
Abstract
The “amyloid cascade” hypothesis of Alzheimer’s disease (AD) pathogenesis invokes the accumulation in the brain of plaques (containing the amyloid-β protein precursor [AβPP] cleavage product amyloid-β [Aβ]) and tangles (containing hyperphosphorylated tau) as drivers of pathogenesis. However, the poor track record of clinical trials based on this hypothesis suggests that the accumulation of these peptides is not the only cause of AD. Here, an alternative hypothesis is proposed in which the AβPP cleavage product C99, not Aβ, is the main culprit, via its role as a regulator of cholesterol metabolism. C99, which is a cholesterol sensor, promotes the formation of mitochondria-associated endoplasmic reticulum (ER) membranes (MAM), a cholesterol-rich lipid raft-like subdomain of the ER that communicates, both physically and biochemically, with mitochondria. We propose that in early-onset AD (EOAD), MAM-localized C99 is elevated above normal levels, resulting in increased transport of cholesterol from the plasma membrane to membranes of intracellular organelles, such as ER/endosomes, thereby upregulating MAM function and driving pathology. By the same token, late-onset AD (LOAD) is triggered by any genetic variant that increases the accumulation of intracellular cholesterol that, in turn, boosts the levels of C99 and again upregulates MAM function. Thus, the functional cause of AD is upregulated MAM function that, in turn, causes the hallmark disease phenotypes, including the plaques and tangles. Accordingly, the MAM hypothesis invokes two key interrelated elements, C99 and cholesterol, that converge at the MAM to drive AD pathogenesis. From this perspective, AD is, at bottom, a lipid disorder.
THE PROBLEM
Alzheimer’s disease (AD) is the most common, and arguably the most devastating, neurodegenerative dementia of the elderly [1]. The two main histopathological hallmarks of AD are the accumulation of extracellular neuritic plaques containing amyloid-β (Aβ) and of intracellular neurofibrillary tangles consisting mainly of hyperphosphorylated forms of the microtubule-associated protein tau. The vast majority of AD is sporadic (SAD), but a genetic variant of apolipoprotein E (APOE), APOE4, predisposes people to the disease [2]. Mutations in three genes have been identified in the autosomal-dominant familial form (FAD): amyloid-β precursor protein (APP), presenilin-1 (PS1; gene PSEN1), and presenilin-2 (PS2; gene PSEN2). Presenilins are components of the γ-secretase complex that, together with α- and β-secretase (BACE1), processes AβPP [3].
In the “non-amyloidogenic” pathway (Fig. 1A, left), full-length AβPP (∼700 aa) is first cleaved by α-secretase at the plasma membrane (PM) to produce a long soluble N-terminal fragment (NTF; sAβPPα) and a short membrane-bound 83-aa C-terminal fragment (CTF), called C83 (α-CTF). C83 is then cleaved by the γ-secretase complex to produce two peptides, P3 and the AβPP intracellular domain (AICD), a nucleus-targeted transcription factor. In the alternative “amyloidogenic” pathway (Fig. 1A, right), full-length AβPP is first cleaved by BACE1 within endosomes to produce a slightly shorter soluble N-terminal fragment (sAβPPβ), and a correspondingly slightly longer 99-aa membrane-bound C-terminal fragment, called C99 (β-CTF) [4]. C99 is then cleaved by the γ-secretase complex to produce two peptides, Aβ (predominantly 40-aa long [Aβ40]) and AICD [5].
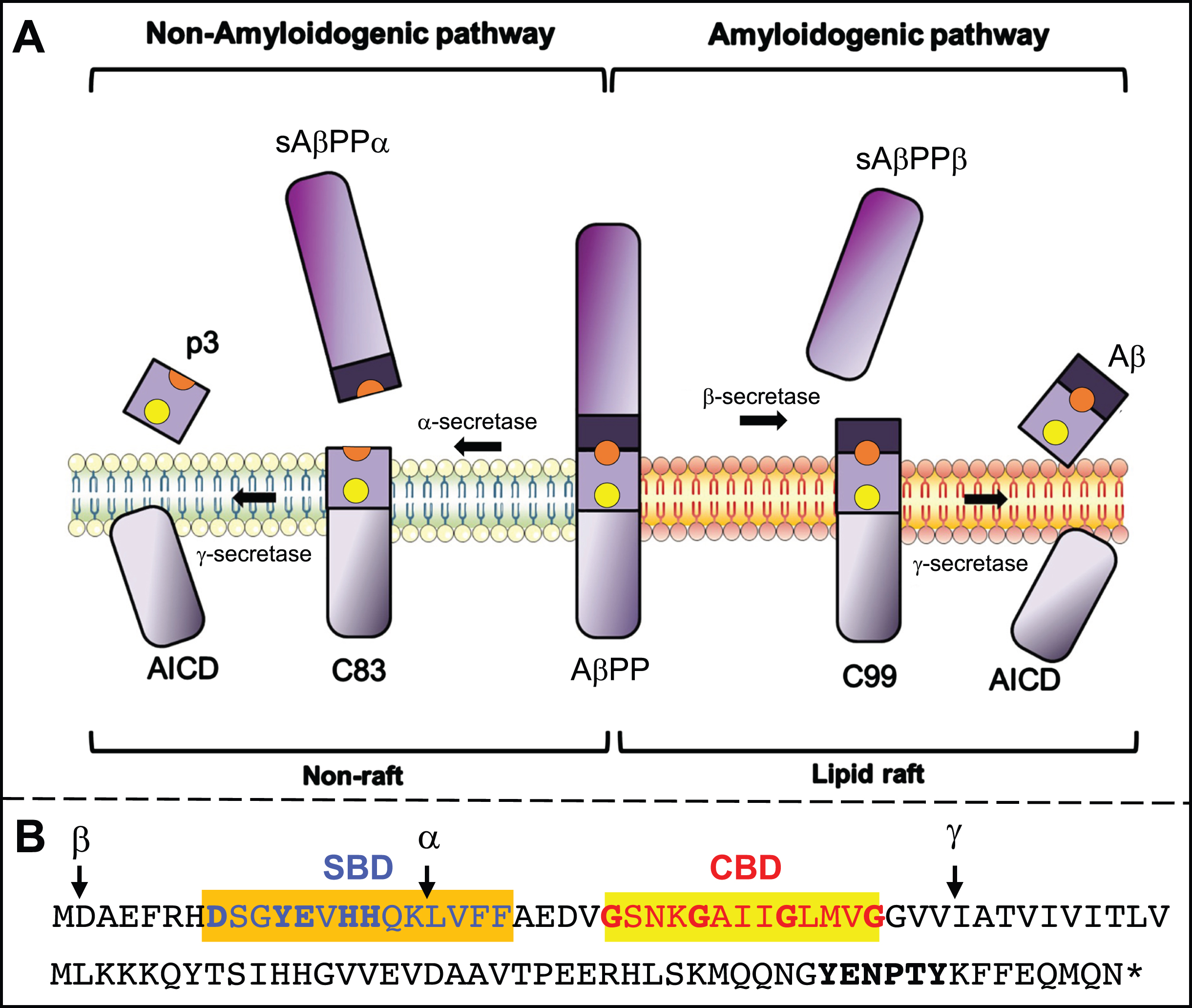
AβPP processing. A) Processing intermediates. Yellow dot, cholesterol-binding domain (CBD); orange dot, sphingolipid-binding domain (SBD). Adapted from [3], with permission (doi.org/10.1016/j.nbd.2020.105062). B) Human C99 sequence; important residues for SBD [149] and CBD (based on [117] and [118]) binding, and the YENPTY motif, in bold. Cleavage sites for α-, β-, and γ-secretase are indicated (arrows). See text for details.
In the most widely-accepted pathogenetic model of AD, both SAD and FAD arise when AβPP is misprocessed to produce a slightly longer, aggregation-prone, form of Aβ (Aβ42) that accumulates in the plaques [1]. The Aβ is toxic to cells, and the resulting stress stimulates tau hyperphosphorylation, leading to the tangles. The senile plaques are proposed to be the cause of the disease. The overall process has been called the “amyloid cascade” [1, 7]. However, there are other data that challenge this perspective. For example, plaques are prevalent in the brains of aged subjects without dementia [8]. Moreover, an AD-protective isoform of APOE3, known as the “Christchurch” variant, was recently shown to delay AD symptoms in an FADPS1 patient in spite of unusually high brain plaque levels [9].
Numerous in vitro studies have shown that high levels of soluble or oligomerized Aβ can induce cell toxicity through multiple mechanisms, such as membrane damage, mitochondrial dysfunction, and tau phosphorylation [10]. However, these results could not be replicated in animal models of AD [11], casting doubt on what looked initially to be a straightforward hypothesis. Nevertheless, the field continued to produce data further cementing amyloid’s putative role as the main cause of neuronal death in AD, which, in turn, stimulated a search for compounds to inhibit Aβ production [12]. Disappointingly, most anti-Aβ strategies and the use of BACE1 inhibitors [13] have had limited success in ameliorating clinical outcome [14], and γ-secretase inhibitors, rather than improving disease outcomes, made them worse [15].
IMPLICATIONS OF THE AMYLOID CASCADE
When amyloid was observed to be abundant in plaques in AD brains, the logical conclusion was that its production was augmented, and that a pathological hallmark as conspicuous as plaques had to be relevant to the disease mechanism. Later, the discovery that γ-secretase is the only enzyme responsible for Aβ production [16] led some to the conclusion that its activity in AD was increased [17], resulting in toxic higher concentrations of Aβ that were deposited in the plaques, and became the source of the idea that AD pathogenesis was caused by altered γ-secretase enzymatic function [18].
However, it is important to note that the efficiency of an enzyme’s function is defined by the rate at which it converts substrate (e.g., C99) to product(s) (e.g., Aβ + AICD). Thus, measuring only the levels of the amount of product as the readout of enzyme activity can lead to misinterpretations. For example, if a given enzyme under normal conditions consumes 1 unit of substrate to produce 1 unit of product per minute (i.e., a conversion efficiency of 1.0), producing 1.5 units of product in a minute could be interpreted as a 50% increase in enzymatic activity. Yet, if those 1.5 units of product were derived from 2 units of substrate (i.e., an efficiency of 0.75), we would conclude the opposite, namely, a 25% decrease in enzyme activity.
