 polyunsaturated fatty acids were found to inhibit astrocyte activation and to maintain AQP4 polarization after Aβ injection [261]. hA review of photobiomodulation therapy indicated that it promoted increased glymphatic clearance of cerebral Aβ via meningeal lymphatic vessels [262]. iContinuous theta-burst stimulation increased glymphatic fluid transport in APP/PS1 mice. This effect was attributed to improved AQP4 polarization [263]. jTreatment of old C57BL/6J mice with VEGF-C, delivered by adenoviral vector or transcranial injection, resulted in increased lymphatic vessel diameter and improved meningeal lymphatic drainage of a tracer into deep cervical lymph nodes. This effect could not be replicated in J20 or 5XFAD mice [246].
Melatonin is used to improve the onset, duration, and quality of sleep [266]. The possibility that it may increase glymphatic drainage deserves further mention. Sleep disturbances are common in AD patients [267] so glymphatic clearance of Aβ might be increased in these individuals if their quality of sleep could be improved [215]. Melatonin concentrations in CSF are decreased in AD patients and decrease further during progression of the disease [268]. Melatonin was suggested as a therapeutic approach for AD (discussed below) based on its effects in the APP/PS1 transgenic mouse model of AD [269].
INTRAMURAL PERIARTERIAL DRAINAGE (IPAD) CLEARANCE OF Aβ
In 1974 Cserr and Ostrach examined clearance of Blue Dextran from mouse brain after injecting the dye into the caudate nucleus [233]. They found that the dye was “transported away from the injection site by bulk flow of cerebral ISF, possibly along the course of cerebral blood vessels.” Weller, Carare, and colleagues explored this pathway further [270–272] and found that drainage of solutes from brain parenchyma occurred along basement membranes of capillaries and arteries, but no intracerebrally injected dye was detected in the walls of veins. In contrast to glymphatic drainage, the drainage pattern observed in these studies was in the opposite direction to that of cerebral blood flow. Studies in experimental animals suggested that solutes draining from the brain along blood vessel walls emptied into cervical lymph nodes [234] via dural lymphatic vessels [273]. An age-related impairment in this mechanism in mice was suggested to be due to decreased amplitude of arterial pulsations and/or changes in the composition of basement membranes [274, 275]. Clearance of Aβ40 from the brain via IPAD is approximately 6-fold slower than its LRP1-mediated clearance across the BBB [62]. IPAD was initially suggested to be driven by arterial pulsation, specifically the refractory or “reflection” wave that follows each main pulse wave [276], but this was challenged by later studies suggesting that the driving force behind IPAD may be vasomotion produced by spontaneous contraction and relaxation of smooth muscle cells [38, 277]. Analysis of a computational model of IPAD led to the conclusion that drainage of solutes by both diffusion and bulk flow occurred in IPAD [278].
Studies in mice with intracerebrally injected Aβ have shown that its drainage follows the same pattern as tracer dyes [279]. A similar pattern of deposition of Aβ along cerebral arteries and capillaries is found in CAA, reflecting failure of Aβ to be cleared from these vessels [279–282]. Clinical sequelae of CAA include lobar intracerebral hemorrhages, cerebral microbleeds, hemorrhagic and ischemic stroke, cognitive impairment, and gait impairment [283–286]. CAA is present in up to 90% of AD patients [287–290] and is associated with more rapid progression of AD [291]. ApoE, particularly apoE4, may impair IPAD-mediated clearance of Aβ [292]. ApoE4-expressing astrocytes secrete more fibrinectin and less laminin than apoE3-expressing astrocytes, promoting aggregation of Aβ on cerebrovascular basement membranes [293]. In CAA, apoE co-localizes with Aβ [294–296].
Several approaches have been suggested or investigated in experimental systems for increasing IPAD clearance of Aβ. These are shown in Table 5.