It is true that in AD altered AβPP cleavage causes an increase in total Aβ and a tendency towards the production of longer neurotoxic species of amyloid (e.g., Aβ42 rather than Aβ40) that are more prone to aggregate into senile plaques [19]. But it is also true that while elevations in Aβ are highly variable among patients, AD tissues typically present with significant increases in the levels of C99 (the substrate for Aβ production) compared to the total amount of amyloid produced [20–22], a result consistent with the idea that γ-secretase activity in most AD patients is decreased, not increased [23]. For example, using an in vitro reconstitution assay admittedly prone to artefacts [24, 25], Sun et al. showed that ∼90% of 138 FAD mutations in PS1 presented with increases in Aβ42:Aβ40 ratio but had reduced total amyloid production [26] (but to be fair, ∼10% showed the opposite result [26, 27], implying that different γ-secretase activities are associated with different PS1 mutations [28]). However, as noted above, enzymatic activity should be reported at the ratio of product:substrate; merely reporting the product (e.g., ratio of Aβ42:Aβ40 [26]), does not say anything about the activity of γ-secretase. Similar results, although not the goal of those studies, were found in other reports [29].
As a result, we can define AD not only by an elevated ratio of longer:shorter Aβ species [30], but also by an increased ratio of C99:total Aβ [31]. This implies that in AD, defects in γ-secretase result in reductions both in the quantity of cleaved product (i.e., Aβ) relative to the amount of available C99 and in the “quality” of its processing towards longer, more aggregation-prone, Aβ species. From this point of view, we can redefine AD as a partial loss of function of γ-secretase [32–35] due to inherited genetic mutations in the case of FAD [36–38], or to undetermined causes in the case of SAD. Based on this, the buildup of uncut C99 could be considered an early pathological hallmark that could elicit many of the molecular features seen in AD cells [28]. This is hardly a new idea, as C99 has been suggested to be a pathogenic trigger of disease by many groups [39–42]. For example, some authors have shown that in animal models of AD with pathogenic mutations in PSEN1 or APP, C99 accumulation occurs in AD-relevant brain areas (cortex and hippocampus) at early stages of development, long before the appearance of any detectable increases in Aβ [39, 42]. These data also highlight the contribution of C99 accumulation to some of the symptoms of AD [32, 41], including endosomal dysfunction, cognitive impairment, and hippocampal degeneration [21, 43–45]. Interestingly, other groups found C99 to be especially toxic to neurons [46, 47], and that the accumulation of C99, rather than alterations in Aβ or AICD, caused symptoms of dementia [41, 48].
HOW DOES ELEVATED C99 RESULT IN LONGER Aβ SPECIES?
As mentioned above, C99 is cleaved in an intracellular compartment by γ-secretase to produce two peptides, Aβ and AICD (Fig. 1). It is important to note that γ-secretase is an unusual “rhomboid-like” protease that cleaves substrates located within membrane domains [49], especially within lipid rafts [50]. Lipid rafts are transient membrane domains formed by local increases in cholesterol, sphingomyelin, and saturated phospholipids, resulting in a more rigid and “liquid-ordered” membrane “island” (i.e., the raft) located within the surrounding “liquid-disordered” membrane [51, 52]. These local elevations in cholesterol (by ∼45% in mouse embryonic fibroblasts [MEFs] [53]) and sphingomyelin (by ∼40% in mouse liver [54]) create highly ordered membrane microdomains that passively segregate and enrich for lipid-binding proteins, thereby facilitating protein-protein interactions for the regulation of numerous signaling pathways [52]. Moreover, the membrane of a lipid raft is thicker than the surrounding liquid-disordered region [55, 56], due mainly to local increases in straight and long saturated phospholipid acyl chains [55–57]. This modification in the structure of the membrane induces changes in the conformation of those transmembrane (TM) proteins embedded within them and modulates their enzymatic activities. Relevant to AD, it has long been known that γ-secretase activity resides in lipid rafts [58, 59], and in fact it has now been shown that these rafts are located intracellularly, in subdomains of the ER [60–62]. Moreover, increases in intracellular cholesterol are known to induce the association between C99 and γ-secretase in membranes, and are required to activate the production of Aβ [63]. Importantly, γ-secretase cleavage is activated by the formation of these lipid rafts and by the regulation of their lipid composition [64].
Equally important, cleavage by γ-secretase is not sequence-specific: it “cuts what it sees” [65] (Fig. 2). Under normal circumstances, following formation of the lipid raft, γ-secretase processes C99 in a “product line” consisting of an initial cleavage event followed by sequential ∼3-aa “bites” to produce a distribution of successively shorter fragments, ranging in length from ∼36 - ∼48 amino acids (i.e., Aβ36-Aβ48) [30, 66–70]. Under these conditions, there is “hydrophobic matching” between the length of C99’s α-helical transmembrane domain [71] and the thickness of the [lipid-raft] membrane in which it resides [72]. In normal cells, the initial cleavage of C99 by γ-secretase [73] favors the production of Aβ40 (Fig. 2A). In AD cells, however, the lipid composition of the raft is altered by increases in the levels of ceramide derived from elevated sphingomyelinase activity [35], which generates a significantly longer [74] and slightly thinner [75–77] lipid raft (but still thicker than the surrounding “free” ER (i.e., our term for ER not apposed to other intracellular organelles) [78–80]), forcing the C99 TM domain to maintain hydrophobic matching by tilting [73, 82]. This tilt changes the alignment of C99 with γ-secretase and alters the positioning/conformation of the initial γ-secretase cleavage site [24, 84] on C99 such that more Aβ42 and less Aβ40 is produced (Fig. 2B) [85–87]. This model is consistent with an altered lipid milieu playing a role in determining the physical apposition of PS1 to C99 [81], and implies that the A β42:A β40 ratio in AD is fundamentally a surrogate marker of the thickness of the membrane in which C99 cleavage occurs [88, 89]. It turns out that Aβ42 happens to have the unfortunate property of being more aggregation-prone, more prevalent in plaques, and more toxic to neurons than is Aβ40 [90], influencing the field to focus on Aβ42 more as a pathogenic culprit and therapeutic target while focusing less on its biophysical implications.
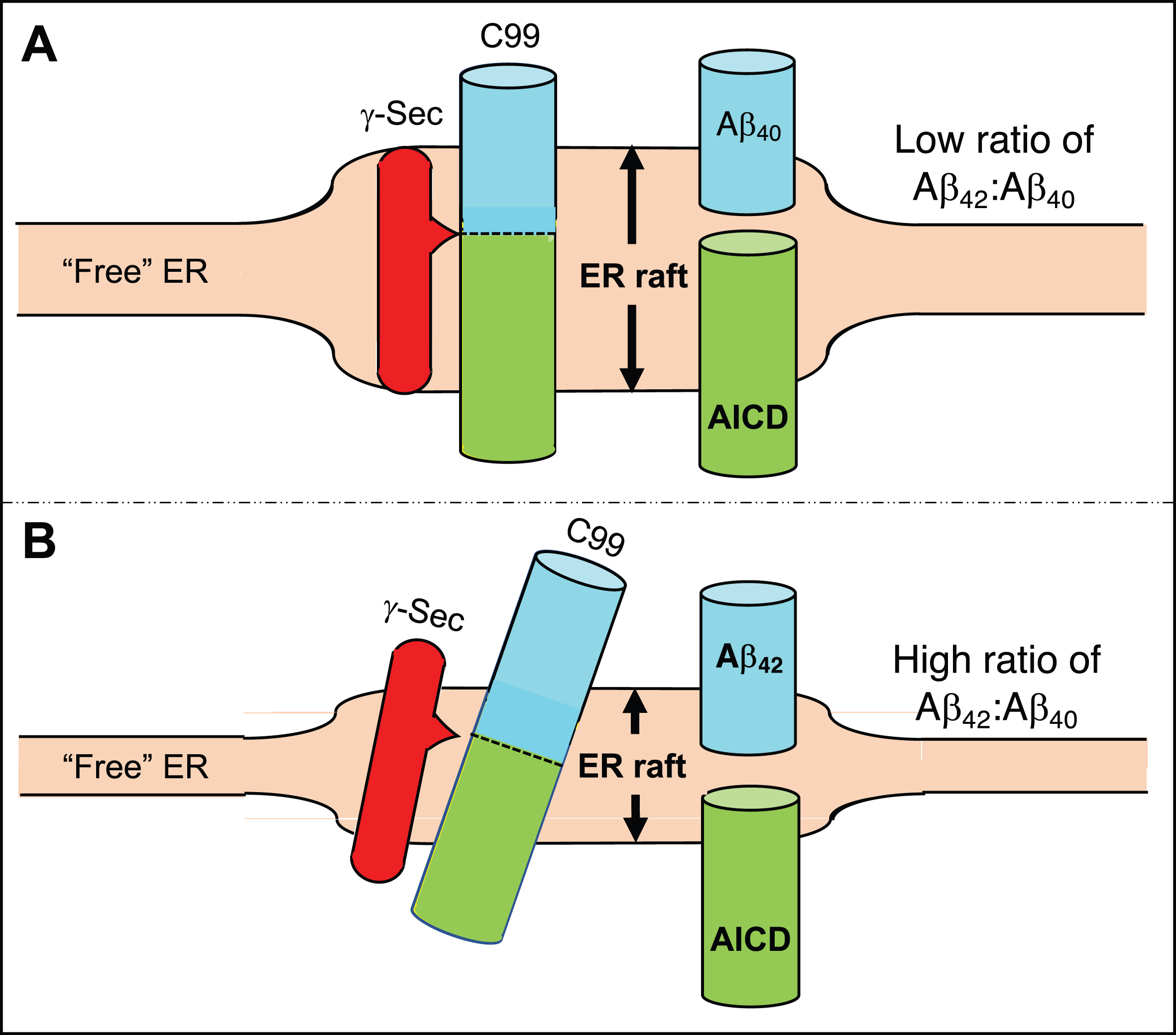
Generation of Aβ40 versus Aβ42. A) In the normal situation the γ-secretase complex aligns with C99 in the ER raft such that its initial cleavage favors a product line that generates Aβ40, and the Aβ42:Aβ40 ratio is relatively low. B) In AD, the ER raft is thinner than normal (shorter double-headed arrow) but is still thicker than the “free” ER. Hydrophobic matching of C99’s α-helical region within the thinner membrane causes it (and perhaps the γ-secretase complex as well) to tilt, resulting in the initial cleavage of C99 displaced by ∼2 amino acids, generating a higher Aβ42:Aβ40 ratio. See text for details.
C99 REGULATES Aβ PRODUCTION
Pathogenic mutations in AβPP [33, 91] and PS1 [92] promote an increase in the Aβ42:Aβ40 ratio associated with increased levels of C99, suggesting that, ironically, the accumulation of C99 is to blame for the decrease in the ability of γ-secretase to cleave its own substrate, namely C99 itself. In support of this idea, in vitro studies have shown that upon exceeding a concentration threshold, C99 peptides associate with each other in membranes [69], inducing a conformational change in them that precludes, or at least delays, their cleavage by γ-secretase [25, 93]. This decrease in enzymatic activity by substrate saturation could explain why inhibition of γ-secretase favors the accumulation of C99 fragments [94]. Taken together, we can conclude that the degree of C99 accumulation is a major determinant of amyloid production via the modulation of its own processing. The mechanism by which C99 affects its own cleavage, ultimately resulting in an increased Aβ42:Aβ40 ratio, is addressed below.
ELEVATED LEVELS OF C99 ARE A LIKELY CAUSE OF AD
Based on the above analysis, we propose that increased C99 is a more accurate indicator of disease progression than is increased amyloid or plaque burden. The correlations between Aβ production, Aβ42:Aβ40 levels, and plaque/tangle burden and the progression of dementia are not significant enough to blame any one of these as the sole cause or indicator of disease [95]. In fact, brain samples from cognitively normal older adults often show significant amyloid deposition [96]. On the other hand, many groups have shown a significant association between C99 levels and the appearance of cognitive impairment, both in animal models and in AD patients [97]. Moreover, an “AD-protective” mutation in AβPP (“Icelandic”) causes a substantial decrease not only in amyloid but also in C99. Conversely, some pathogenic mutations in PSEN1 associated with aggressive forms of AD [98] do not cause higher amyloid levels, senile plaques, or neurofibrillary tangle formation [99, 100].
This raises a conundrum: if C99 is so important in AD pathogenesis, why hasn’t the field come to the conclusion that C99, and not Aβ, is the culprit, or at least concluded that γ-secretase activity is reduced, not increased, in AD? One explanation is that experimental limitations, as well as the definition of AD itself, have contributed to difficulties in data interpretation.
With respect to experimental design, we note that C99 encompasses Aβ (at its N-terminal “half”) and AICD (at its C-terminal “half”), and therefore contains the same [partial and/or overlapping] amino acid sequences as are present on both fragments (see Fig. 1). Thus, in the absence of detailed controls, it is easy to envision difficulties in interpreting experimental data that might unequivocally implicate any particular AβPP fragment. In fact, many antibodies used to detect Aβ in cells or tissues by imaging approaches or by enzyme-linked immunosorbent assay (ELISA) cannot discriminate between C99 and other AβPP fragments [101], a pitfall that raises the interesting possibility that, guided by the amyloid cascade hypothesis, some histological studies had interpreted elevations in C99 as deposition of Aβ [102, 103].
Second, AD is defined operationally: to receive a diagnosis of AD requires the presence of a threshold number of plaques and tangles in postmortem brain samples from patients with cognitive impairment [104]. Therefore, those cases diagnosed with AD-like dementia, but presenting with low numbers of extracellular deposits of Aβ, will not be diagnosed as AD after histological analysis, even if they carry mutations in PSEN1 [36, 106].
That C99, and not Aβ, is a trigger of disease can also explain why treatments aimed at inhibiting γ-secretase activity or minimizing Aβ production/size have yielded disappointing results [107]. However, at this point, a logical question arises: if C99 is so bad, why have anti-BACE1 therapies (that prevent C99 formation in the first place) not been successful either? There are really only two responses to this question: (1) The C99 idea is wrong, or (2) C99 and/or the regulation of AβPP cleavage to produce the “right” amount of C99 is necessary to fulfill a currently-unknown cellular function. Which brings us to the question of:
WHAT IS THE FUNCTION OF C99?
Full-length AβPP has been proposed to play a role in neurite outgrowth [108] and cell signaling [109], while the C99 fragment has been shown to modulate voltage-gated potassium channels [110] and to play a role in mitophagy [111]. However, the most frequent and supported hypothesis for the normal function of AβPP (and by inference, of C99) is that it is associated with the regulation of cellular cholesterol [112]. The expression, processing, and activity of AβPP are modulated by changes in the levels and distribution of cholesterol [113, 114]. Conversely, alterations in the expression, distribution, and cleavage of AβPP results in changes in cholesterol homeostasis [115]. Extensive data suggest that one function of AβPP could be to serve as a cholesterol sensor [116] and/or a regulator of cholesterol distribution in the cell [35, 53]. To make the connection between AβPP and cholesterol even more direct, AβPP contains a cholesterol-binding domain (CBD; yellow dot in Fig. 1A and highlighted sequence in Fig. 1B) consisting of tandem GxxxG motifs located within C99 immediately upstream of the γ-secretase cleavage site [116–119], and whose abrogation prevents AβPP’s and C99’s ability to regulate this lipid [53]. (The GxxxG motifs have also been proposed to play an alternative role, as a mediator of C99 homodimerization, but this is controversial [120, 121].) Thus, a likely role of C99 is to maintain cellular cholesterol homeostasis. But how is this accomplished?
Cholesterol is produced within animal cells, in the endoplasmic reticulum (ER) [122], but is also taken up from outside the cell, primarily from circulating lipoproteins containing free cholesterol and cholesteryl esters (CEs), with most of the cholesterol released to the plasma membrane [122]. Compared to the PM, the ER contains very low, but biologically important, levels of cholesterol [123]. For example, the amount of cholesterol in ER membranes regulates the activation of the sterol-regulatory element binding proteins (SREBP1 and 2), transcription factors that are localized to the ER membrane in an inactive state. When the level of intracellular cholesterol is low, SREBP is released and eventually travels to the nucleus, where it switches on genes, such as those encoding the low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutary-CoA reductase (HMGCR), that cause the cell to generate more cholesterol. Conversely, when the level of ER cholesterol is high, SREBP remains within the ER and cannot translocate to the nucleus, thereby reducing the production of cholesterol by reducing HMGCR levels [124].
These pathways imply that normal cholesterol homeostasis in the cell is maintained by the crosstalk between the PM, where most of the cholesterol resides, and the ER, where most of the cholesterol-regulatory enzymes reside [124]. This PM-ER communication is a regulated process, with cholesterol trafficking mediated by both vesicular (e.g., via the LDL receptor and endosomes) [125] and non-vesicular (e.g., via Aster/GRAMD [GRAM domain-containing protein] proteins [126]) [127, 128] processes; we will focus here mainly on the vesicular pathway. Excess cholesterol in a PM-localized raft induces the invagination of the plasma membrane and the formation of a cholesterol-rich endosome that pinches off from the PM, enters the cell proper, and eventually fuses with a lysosome [129]. The lower pH in the resulting endolysosome induces the activation of cholesterol-regulatory enzymes, such as Niemann-Pick type C protein 1 (NPC1) [130], which contributes to the fusion of the endolysosome with the ER (likely via inter-organelle contact sites [131]) and the release of cholesterol to ER membranes [132].
We note in this context that problems in endosome function [133] and in lysosome acidification [134], driven by elevated levels of C99 [135], play a key role in AD pathogenesis [136]. In particular, C99 accumulates in lysosomes that are defective in acidification [134]. This result seems counter-intuitive, as elevated endolysosomal pH should decrease, not increase, BACE1 enzymatic activity and hence C99 production. Remarkably, however, the vacuolar ATP-synthase (v-ATPase) required for endosomal acidification is inhibited by phosphorylation of C99 (at Tyr-682 in the highly-conserved YENPTY motif [135] [see Fig. 1]). Thus, it is possible that the degree of BACE1-induced production of C99 [43, 137] is regulated by v-ATPase-mediated lysosome acidification via an equilibrium between phosphorylated and dephosphorylated C99 [138].
Importantly, this PM-derived cholesterol changes the structure of the ER membrane by forming an intracellular lipid raft (Fig. 3) [139]. Formation of the raft is maintained by proteins with affinity for cholesterol, in particular C99, that are capable of clustering cholesterol into local islands. This membrane reorganization results in changes in the distribution and conformation of ER proteins embedded within these islands (i.e., rafts) and in the modulation of the activities they regulate. For example, C99-mediated raft formation induces the activity of acyl-CoA:cholesterol acyltransferase (ACAT1; gene SOAT1), the enzyme that converts cholesterol into cholesteryl esters that are then deposited as lipid droplets (LDs), in order to detoxify the cell of excess cholesterol and return cholesterol to normal levels in a homeostatic loop [53, 140]. Alterations in the regulation of any step in this pathway leads to changes in the distribution of cholesterol and in the composition of lipid membranes, leading to altered raft “setpoints” (discussed below).