Experimental approaches for increasing IPAD clearance of Aβ
APOA-I, apolipoprotein A-I; ApoE4, apolipoprotein E4. aThe possibility was suggested that direct oral anticoagulants (DOAC) might mitigate hypoperfusion-enhanced neurodegenerative processes including reduced perivascular clearance of Aβ [311]. The DOAC dabigatran etexilate lowered cerebral Aβ in TgCRND8 mice [297]. bKnockout of the APOA-I gene, which encodes for apolipoprotein A-I (apoA-I), increased IPAD-mediated clearance of cerebral Aβ in Tg2576 mice, and decreased cerebrovascular and parenchymal levels of insoluble Aβ [298]. ApoA-I is the main structural protein of high-density lipoprotein and is present in CSF [312]. cIntracerebroventricular injection of chitin in TgCRND8 mice stimulated perivascular macrophages and reduced Aβ42-labeled cortical blood vessels [299]. dThe selective type 3 phosphodiesterase inhibitor cilostazol increased perivascular drainage (i.e., IPAD-mediated clearance) of Aβ1 - 40 in Tg-SwDI mice [300]. eApoE was found to co-localize with Aβ in basement membrane drainage pathways in arterial walls. The attachment of apoE/Aβ complexes to laminin in the basement membrane was weaker for apoE4/Aβ complexes than for apoE3/Aβ complexes, suggesting that perivascular clearance of apoE4/Aβ complexes may be less efficient than for other isoforms of apoE. Therapeutic correction for apoE4/Aβ/laminin interactions was suggested as a possible approach to increase the efficiency of Aβ clearance [301]. fTreatment with the antioxidant ergothioneine reduced the concentration of Aβ in neuroretinas of 5XFAD mice. Ergothioneine was suggested to increase Aβ clearance by blood-derived phagocytic macrophages and via perivascular drainage [302]. gFasudil hydrochloride is a selective Rho- associated, coiled-coil containing protein kinase (ROCK) inhibitor which stimulates the PI3K/Akt/eNOS pathway [313]. Similar to the effect of acetylcholine, stimulation of this pathway induces vasodilation by activating endothelial nitric oxide synthase, promoting synthesis of nitric oxide. Treatment with fasudil hydrochloride increased IPAD in the hippocampus of both control mice (C57BL/6) and mice previously treated with mu-saporin, an immunotoxin which targets cholinergic neurons [303]. hTreatment of 3xTg-AD mice with the gastrointestinal hormone ghrelin upregulated capillary microRNAs miR126 and 145 in hippocampus and cerebral cortex, and lowered cerebral Aβ oligomer concentrations. This reduction was suggested to be due to increased perivascular clearance of Aβ, possibly due to activated endothelium and increased pericyte coverage of capillaries. Decreased expression of RAGE may also have been a contributing factor [50]. Treatment of 5XFAD mice with the ghrelin agonist MK-0677 reduced Aβ deposition in the frontal cortex [305] but in another study no decrease in cerebral Aβ was found when ghrelin was administered to this transgenic mouse model of AD [306]. (See also: Moon et al., 2014 [304]). iLysyl oxidase participates in remodeling of extracellular matrix (ECM) by catalyzing crosslinking of ECM components including collagen IV, laminin, and fibronectin. Inhibition of lysyl oxidase was suggested to improve IPAD clearance of Aβ [307]. jChronic administration of the anti-Aβ monoclonal antibody Ponezumab, which binds a C-terminal epitope of Aβ40 (Aβ33 - 40) and is thought to promote “peripheral sink” Aβ clearance [314], to PSAPP mice reduced CAA-type pathology [308]. kTaxifolin is a plant flavonoid with anti-oxidant and anti-inflammatory effects [310]. Taxifolin treatment of a mouse model of CAA, Tg-SwDI mice, inhibited formation of oligomeric Aβ and reduced cerebrovascular Aβ immunoreactivity [309]. Taxifolin was suggested in that study to have increased Aβ clearance via IPAD and the BBB.
Enhancing the efflux mechanisms discussed in this review offers potential therapeutic options for lowering the concentrations of soluble Aβ conformations in the brain. Whether this approach would slow AD progression is unknown. Previous attempts to slow AD progression by lowering brain levels of soluble Aβ failed (with the exception of lecanemab, which preferentially binds to soluble aggregates (protofibrils) as discussed below); these include the monoclonal antibodies solanezumab [15] and ponezumab [314], both of which targeted monomeric Aβ, as well as BACE1 inhibitors [315] and γ-secretase inhibitors [316]. The failure of these approaches suggests that increasing cerebral efflux of Aβ is unlikely to slow disease progression if used as monotherapy. But if used as an adjunct to lecanemab or donanemab, this might allow greater slowing of AD progression than can be achieved by treatment with either antibody alone. Antibody binding to Aβ can promote its clearance from the brain by several mechanisms: (a) activation of microglia via binding of antibody Fc fragments to the microglial Fc receptor (FcR), thereby increasing phagocytic uptake (and subsequent proteolytic degradation) of Aβ [317] (although this is true for fibrillar Aβ, microglial uptake of soluble Aβ may be via fluid-phase macropinocytosis [318], although whether it occurs by phagocytosis or fluid-phase macropinocytosis is unclear [319]), (b) activation of the classical complement pathway by crosslinking C1q between adjacent Fc fragments of IgG molecules (bound, in this case, to Aβ) [320] (complement activation results in cleavage of C3 to generate C3b; when bound by microglial complement receptor CR1, C3b is cleaved to iC3b, which functions as an opsonin, promoting phagocytosis when bound to the microglial complement receptor CR3 [321], (c) binding of antibody-Aβ immune complexes to the BBB’s neonatal FcR, which facilitates their crossing the BBB [34], (d) antibody-mediated dissolving of fibrillar Aβ [322], and (e) a “peripheral sink” mechanism in which antibody binding to Aβ in peripheral blood alters the equilibrium between systemic and brain Aβ levels, inducing increased BBB-mediated efflux of Aβ from the brain [323]. (Some investigators have challenged the validity of the peripheral sink hypothesis [324, 325). Donanemab binds to a fibril-specific N-terminal epitope on pyroglutamate-modified Aβ 326], promoting microglial phagocytosis of Aβ [327]. Lecanemab binds large soluble Aβ aggregates (protofibrils) and has lower selectivity for fibrils [328]; the mechanism(s) by which it increases clearance of cerebral Aβ is less clear, although BBB clearance (via binding of lecanemab-Aβ complexes to the BBB’s neonatal FcR) might be involved.
A related question is whether therapeutic increasing of cerebral Aβ efflux would lower the risk of developing amyloid-related imaging abnormalities (ARIA) in subjects treated with lecanemab or donanemab. ARIA is indicated by detection, on MRI, of either edema and effusion (ARIA-E; vasogenic edema in the parenchyma or sulcal effusions in leptomeninges [329]) or bleeding, as indicated by microhemorrhages and/ hemosiderin deposition (ARIA-H). ARIA-E reflects extravasation of proteinaceous fluid while ARIA-H reflects extravasation of erythrocytes [330]. The incidence of ARIA increases when subjects are treated with monoclonal antibodies targeting Aβ’s N-terminal amino acids, and apoE4-positive subjects have an increased risk for developing ARIA when treated with these antibodies [331]. In lecanemab’s phase 3 trial, the incidence of ARIA-E was 12.6% for lecanemab-treated subjects and 1.7% for placebo-treated subjects [17] while in donanemab’s phase 3 trial ARIA-E was detected in 24.0% of donanemab-treated and 2.1% of placebo-treated subjects, and ARIA-H was found in 31.4% of donanemab-treated and 13.6% of placebo-treated subjects [20]. The mechanisms underlying ARIA are incompletely understood; it has been suggested to be related to a temporary increase in cerebrovascular Aβ due to antibody mobilization of Aβ from SPs, causing increased vascular fragility and permeability [332, 333]. Increased