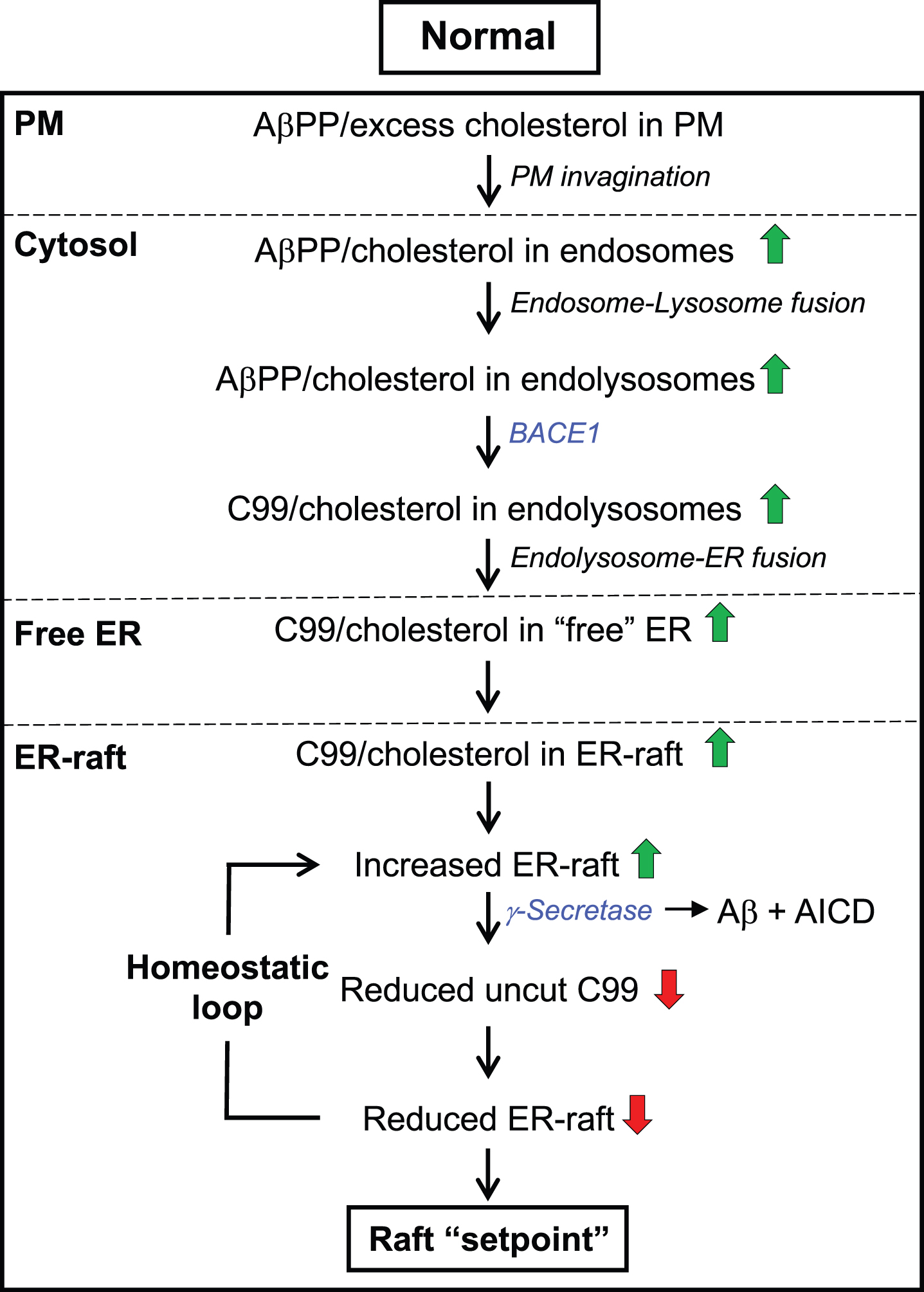
The C99-cholesterol axis in the normal situation. Role of cholesterol: Excess plasma membrane (PM) cholesterol derived from extracellular lipoproteins induces the invagination of the PM and the formation of cholesterol-rich endosomes, which pinch off from the PM, enter the cell, and eventually fuse with lysosomes. The resulting endolysosomes fuse with the ER and release cholesterol to ER membranes, forming an intracellular lipid raft. Cholesterol-regulatory proteins detoxify the cell of excess cholesterol and return cholesterol to normal levels in a homeostatic loop. Role of C99: At the same time, the excess PM cholesterol binds to AβPP (via the CBD) that is also internalized in endolysosomes, where it is cleaved by BACE1 at low pH, forming C99. C99 is then delivered to the ER raft where it is cleaved by γ-secretase in an amount sufficient to reduce the initial raft C99 content to a “setpoint” level. Once C99 is cleaved (releasing Aβ containing the CBD), clustering of cholesterol in lipid rafts is reduced dramatically. Note that the ability of C99 to bind cholesterol (via the CBD) allows for a homeostatic loop to be set up in which the equilibrium between the amount of cut versus uncut C99, and the corresponding amount of cholesterol bound to uncut C99, determines the raft setpoint. Green/red arrows, increased/decreased levels. See text for details.
This trafficking of cholesterol is paralleled by a similar trafficking of AβPP and C99 (Fig. 3). As noted above, in the amyloidogenic pathway AβPP is internalized in cholesterol-rich endosomes [141], where it is then cleaved by BACE1 [142, 143], an enzyme activated at low pH [144]. This results in the production of C99 that is then delivered from endosomes to the ER (by a currently unknown mechanism [131], but possibly via endosome-ER contacts [145–147] and/or the retromer complex that recycles proteins from endosomes to the plasma membrane via the Golgi [148]), where it “attracts” cholesterol [116, 117] and sphingomyelin [149] (see below), resulting in the formation of the lipid raft domain described above. In the raft, C99 associates with γ-secretase (by physical proximity [63]) and is cleaved into Aβ (which is then exported from the raft via exosomes [150, 151]) and AICD. Once C99 is cleaved, releasing Aβ containing the CBD (see below and Fig. 1), clustering of cholesterol in lipid rafts is reduced dramatically. Thus, like cholesterol, C99, and raft formation, is regulated in a homeostatic loop that is mediated by the level of C99 and the degree of its cleavage by γ-secretase (i.e., C99 controls raft formation and hence γ-secretase levels; γ-secretase controls C99 levels and hence raft formation [53]). In indirect support of this scenario, we note that of the 114 documented pathogenic point mutations in AβPP (https://www.alzforum.org) 25 are located in the C99 region, but none are at the critical glycines in the CBD GxxxG motifs (see Fig. 1B), implying that the presumed loss of C99’s ability to bind cholesterol disrupts the relationship between C99 and cholesterol to the point where C99 is no longer pathogenic (a conclusion also supported by mutagenesis of the CBD [53]).
In broad view, both cholesterol trafficking and AβPP trafficking follow essentially the same topographical pathway (PM → endosome → endolysosome → ER → lipid raft) (Fig. 3). We believe that this is not a coincidence: the fact that the cholesterol-internalization and AβPP-cleavage pathways operate in parallel could help explain the effects of cholesterol on AβPP metabolism [152] and the fact that cholesterol turnover [153, 154]—mainly the oxidation of cholesterol to 24(S)-hydroxycholesterol by CYP46A1 (cytochrome P450 46A1; cholesterol 24-hydroxylase) [155] and its negative regulator ATAD3A (ATPase family AAA domain-containing protein 3A) [156], both of which are localized to ER rafts [156, 157]—is necessary for Aβ production [115, 158–160]. Moreover, the “superposition” of both pathways reveals a picture in which AβPP is a reporter of the lipid environment of the cell, by regulating the transport of cholesterol between PM and ER, and that AβPP’s internalization and cleavage by BACE1 and γ-secretase are essential steps in the regulation of cellular cholesterol and overall lipid homeostasis.
Accordingly, we propose that C99 is required to maintain intracellular cholesterol and raft homeostasis. Specifically, in the normal situation (Fig. 3), after a threshold is exceeded, the excess of cholesterol in the plasma membrane [161] binds to full-length AβPP [162, 163] and induces its internalization by the formation of cholesterol-rich endosomes. Following cleavage of AβPP by BACE1 within endolysosomes to produce C99, C99 traffics to the ER where it “attracts” cholesterol (at 1:1 stoichiometry [117]) [116, 117] and sphingomyelin [149], resulting in the formation of a lipid raft domain. Formation of the raft promotes the association of γ-secretase with C99 and its cleavage into Aβ and AICD. Once C99 is cleaved and Aβ is released, clustering of cholesterol in lipid rafts is reduced dramatically.
Thus, the interplay between C99 and cholesterol—a “C99-cholesterol axis”—sets up a self-correcting homeostatic feedback loop such that, under normal circumstances, the amount of uncleaved C99 is maintained at extremely low levels, in order to help maintain intracellular cholesterol and raft levels at a desired “setpoint”, both quantitatively (e.g., raft amount) and qualitatively (e.g., raft function) (Fig. 3). Moreover, this setpoint likely varies both spatially (e.g., in different cells and tissues) and temporally (e.g., during development or in response to changing environmental conditions). In AD, however, the steady-state levels of C99 and cholesterol are increased significantly, disrupting this feedback loop and setting off a chain of events that results in the disease, as described below.
FAMILIAL AND SPORADIC AD: A SHARED PHENOTYPE WITH DISTINCT TRIGGERS
As in FAD, tissues from SAD patients also have increased C99 levels [97], by various proposed mechanisms, including lysosomal dysfunction [134] and altered activation of γ-secretase [164]. Relevantly, increased exposure of cells to high levels of extracellular cholesterol can induce an increased rate of cholesterol internalization and AβPP cleavage, and an increase in C99 and in the Aβ42:Aβ40 ratio [165, 166]. Therefore, any condition or gene variant that provokes an elevation in cholesterol uptake and traffic to the ER will be capable of inducing high levels of C99 in sporadic forms of the disease [167].
Notably, the ɛ4 allele of APOE (ApoE4) is one of these SAD genetic risk factors, as it is recycled from endolysosomes much less efficiently than is ApoE3 [168–170]. This results in an accumulation of intracellular cholesterol, thereby stimulating an increase in C99, similar to what was described above for FAD. In fact, of the 34 AD risk-factor genes with coding-region non-synonymous mutations identified by us (Table 1), nine are associated with altered cholesterol trafficking or metabolism (marked in red in Table 1; see also [171]).
AD risk genes associated with coding-region non-synonymous mutations
Gene functions connected to cholesterol (entries in red), MAM (blue), both cholesterol and MAM (green), or neither (black).
Thus, an increase in the steady-state level of C99 in AD can come from many sources (Table 1), including: 1) reduced cleavage of C99 by γ-secretase [32]; 2) increased BACE1 cleavage of AβPP [172–174], as is found in the AβPP “Swedish” mutation [175]; 3) increased steady-state levels of AβPP, as is seen in Down syndrome [176] (APP is on chromosome 21) or in patients with mutations in the sortilin-related receptor (SORL1; an AD risk factor [167]) that reduces AβPP recycling via the retromer pathway [177, 178]; 4) increased intracellular cholesterol levels [165, 179] due to, for example, mutations in ATP-binding cassette sub-family A member 1 (ABCA1), a cholesterol exporter [180] and AD risk factor [181]; and 5) increased intracellular cholesterol due to inefficient recycling of ApoE4-derived lipoproteins [166]. Clearly, elevated C99 is toxic to cells, no matter the source. Thus, another question immediately arises: what is the nature of that toxicity? To answer this question, we must take a slight detour into the structure and function of the lipid raft subdomain of the ER.
MAM: A LIPID RAFT IN THE ER
All the evidence marshalled above points to elevated C99 as the trigger of the cellular defects seen in AD, mediated by C99’s association with cholesterol (and, a fortiori, with sphingomyelin; see below). In fact, C99’s toxicity is proportional to its capacity to form lipid rafts in the ER [53]. These ER-localized rafts have a name reflective of their special nature and special status in the cell: mitochondria-associated ER membranes, or MAM [182].
MAM is a highly dynamic and transient subdomain of the ER that communicates, both physically and biochemically, with mitochondria [51, 182]. MAM lies in close apposition to mitochondria (ranging from ∼10–80 nm [183–185]) (Fig. 4A), facilitating bidirectional crosstalk between the two organelles, with specific molecules travelling relatively short distances from the ER to mitochondria for some molecules and in the opposite direction for others. This apposition is mediated physically by pairs of tethers, one on the ER cytoplasmic face and one on the mitochondrial outer membrane (MOM), that help bring the two organelles together. For example, mitofusin-2 (MFN2), located both at the ER and the MOM, dimerizes to align the two organelles [186], as do the pairs VAPB (vesicle-associated membrane protein-associated protein B/C) - PTPIP51 (protein tyrosine phosphatase interacting protein 51) [187], and SIGMAR1 (sigma-1 receptor) - VDAC (voltage-dependent anion channel) [51], with tethering regulated, in an unknown fashion, by MAM-localized PACS2 (phosphofurin acidic cluster sorting protein 2) [188, 189]. By contrast, presenilins are not physical tethers but are negative regulators of apposition such that pathogenic mutations modify presenilin structure to promote tethering via alteration in the lipid composition of the MAM, as described here. Importantly, both C99 [35] and γ-secretase activity [60–62] are localized to the MAM, as is γ-secretase activating protein (GSAP) [190]. Thus, the final step in the amyloidogenic pathway to produce A β can take place only in the MAM [191].
MAM functions include phospholipid synthesis [182, 192], cholesterol and steroid metabolism [193, 194], sphingolipid (e.g., sphingomyelin and ceramide) metabolism [35], glucose homeostasis [195], calcium trafficking [51, 197], and mitochondrial oxidative phosphorylation (OxPhos) [35] and dynamics [186, 198]. MAM is also central to the modulation of various other key cellular processes, including the regulation of hypoxia [199], iron homeostasis [199, 200], ER stress [201], autophagy [202], and inflammasome signaling [203]. We note that all of these processes are disturbed in AD [28], implying an intimate relationship between MAM function and AD pathology [89, 204]. For example, calcium trafficking between the ER and mitochondria, which is perturbed in AD [205], involves more than 30 MAM-localized proteins [206]. Of these, perhaps the most prominent is the sigma-1 receptor (gene SIGMAR1) [207], which is a key “gatekeeper” for the transfer of Ca2 + from the ER to mitochondria [208], with an interorganellar gap distance of only ∼7–15 nm required for maximum transfer [196]. Similarly, 9 of the 10 enzymes of glycolysis, which is impaired in AD [209], are localized to MAM [210] and the tenth, hexokinase, is localized to the mitochondrial outer membrane [211]. Autophagy is another cellular function intimately reliant on MAM [202, 212]: not only are at least 5 autophagy-related proteins localized to MAM, but MAM is required for the synthesis of the phosphatidylethanolamine (see below) that is required to build the phagophore [202]. Finally, mitochondrial bioenergetics [35] and organelle morphology and dynamics—most notably the balance between organellar fusion and fission—which are also altered in AD [213, 214], are MAM-mediated functions. Also relevant to mitochondrial function, hypoxia, which is a feature of AD [215], upregulates BACE1 at the transcriptional level [216]. Given this multiplicity of functions, it is highly likely that MAM composition, not only its constituent proteins and lipids, but even microRNAs [217], differs in different cell types, at different times, and in response to different environments [218].
With respect to AD, C99 induces the formation of MAM via its ability to bind and “cluster” cholesterol, forming the raft, as described above. In turn, several proteins are recruited to the MAM so as to co-regulate multiple homeostatic pathways, some of which have been described above (e.g., cholesteryl ester synthesis via ACAT1 [219]). Alterations in the regulation of MAM formation, as occurs in AD, perturbs these pathways. As examples, we will focus on three key MAM functions relevant to AD: regulation of phospholipids, sphingolipids, and cholesterol.
Altered phospholipid metabolism is a feature of AD [220, 221]. Most of the cell’s phosphatidylethanolamine (PE) is synthesized in a salvage pathway located at the ER-mitochondrial interface (i.e., MAM): phosphatidylserine (PS) is synthesized in the MAM by phosphatidylserine synthases [222]; PS then translocates to the mitochondria [223, 224] where it is decarboxylated by phosphatidylserine decarboxylase (PISD) [225] to form PE; PE then travels back to the MAM, where it traffics to the rest of the cell or undergoes further modifications [226] (see Fig. 4B). The trafficking of PS from MAM to mitochondria is a well-recognized measure of MAM function [227]. In cells from both SAD and FAD patients this pathway, and cellular PS and PE production, are upregulated significantly [74].
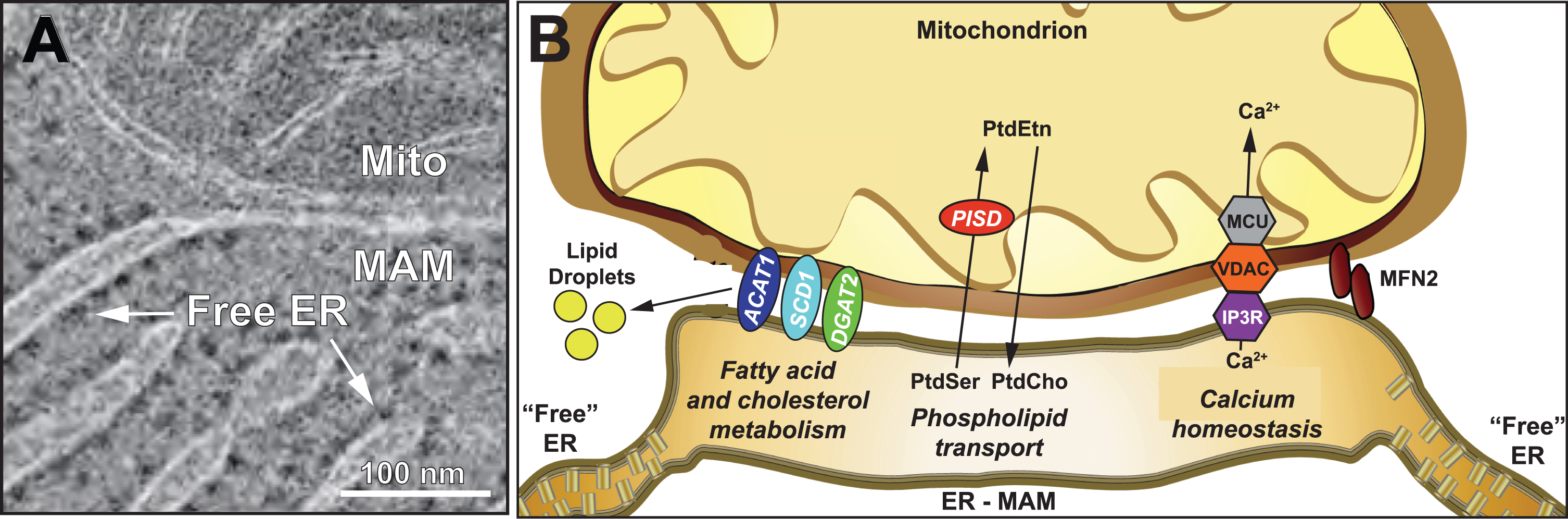
Schematic representation of the MAM subdomain of the ER. A) EM of rat hepatocytes showing MAM compared to “free” ER (adapted from [185], with permission). B) Key MAM functions. ACAT1, acyl-CoA cholesterol acyltransferase-1; DGAT2, diacyglycerol-O-acyltransferase-2; IP3R, inositol triphosphate receptor; MFN2, mitofusin-2; MCU, mitochondrial calcium uniporter; PISD, phosphatidylserine decarboxylase; SCD1, stearoyl-CoA desaturase-1; VDAC, voltage-dependent anion channel. See text for details.
Sphingolipid pathways are also altered in AD [228, 229]. In particular, the ceramide content in brains and cells from AD patients and in AD cell models is increased [55, 230–233] as a result of the upregulation of de novo ceramide synthesis [234] and of the activity of sphingomyelinases (SMases) that convert sphingomyelin (SM) to ceramide [231, 235]. MAM is involved in the regulation of sphingolipid metabolism [236] and at least one neutral SMase in mice likely resides at the MAM [237] (although the homologous gene in humans is apparently a pseudogene [238]). The ceramide that is enriched at MAM [35, 239] (by ∼25% in mouse liver [54]), can displace raft cholesterol [240, 241] while still maintaining the liquid-ordered nature of these rafts [240]. In AD, MAM-localized SMase activity is upregulated [35], with a concomitant local increase in ceramide [35] that increases MAM [242] and Aβ42 [243] formation and reduces mitochondrial bioenergetics [35, 244–248].
Finally, elevated serum cholesterol [249] and altered cholesterol metabolism [230, 249–251] have been well-documented in AD, as have been the effects of increased cholesterol on AβPP metabolism [152], including increased Aβ production [165, 252]. Importantly, elements of the cellular cholesterol homeostatic machinery, including SREBP1 [253], which interacts with C99 [254], and ACAT1 [255] are localized in MAM. In cells from both SAD and FAD patients, ACAT1 activity is upregulated significantly [74], as is the accumulation of lipid droplets [74, 257], including LDs within neurons [258]. Remarkably, ACAT1 activity is required for Aβ production [115, 260]. Moreover, ACAT1-derived CEs regulate not only Aβ production [159, 261] but also tau production, by inhibiting the degradation of phosphorylated tau [261]. Thus, MAM-localized cholesterol metabolism plays a fundamental role in the development of the two key neuropathological hallmarks of AD: plaques and tangles.
The parallel alteration of cholesterol and sphingolipids is not coincidental, as the levels of these two classes of lipids are positively co-regulated in the cell [256, 262–265] and help to establish the formation of lipid rafts [56]. Specifically, sphingomyelin forms an “umbrella” around cholesterol (at a 1:1-1:2 ratio [263]), protecting it from interactions with water at the membrane/cytoplasm interface [266, 267], thereby initiating lipid raft formation (e.g., MAM). This relationship between sphingolipids and sterols is relevant to AD, as AβPP processing has been shown to occur preferentially in lipid rafts [268] that also contain C99 [269].
As mentioned above, C99 contains a cholesterol-binding domain. Perhaps of equal significance, C99 also contains a sphingolipid-binding domain (SBD; orange dot in Fig. 1A and sequence in Fig. 1B) that binds to sphingomyelin [149]. Thus, these two lipid-binding domains can potentially cooperate to promote raft formation by attracting equal amounts of cholesterol and sphingomyelin to C99. Moreover, and perhaps not coincidentally, in keeping with the dynamic nature of raft assembly and disassembly, both the CBD and the SBD are located within the N-terminal region of C99 such that cleavage of C99 by γ-secretase releases both domains as part of Aβ (see Fig. 1). From this point of view, C99 helps assemble the raft, whereas γ-secretase, via C99 cleavage and Aβ release and export [151], helps disassemble it. Perhaps Aβ has its own role to play in the cell after all [116, 271].