production of inflammatory cytokines as a consequence of antibody activation of microglia was also suggested to contribute to development of ARIA [334, 335]. Treatment-induced ARIA-E is generally asymptomatic and resolves without treatment [336], although it has been associated with confusion, visual disturbances, headache, and gait abnormalities [333]. Whether the cerebral microhemorrhages indicated by ARIA-H can influence cognitive performance is not clear [337]. Barakos et al. [338], discussing the development of ARIA in patients treated with anti-Aβ monoclonal antibodies, noted that “there may be a recovery of vessel wall integrity and an increase in effective perivascular drainage with continued treatment, and, therefore, some clinical trials have observed that the risk of ARIA is highest early in treatment and subsequently decreases in patients who continue dosing.” Therefore, approaches that increase IPAD (perivascular) - mediated efflux of Aβ (Table 5) might reduce the incidence of ARIA in patients treated with lecanemab or donanemab. As stated above, CAA is often present in AD [287–290]. MRI findings of ARIA-H are similar to those of CAA [330], so increasing IPAD-mediated clearance of cerebral Aβ might also reduce CAA-associated pathology in AD patients.
Some of the experimental approaches discussed above would be problematic in AD patients. For example, the use of viral vectors in human gene therapy trials poses substantial risks [339, 340], intracerebroventricular administration of therapeutic agents would be less desirable than systemically-administered agents, and the use of some anticoagulants has been associated with increased risk for intracerebral hemorrhage [341]. Of note with regard to the possibility of treating AD patients with anti-coagulants, cerebral microbleeding was reported in 29% of AD patients [337] so it would be a concern with this approach [342]. Nevertheless, most of the experimental approaches listed in Tables 2–5 have been used in clinical settings. Clinical applications found for the experimental approaches discussed above are shown in Tables 6–9.
Clinical applications of experimental approaches for improving BBB clearance of cerebral Aβ. Clinical trials are identified by their ClinicalTrials.gov Identifier
AD, Alzheimer’s disease; ADAM10, A disintegrin-like and metalloproteases 10; LDLR, Low-density lipoprotein receptor; MCI, mild cognitive impairment; MMP-9, matrix metalloproteinase-9; PICALM, phosphatidylinositol binding clathrin assembly protein). aSix-month treatment of MCI patients with extra-virgin olive oil (NCT03824197) reduced BBB permeability and improved clinical dementia rating (CDR) and behavioral scores [364]. bThe antimalarial drug artesunate increases PICALM expression at the BBB [122]. cSomatostatin is not used clinically because of its short half-life. Somatostatin analogs, which have longer half-lives, are used [375]. dIn clinical trials, Sano et al. [376] concluded that treatment with α-tocopherol slowed AD progression, and a similar conclusion was reached by Dysken et al. [377].
Clinical applications of experimental approaches for improving BCSFB clearance of cerebral Aβ. Clinical trials are identified by their ClinicalTrials.gov Identifier
CSF, cerebrospinal fluid; Foxp3, forkhead box P3; LRP2, low-density lipoprotein receptor-related protein 2; MMP, matrix metalloproteinase; TNF, tumor necrosis factor; TTR, transthyretin. aNo clinical trials or human applications involving transplant of choroid plexus epithelial cells were found, although this approach continues to be investigated in experimental systems (see reviews by Jang and Lehtinen, 2022 [409], and Liu et al., 2022 [410]). bNo clinical trials or human applications were found in which the intent was to increase CSF production; however, the effects of caffeine, whose chronical consumption increased CSF production in rats [199], have been well studied (reviewed by Cappelletti et al., 2015 [411]). cLigustilide is the main active ingredient in the root of the herb Dong quai. Pure ligustilide has not been tested in humans due to its poor stability and bioavailability [412].
Clinical applications of experimental approaches for improving glymphatic/paravascular clearance of cerebral Aβ. Note the large number of clinical applications of melatonin and omega-3 polyunsaturated fatty acids. Clinical trials are identified by their ClinicalTrials.gov Identifier
AD, Alzheimer’s disease; ADAS-cog, Alzheimer’s Disease Assessment Scale; ADCS-ADL, Alzheimer’s Disease Cooperative Study – Activities of Daily Living; CDR-SB, Clinical Dementia Rating sum of boxes; CIBIC-Plus, Clinician’s Interview-Based Impression of Change Plus caregiver input; COVID-19, Coronavirus Disease of 2019; PUFA, polyunsaturated fatty acids; VEGF-C, vascular endothelial growth factor C. aWang et al. [415] treated patients with mild-to-moderate AD with dl-3-n-butylphthalide plus donepezil (n = 49) or donepezil alone (n = 43) for 48 weeks (ClinicalTrials.gov Identifier: NCT02711683). Changes in scores for Alzheimer’s Disease Assessment Scale (ADAS-cog) and Alzheimer’s Disease Cooperative Study - Activities of Daily Living (ADCS-ADL) were significantly different between the two treatment groups. Wang et al. concluded that the patients treated with dl-3-n-butylphthalide plus donepezil had slower cognitive decline and better performance of activities of daily living than the patients treated with donepezil alone. bJia et al. [416] conducted a randomized, double-blind, placebo-controlled trial in which patients with subcortical vascular cognitive impairment without dementia (n = 281) were treated for six months with dl-3-n-butylphthalide or placebo. Statistically significant differences (improved functioning) vs. placebo were found for ADAS-cog and Clinician’s Interview-Based Impression of Change Plus caregiver input (CIBIC-Plus) scores. cA meta-analysis of seven randomized controlled clinical trials in which AD patients were treated with melatonin found no improvement in measures of cognitive functioning [421]. dFreund-Levi et al. [430] conducted a randomized, double-blind, placebo-controlled clinical trial involving omega-3 supplementation of patients with mild-to-moderate AD. Statistically significant treatment effects were found for depressive symptoms in non-apoE4 carriers, and for symptoms of agitation in apoE4 carriers. Chiu et al. [431] performed a similar study in which subjects with mild-to-moderate AD or MCI were treated with omega-3 PUFA or placebo. Statistically significant improvement for treatment vs. placebo groups was found in ADAS-cog scores for the MCI patients. In contrast, a clinical trial by Quinn et al. [432] involving supplementation of mild-to-moderate AD patients with the omega-3 fatty acid docosahexaenoic acid found no statistically significant effects on ADAS-cog or Clinical Dementia Rating sum of boxes (CDR-SB) scores. Burckhardt et al. [433], in a systematic review, concluded that there was “no convincing evidence for the efficacy of omega-3 PUFA supplements in the treatment of mild to moderate AD.” eNabavi et al. [439] reviewed the effects of omega-3 therapy on breast cancer, colorectal cancer, leukemia, gastric cancer, pancreatic cancer, esophageal cancer, prostate cancer, lung cancer, head and neck cancer, and cancer-related cachexia.