GENETIC RISK FACTORS IN SAD ARE ALSO CONSISTENT WITH ALTERED MAM FUNCTION
It is clear that C99 levels can be elevated in the familial form of AD, due to genetic mutations in PSEN1, PSEN2, and APP that conspire to increase C99, and hence cholesterol levels. But what about sporadic AD, where these genes are normal? What is the underlying pathogenetic process in SAD, and does it differ from that in FAD [28]?
Besides the mutations in the nine genes associated with cholesterol metabolism noted above (marked in red in Table 1), there are mutations in nine additional SAD risk genes that are associated with MAM function (marked in blue in Table 1) and in eleven other genes associated with both cholesterol metabolism and MAM function (marked in green in Table 1). Thus, of the 34 SAD risk factor genes on the list, 29 are consistent with the mechanism of AD pathogenesis that we propose here.
THE C99-CHOLESTEROL AXIS IN AD
These findings have led us to propose a model that invokes perturbations in C99 and cholesterol (i.e., disruption of the “C99-cholesterol axis”) as potential players in a linked pathogenetic mechanism leading to AD (Fig. 5). As in the normal situation (Fig. 3), after a threshold is exceeded [272], excess PM cholesterol binds to full-length AβPP and induces its internalization by endosomes [162] and cleavage by BACE1 within endolysosomes to produce C99 which, owing to increased BACE1 activity [273–275], is now elevated above normal levels [97]. At this point, the pathogenic pathways resulting in AD diverge, but not along classical FAD-SAD lines, as is commonly described. Rather, the divergence is between the early-onset (EOAD) and late-onset (LOAD) forms of the disease.
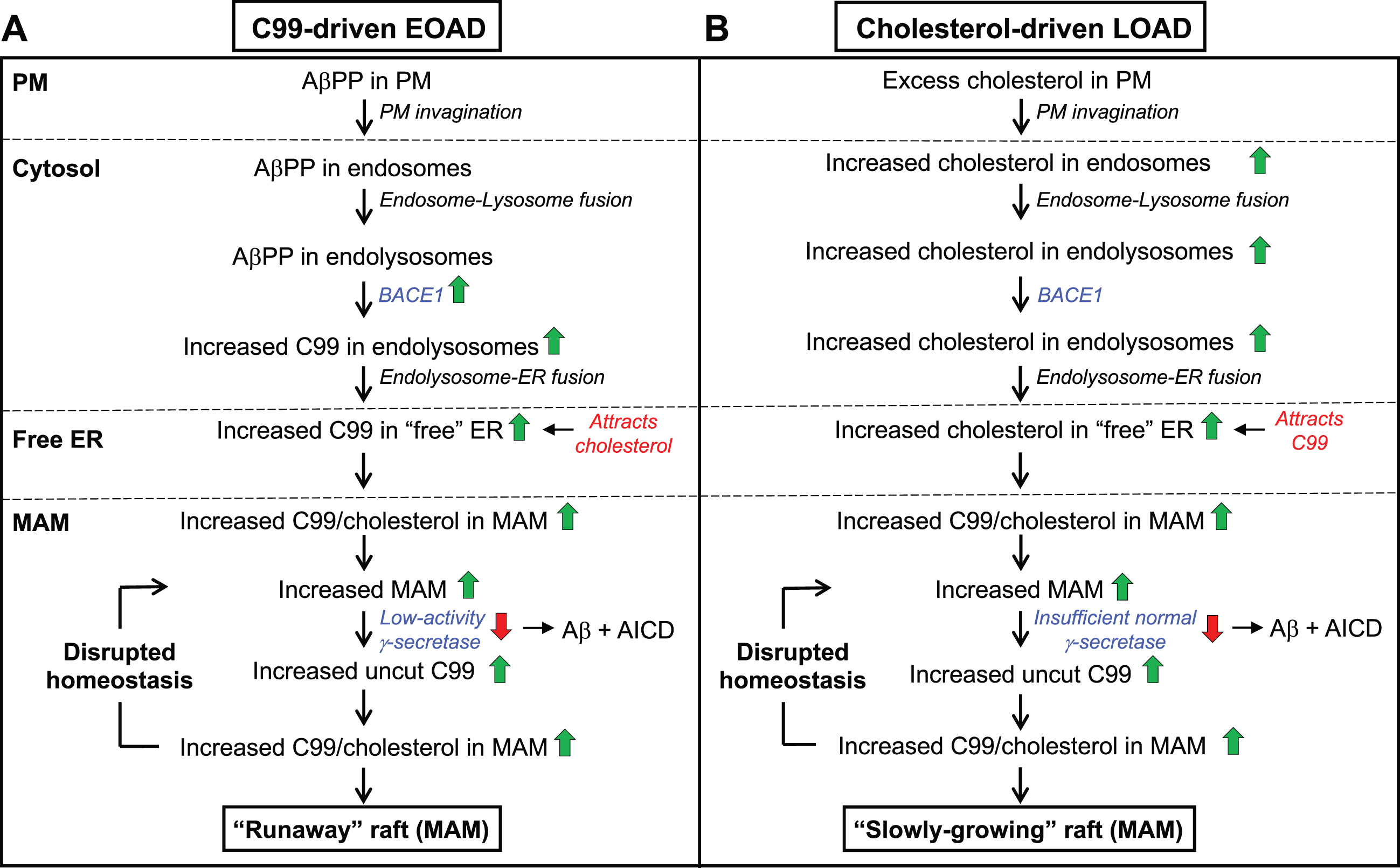
The C99-cholesterol axis in AD. In AD, cholesterol and AβPP enter the cell as in the normal situation (see Fig. 3). However, (A) in C99-driven early-onset AD (EOAD) - both FAD and those forms of SAD associated with underlying C99-related risk factors (see Table 1) - impaired γ-secretase activity is insufficient to cleave all of the MAM-localized C99, leaving “excess” uncut C99 to accumulate in the MAM raft above the “setpoint” level. The elevated uncut C99 attracts more cholesterol (and sphingomyelin), enlarging the raft that, in turn, attracts even more C99 that, too, is cleaved sub-optimally. In this way, MAM homeostasis is disrupted in a vicious cycle in which the raft grows inexorably and relatively rapidly (a “runaway” raft). B) In cholesterol-driven late-onset AD (LOAD) associated with underlying cholesterol-related risk factors (see Table 1), the raft also grows, but for a different reason: the accumulation of excess intracellular cholesterol binds to C99 and causes the raft to grow. Although γ-secretase is functioning relatively normally and efficiently, it is present in an amount insufficient to cleave all the C99 present in the MAM. The excess uncut C99 attracts more cholesterol, enlarging the raft while also generating yet more uncut C99, resulting in the same type of vicious cycle seen in EOAD, albeit in a more slowly-growing raft. Green/red arrows, increased/decreased levels. See text for details.
In EOAD (Fig. 5A), which includes all forms of FAD (including Down syndrome) and those forms of SAD associated with risk factors affecting C99 (e.g., the “blue” genes in Table 1), elevated C99 traffics to the ER-raft (i.e., MAM) where, owing to FAD mutations or to C99-related SAD risk factors, its cleavage is incomplete (i.e., the mismatch between C99 level and γ-secretase activity is such that uncut C99 accumulates above the desired setpoint level). The excess of uncut C99 now attracts more cholesterol (by ∼40% in PS1/PS2-double knockout [DKO] MEFs compared to wild-type MEFs [53]), resulting in the formation of a more extensive raft domain containing more “incoming” C99 that is also cleaved inefficiently, Moreover, the chronic formation of MAM and the increased absolute amount of Aβ triggers the upregulation of SMase activities [270], resulting in reduced sphingomyelin and elevated ceramide levels [35], and a concomitant reduction in cholesterol at the MAM, leading to the formation of longer [74] and thinner [75, 276] MAM structures. Thus, in “C99-driven” EOAD, the normal homeostatic loop is replaced by a vicious cycle in which excess C99 drives raft formation while the growing “runaway” raft recruits more γ-secretase that is unable (or in the case of Down syndrome, insufficient) to cleave all of the C99 presented to it.
In LOAD (Fig. 5B), the C99-cholesterol axis is also perturbed, but in a slightly different way. In the majority of these patients, the disease is “cholesterol-driven,” that is, risk factors (e.g., ApoE4) conspire to increase the level of intracellular cholesterol above normal levels [53, 277] and a subsequent increase in MAM-localized cholesterol (by ∼100% in PS1/PS2-DKO MEFs [53]) that in turn provokes an increase in raft-localized C99. Note that in LOAD, γ-secretase is structurally normal but, coupled with a potential insufficiency in its amount, activity, and/or orientation within the lipid raft, is nevertheless unable to cleave the large amount of cholesterol-recruited C99 present, resulting in a slow but steady accumulation of uncut C99. Once again there is a mismatch between C99 level and γ-secretase activity, only this time it results in a “slowly-growing” raft that exceeds the desired setpoint level.
While we believe that C99 plays a key role in delivering cholesterol to the MAM via the vesicular pathway, we note that a number of proteins involved in the non-vesicular pathway for cholesterol trafficking from the plasma membrane to the ER [272, 279] are also localized to the MAM. These include ADP-ribosylation factor 1 (ARF1) [280], oxysterol-binding protein-related proteins-5 (ORP5) and –8 (ORP8) [223, 281], and Aster-B/GRAMD1B [282] (which is abundant in brain [278]). Moreover, the Aster proteins rely on phosphatidylserine for their function [223, 284], implying that no matter how cholesterol enters the cell, MAM (and therefore C99) likely plays a central role in intracellular cholesterol trafficking [285].
Note that this analysis supports the idea that FAD and SAD (or more properly, EOAD and LOAD) are essentially two subtypes of the same disease [286], and that both disorders share a common pathomechanism that differs only in age of onset, severity of symptoms, and rate of progression. In sum, from a mechanistic point of view, F AD and SAD are the same disease.
In both cases, the cell makes heroic efforts to reduce the inexorable increase in raft size (e.g., by shutting down de novo cholesterol synthesis [53]; by upregulating SMase activity [35] to both increase ceramide and reduce sphingomyelin levels (thereby reducing cholesterol levels); by converting cholesterol to cholesteryl esters that are stored in lipid droplets [74]; by oxidizing cholesterol [287] [at least in the early stages [288], if not the late stages [289], of the disease]; by exporting cholesterol [290]), and succeeds admirably in staving off the inevitable for decades.
WHAT ABOUT TAU?