Clinical applications of experimental approaches for improving IPAD clearance of cerebral Aβ. Note the large number of clinical applications of cilostazol. Clinical trials are identified by their ClinicalTrials.gov Identifier
Aβ, amyloid β-protein; AD, Alzheimer’s disease; apoE4, apolipoprotein E4; CAA, cerebral amyloid angiopathy; MCI, mild cognitive impairment. aTaguchi et al. [449] performed a retrospective analysis to examine the effects of cilostazol on cognitive functioning. The analysis included all patients treated at their hospital with cilostazol. Changes in Mini-Mental State Examination (MMSE) scores over > 6 months were compared between subjects who continued to receive cilostazol and subjects who stopped cilostazol treatment. Patients taking acetylcholinesterase inhibitors were excluded. Slowing in the decrease in MMSE scores was found for MCI patients but not for non-cognitively impaired individuals or AD patients. bIhara et al. [450] also performed a retrospective study, comparing changes in MMSE scores over 28–30 months in patients with dementia (MMSE < 27; type of dementia not specified) between subjects treated with donepezil and cilostazol vs. those receiving donepezil alone. Subjects were subdivided into mild dementia (MMSE 22–26) and moderate-to-severe dementia (MMSE < 21). Slowing of the decrease in MMSE scores was found for the patients with mild dementia. Tai et al. [460] performed a small prospective study, comparing 12-month changes in MMSE scores in patients with “stable AD” between subjects taking cilostazol and acetylcholinesterase inhibitors and subjects not receiving cilostazol. The “cilostazol add-on” group was found to have less decrease in MMSE scores than the subjects not taking cilostazol, although no effect of cilostazol was found on CDR-SB scores. Cilostazol-associated slowing of cognitive decline was also reported in small pilot studies for patients with moderate AD [461] and patients with both AD and cerebrovascular disease [462]. In contrast, a 24-week prospective study by Lee et al. [463] found no significant differences in measures of cognition or global functioning between AD patients treated with cilostazol plus donepezil vs. those treated with donepezil alone, although cilostazol was suggested to slow the decrease in regional cerebral metabolism in the AD patients. cA double-blind clinical trial investigating the effects of the ghrelin agonist MK677 in patients with mild-to-moderate AD found no slowing of disease progression [454]. dA one-year clinical trial with Ponezumab in patients with mild-to-moderate AD found no effects on disease progression [314].
A few approaches for increasing cerebral Aβ efflux have been investigated for their effects on slowing of cognitive decline in AD and/or MCI patients. Clinical trials have suggested that α-tocopherol (Table 6) may slow AD progression [376, 377]. Treatment of AD patients with L-3-n-butylphthalide (Table 8) as an adjunct to donepezil was found to slow cognitive decline and loss of the ability to perform activities of daily living to a greater extent than treatment with donepezil alone [415]. A meta-analysis of findings in clinical trials of melatonin (Table 8) in AD patients found no evidence for slowing of cognitive decline [421] and a similar conclusion was reached for omega-3 PUFA (Table 8) in a systematic review [433]. Cilostazol (Table 9) was reported to slow AD-related cognitive decline in prospective studies [460–462] but another study found no effects of cilostazol on cognition or global functioning in AD patients [463]. A phase 2 trial with cilostazol in patients with MCI (NCT02491268) started in 2015 with an expected completion date of December 1, 2020 but results have not been posted on ClinicalTrials.gov. In a retrospective study, treatment with cilostazol was associated with slowing of cognitive decline in MCI patients but not AD patients [449] while in another retrospective study cilostazol slowed the decrease in MMSE scores in patients with mild dementia but not moderate-to-severe dementia (type of dementia not specified) [450]. No slowing of AD clinical progression was found in clinical trials involving the ghrelin agonist MK-677 [454] or the anti-Aβ monoclonal antibody Ponezumab (Table 9) [314].
CONCLUSIONS
Many approaches have been used in experimental systems to increase the efflux of Aβ from the brain. Most of these approaches are used to treat other conditions but few of them have been investigated for possible benefits in AD patients. Although lecanemab and donanemab slow the clinical progression of early-stage AD, there may be a ceiling to their ability to do so. Therapeutic interventions to increase Aβ efflux from the brain, if used as an adjunct to treatment with these antibodies, might allow further slowing of AD progression.
AUTHOR CONTRIBUTIONS
David Loeffler (Conceptualization; Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The author has no acknowledgements to report.
FUNDING
Funding was provided by donations to the Beaumont Foundation to support Alzheimer’s research in the Beaumont Research Institute.
CONFLICT OF INTEREST
The author has no conflict of interest to report.