Hyperphosphorylated tau, a common but inconsistent feature of AD (similar to Aβ), can aggregate into tangles and disrupt cytoskeletal stability and axonal trafficking. Cholesterol appears to play a key role in inducing tau hyperphosphorylation, but the exact mechanism is unclear, as it was reported that tau hyperphosphorylation was stimulated by levels of intracellular cholesterol that were both increased (e.g., via the MAM-localized [291] transient receptor potential cation channel TRPV4 [292]) and decreased (e.g., via the cholesterol biosynthesis enzyme 24-dehydrocholesterol reductase [DHCR24; also called Seladin-1] [293]). Conversely, promotion of cholesterol efflux reduced tau phosphorylation [294], as did reduced MAM-mediated [255] cholesterol esterification [261, 295]. More recently, it was found that tau hyperphosphorylation disrupts ER-mitochondria contacts at MAM [296]. Furthermore, inhibition of BACE1, but not γ-secretase, reduced phosphorylated tau levels in FAD neurons [297, 298], indicating that C99 was at the heart of this effect. While the exact details still need to be worked out, these results clearly speak to important roles for cholesterol metabolism and MAM functionality in promoting and/or responding to tau pathology.
AN OVERALL MODEL OF AD PATHOGENESIS: THE “MAM HYPOTHESIS”
Based on the data and arguments presented above, we propose a “MAM hypothesis” of AD pathogenesis [89, 299–301]. It posits that in both familial and sporadic AD, the functional cause of the disease is massively increased ER-mitochondrial communication and up-regulated MAM function that, in turn, results in the phenotypes seen in AD, including the plaques and tangles. The biochemical cause of AD is MAM-mediated lipid dyshomeostasis driven by increased intracellular cholesterol levels that result from increased steady-state levels of the MAM-localized cholesterol sensor C99 (in the case of EOAD) or from the unregulated accumulation of intracellular cholesterol that interacts with C99 (in the case of LOAD).
An overall model of the MAM hypothesis of AD pathogenesis is shown in Fig. 6. Specifically, in FAD, AβPP processing is affected directly, with a profound interrelationship between AβPP misprocessing (i.e., increased C99 levels) and deranged cholesterol metabolism, as described above. In SAD, the same relationship between C99 and cholesterol also holds true, but in a different way, as the genetic predisposition to develop SAD runs on two parallel tracks, one promoting the increase in cellular C99 levels (causing EOAD, as in FAD) and the other promoting elevated intracellular cholesterol (causing LOAD). As in FAD, both perturbations operate together and interact with each other to generate the downstream MAM-mediated phenotypes, including the plaques and tangles [261].
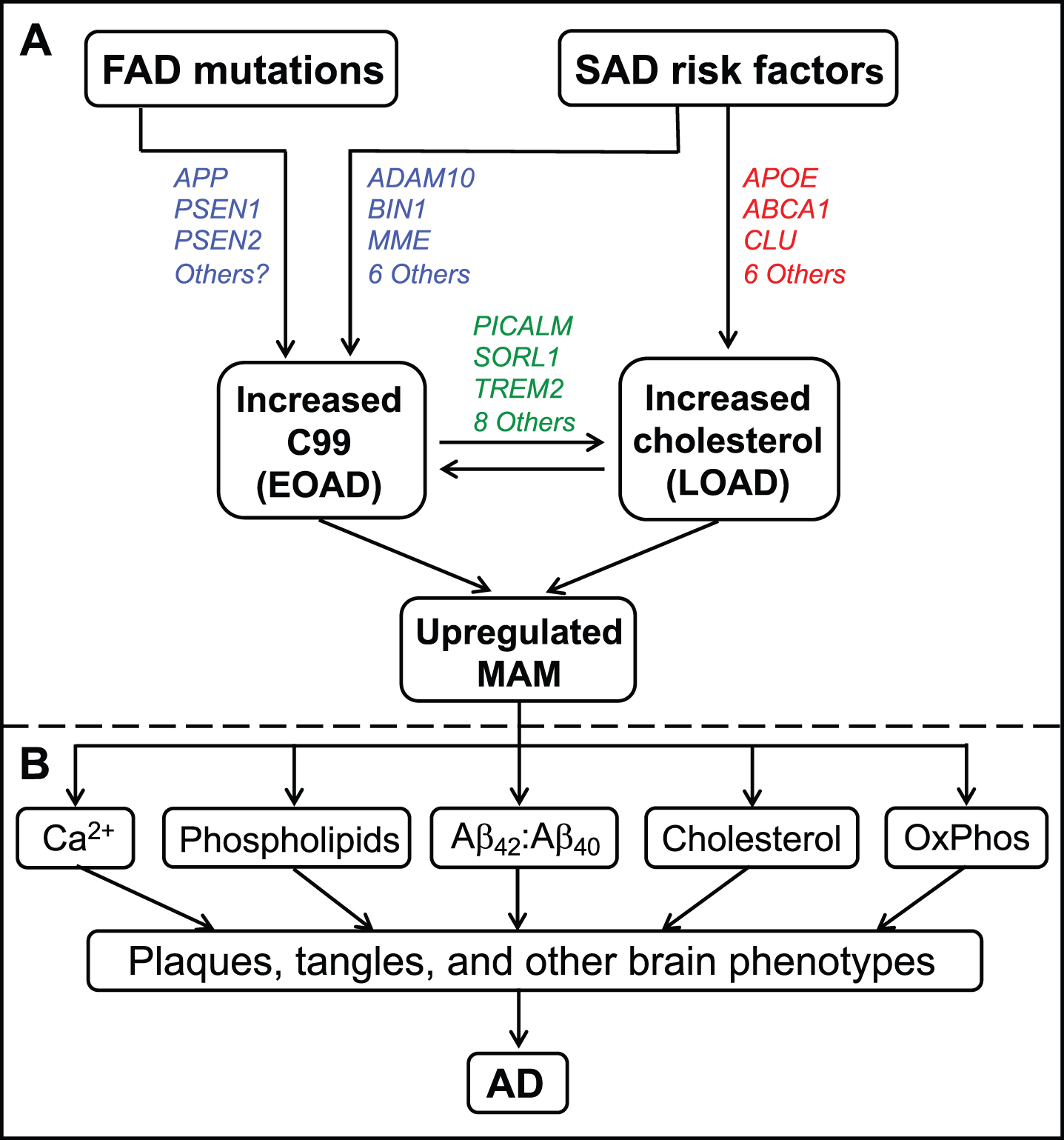
A model of AD pathogenesis. A) Inherited mutations in FAD (affecting C99 [in blue]), or genetic risk factors in SAD (affecting either C99 [in blue] or intracellular cholesterol levels [in red], or both [in green]; colors as in Table 1), converge to increase ER-mitochondrial communication at the MAM. B) These perturbations eventually give rise to the features of AD, as shown. See text for further details.
Note that both elevated C99 and elevated intracellular cholesterol conspire to increase ER-mitochondrial communication at the MAM (Fig. 6A) that, in turn, gives rise to the biochemical, morphological, and clinical features of the disease (Fig. 6B). Importantly, all of those features, including the plaques and tangles, are downstream consequences of the primary functional problem, which is upregulated MAM function. This model helps explain why clinical trials based on removing plaques and/or Aβ have fared so poorly.
We noted above that the SAD risk factors (Fig. 6A) fell into two categories: those that mainly affect MAM function and those that mainly affect cholesterol metabolism. Thus, one might expect that clinically, SAD patients expressing MAM-related risk factor genes would resemble FAD patients, with a relatively early age of onset and a rapid disease progression (i.e., EOAD), whereas those expressing cholesterol-related risk factors would have a later age of onset and slower progression (i.e., LOAD). Remarkably, this may actually be the case, as it was recently shown that the distribution of the age of onset in ɛ4-negative SAD patients is indeed bimodal, with one onset peaking at age ∼57 years (i.e., EOAD) and the other peaking at age ∼77 years (i.e., LOAD) [302], consistent with, but of course not proof of, this supposition.
Thus, the MAM hypothesis invokes two key interrelated elements that drive AD pathogenesis, C99 and cholesterol, that converge at the MAM. Taken together, we propose that both FAD and SAD are caused by the same underlying and linked pathogenetic processes—elevated C99 and increased intracellular cholesterol. From this perspective, AD is, at bottom, a lipid disorder, similar to what has been proposed by others [115, 303–305] but described here in somewhat greater detail.
CLINICAL IMPLICATIONS
How does upregulated MAM function lead to a neurological disorder? In other words, how does MAM upregulation impair neuronal functionality to the point where cognitive functions are compromised? We believe that the answer to this question lies in MAM’s role as a center of cellular lipid metabolic regulation, where multiple lipid classes are metabolized by MAM-localized enzymes. Lipids regulate multiple basic brain functions [306], including action potentials, synaptic vesicle release, endocytic transport, cellular signaling, and pathogen recognition [307]. Many of these roles arise by virtue of the fact that lipids are the principal constituent of cellular membranes, where their different charges and shapes modulate membrane properties. As such, lipids can control the recruitment, conformation, and interaction of membrane-associated proteins like receptors, channels, and enzymes [308]. Lipids also regulate membrane curvature, important for the budding and fusion of vesicles containing neurotransmitters and other cargo [309, 310]. They also regulate membrane permeability and fluidity, which are largely dependent on the fatty acid composition and degree of saturation of constituent phospholipids [311]. Furthermore, lipids, and cholesterol in particular, are the primary constituents of myelin [312], which ensheathes axons and enables efficient conductance of action potentials. Finally, lipids represent a source of energy storage and transport and can serve as reservoirs of bioactive signaling molecules. It is therefore not surprising that lipid alterations can be drivers of neurodegenerative diseases [313], which we propose to be the case in AD.
THE MAM HYPOTHESIS HAS EXPLANATORY POWER
The MAM hypothesis answers or explains many (but by no means all) of the features of AD that are not easily explained by the amyloid cascade. This includes the elevated Aβ42:Aβ40 ratio, the altered cholesterol, phospholipid, sphingolipid, calcium, and glucose levels, the increased lipid droplet formation, the reduced bioenergetics and altered mitochondrial morphology, the role of many SAD genetic risk factors and of Down syndrome, the different severities of various AβPP and presenilin mutations, the partial elucidation of the dominant nature of the FAD mutations and whether FAD and SAD are manifestations of a single underlying disease process, the secondary role of plaques and tangles, and the poor track record of amyloid-based clinical trials.
The MAM hypothesis could also provide insight regarding the preponderance of affected females over males [314]. Both estrogen and progesterone decrease profoundly in post-menopausal women [315], and also decline in elderly men, but to a lesser degree [316]. Notably, steroid synthesis, like phospholipid synthesis, is a collaboration between MAM and mitochondria: MAM-localized cholesterol translocates to mitochondria [194, 318], where it is converted to pregnenolone. Pregnenolone then translocates back to the MAM for synthesis of the various steroid hormones [309], including estrogen and progesterone, and for the synthesis of pregnenolone esters by ACAT1 [319]. It turns out that estrogen inhibits the transcription of BACE1 [320, 321], so its loss de-represses BACE1 activity [322], increasing C99. Similarly, progesterone inhibits the expression of ACAT1 and reduces MAM apposition length [323, 324], so its loss de-represses ACAT1 activity, thereby perturbing cholesterol homeostasis and also increasing C99. Thus, the age-related decline in both hormones in brain [325, 326] in women (and to a lesser degree in men) can upregulate MAM, triggering AD in a “sex-biased” manner, with obvious implications for hormone replacement therapy in AD, a highly contentious topic [327, 328]. Moreover, the decline in these hormones might help explain why advancing age is the single most important determinant of AD susceptibility [329].
Finally, the MAM hypothesis helps explain why inhibiting either BACE1 [13] or γ-secretase activity [15] as approaches to treat AD would be ill-advised. In the former case, BACE1 inhibition would result in a severe reduction in C99 levels, thereby constraining normal raft production, likely with deleterious consequences. In the latter case, inhibiting γ-secretase would increase C99 production, the very outcome that we should be trying to avoid. On the other hand, if AD is caused by disruption of the C99-cholesterol axis, reducing or preventing the accumulation of intracellular cholesterol might be of therapeutic value, for example, via the use of statins, a controversial topic [330, 331]. However, we believe that statins will likely be of limited utility, given that they are designed to inhibit the activity of HMGCR, which we have shown is already down-regulated in AD cells [53]; also, statins must cross the blood-brain barrier in order to be effective.
As is with works in progress, the MAM hypothesis currently does not explain other features of the disease. These include the relative paucity of FAD mutations in PS2 compared to those in PS1; the loss of olfaction as an early indicator of AD; the brain-specificity and brain subregion-specificity (e.g., hippocampus versus cerebellum; motor neurons versus cortical neurons) of the clinical phenotype (but see below); and the identification of risk genes that currently do not fit into the MAM paradigm (e.g., entries in black in Table 1), to name but a few. Nevertheless, it is reasonable, if optimistic, to state that it is not inconceivable that they too will eventually be explained in a manner consistent with the MAM hypothesis.
For example, for currently unclear reasons, there is a discrepancy between the number of pathogenic mutations in PS1 (more than 100) compared to those in PS2 (only ∼5) (https://www.alzforum.org). Although PS1 and PS2 are highly similar in structure (64% amino acid identity), they differ in their γ-secretase activities, substrates, and functions [28, 332]. Moreover, contrary to what is seen in the vast majority of PS1 mutations [26], pathogenic mutations in PS2 increase γ-secretase activity [28] and presumably reduce C99 levels, thereby posing a challenge to the MAM hypothesis. However, PS2, but not PS1, binds to MFN2, thereby promoting tethering of ER to mitochondria, and FAD-mutant PS2 binds MFN2 even more avidly and enhances the degree of tethering above normal levels [333, 334]. Thus, mutations in PS2, coupled with the potential interactions of PS2 with full-length MFN2 and/or its splice variants [335], could indeed mimic the effects of PS1 on enhanced ER-mitochondrial communication and on the C99-cholesterol axis (brain cholesterol levels are elevated in FADPS2 patients [336]), albeit via a more circuitous route. Taken together, these findings could explain why pathogenic mutations in PS2 are not only relatively rare, but also result in a “milder” form of AD compared to mutations in PS1 [28].
TISSUE SPECIFICITY
One problem that has dogged the field is that of tissue specificity: APP, PSEN1, and PSEN2 are housekeeping genes that are expressed in essentially all tissues [257, 338] (see also genecards.org), and yet, FAD-causative mutations in these genes cause overt pathology only in the brain. In one sense, the issue of tissue specificity may be a red herring, as lipid droplets accumulate in peripheral blood cells from AD patients [257]. Moreover, AD-model mice expressing ApoE4 exclusively in their livers evinced enhanced AD-like amyloid pathology compared to mice expressing ApoE3 [339], implying that there is communication between the periphery and the brain in AD, likely via a breach in the blood-brain barrier. In addition, while MAM contacts ER “loosely” (gap distance of ≥10 nm), liver mitochondria are also contacted by tightly-adhering ER (“wrappER”) that is distinct from MAM [340], with essentially no gaps between the two organelles. WrappER mediates the biogenesis of lipids and lipoproteins [341] and plays an important role in cholesterol synthesis and trafficking via three-way ER-mitochondria-peroxisome contacts [342]. Taken together, these results are not only consistent with the concept that AD is indeed a lipid disorder, but also imply that in the context of AD pathology, the C99-cholesterol axis may not be confined to the brain.
And even within the brain there is regional specificity, as, for example, cerebellum is far less affected in AD than is hippocampus [343], with tau pathology spreading outwards from the entorhinal cortex in a prion-like manner [344, 345]. Can the MAM hypothesis shed any light on this paradox? One clue may lie in the emphasis of the MAM hypothesis on lipids, and especially on the role of cholesterol and sphingomyelin in lipid raft formation. We note that two of the earliest signs of AD are deficits in olfaction [346] and episodic memory [347]. While the two phenomena are apparently unconnected, there may be a relationship between them at the cellular level, as the olfactory bulb and the hippocampus are apparently the only loci in the adult human brain that are capable of renewal by stem cells [348] (a subject of some debate [349]). Given that 1) these newly-born cells are the only ones that require de novo myelination by oligodendrocytes, 2) two of the most abundant components of myelin are cholesterol and sphingolipids [350], and 3) both cholesterol biosynthesis [351] and progesterone [352] are required for myelination, it is possible that the aberrant MAM function described here is particularly pronounced in oligodendrocytes [353], either quantitatively (e.g., too little or too much myelin) or qualitatively (e.g., altered myelin composition) or both [354]. This may lead to altered neuronal conductance that is particularly pronounced in, but clearly not limited to, these newly-born cells.
THE MAM HYPOTHESIS AND NEUROINFLAMMATION
Our hypothesis proposes that higher levels of internalized cholesterol trigger MAM formation and subsequent modulation of metabolic enzymes. In the context of microglia, we note that immune challenges induce the transport of cholesterol from the plasma membrane to the ER [355], resulting in the formation of MAM domains. In turn, MAM formation in microglia induces the recruitment and clustering of enzymes involved in the regulation of bioenergetics (i.e., switching from OxPhos to glycolysis [356, 357]) and lipids (including regulation by TREM2 [triggering receptor expressed on myeloid cells 2], a known AD risk factor that is expressed exclusively in microglia [358]), thereby adapting microglial metabolism and cellular membranes to a pro-inflammatory state [357]. Once the activation subsides, cholesterol removal from the ER dissolves MAM’s structure, leading to a reversal of the changes in mitochondrial metabolism and the resolution of inflammation. From this point of view, the upregulation of MAM domains could explain the pro-inflammatory phenotypes associated with AD and help explain why cholesterol alterations are frequently associated with the emergence of damage-associate microglia (DAM) phenotypes.
THE MAM HYPOTHESIS HAS PREDICTIVE POWER
The hypothesis predicts that since AD is fundamentally a lipid disorder due to altered lipid homeostasis (most notably at the MAM), levels of specific lipids should be altered in predictable ways. To take but one example, MAM-localized ACAT1 [255] has a marked predilection for generating cholesteryl esters containing unsaturated fatty acids, especially those containing oleate (C18:1) [359, 360]. Similarly, MAM-localized long chain fatty acid-CoA ligase 4 (FACL4; gene ACSL4) [361], used in the transfer of fatty acids to phospholipids and sphingolipids, has a preference for C20:4 and C20:5 [362]. On the AβPP processing side, the elevation in C99, and the corollary reduction in Aβ, implies that there should be an AD-specific increase not only in the ratio of Aβ42:Aβ40, but also in the ratio of C99:total Aβ. Thus, alterations in specific lipids and in C99 levels should be pathognomonic of AD.
IMPLICATIONS FOR DIAGNOSIS AND TREATMENT
The specific alterations in both lipid profiles and in the C99:total Aβ ratio predicted by the MAM hypothesis should be detectable in more easily-accessible tissues, such as blood (e.g., in plasma/serum for the lipids and the Aβ; in peripheral blood mononuclear cells for the C99). In fact, analysis of lipids in AD blood has already been reported [363–365] but the specificity and mechanistic interpretation of those “agnostic” shotgun approaches have limited their use as true and robust diagnostics. On the other hand, a mechanism-based diagnostic scheme based on the MAM hypothesis should be both more accurate and more precise, by focusing on specific MAM-mediated lipid species, combined with the predicted elevated C99:Aβ ratio.
From the MAM point of view, it is clear that efforts to treat AD by ameliorating downstream effects (e.g., by modulating calcium levels, increasing mitochondrial bioenergetics, reducing total Aβ, and the like) will probably not work. On the other hand, two different approaches to treatment immediately present themselves: 1) re-normalizing cholesterol homeostasis, and 2) re-normalizing C99 levels. One can imagine techniques to execute both approaches [366], but these are beyond the scope of this review.
AUTHOR CONTRIBUTIONS
Estela Area-Gomez (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing); Eric A. Schon (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing)
Footnotes
ACKNOWLEDGMENTS
We thank the many contributors to the development of the MAM hypothesis over the years, including Rishi Agrawal, H. Orhan Akman, Thomas Bird, Istvan Boldogh, Eduardo Bonilla, Gloria Patricia Cardona, Robin Chan, Adrianus de Groof, Gilbert Di Paolo, Karen Duff, Gary Gibson, David Gorin, Cristina Guardia-Laguarta, David Holtzman, Junichi Ikenouchi, Khushbu Kabra, Carla Koehler, Mari Carmen Lara Castillo, Delfina Larrea, Francisco Lopera, Mark Mattson, Mark Mehler, Tamar Michaeli, Jorge Montesinos, Patricia Morcillo, Renu Nandakumar, Marta Pera, Liza Pon, Serge Przedborski, Irina Stavrovskaya, Gary Struhl, Mark Tambini, Kirstin Tamucci, Kurenai Tanji, Masato Umeda, Kevin Velasco, Michael Yaffe, Hua Yang, Wai Haung Yu, Taekyung Yun, and Xiongwei Zhu.
FUNDING
This work was supported by grants from the U.S. National Institutes of Health (to EAG [1R01AG056387] and EAS [![]() ]) and the J. Willard and Alice S. Marriott Foundation (to EAS).
]) and the J. Willard and Alice S. Marriott Foundation (to EAS).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.