
Abstract
Vaccines such as Bacille Calmette–Guérin (BCG) can apparently defer dementia onset with an efficacy better than all drugs known to date, as initially reported by Gofrit et al. (PLoS One
Keywords
INTRODUCTION
Vaccines have non-specific effects that can have a profound benefit for health systems. Is it possible that such immune boosting can be applied to a variety of infectious and neurodegenerative diseases, especially those of the elderly? This concept emerges from the beneficial role of Mycobacterium-derived Bacille Calmette–Guérin vaccine (BCG) in cancers as well as in recent studies on juvenile diabetes, multiple sclerosis, and most recently on Alzheimer’s (AD) and Parkinson’s diseases (PD) [1–6]. The effect of BCG on the prevention of recurrent pneumonia in discharged elderly patients is also remarkable [7]. For this reason alone, BCG for the elderly should be given serious consideration. BCG is central to these stories of immunotherapy, but other vaccines have also been reported to be beneficial in the prevention of AD and PD. Given the burden in suffering [8], as well as funding, it seems obvious that a public health program of vaccination with BCG for neurodegenerative diseases, perhaps by the oral route, should not be overlooked.
We have divided this work into two parts. We recognize that the neuroscientist, neurologist, or gerontologist may not be familiar with the underlying concepts, and we therefore provide a historical introduction. We introduce the reader to the concepts of adjuvants and non-specific immunity, and follow the origins of BCG, how mycobacteria entered into adjuvants, and their mechanism(s) of action, as a prelude to the discovery that BCG may protect against dementia development (Part II [9]), and a possible path to wider implementation. These are immensely complex and fascinating topics and cannot be covered here in the full depth they deserve, and we therefore refer wherever possible to authoritative reviews. However, despite not covering all relevant aspects in full detail (notably the important concept of trained immunity), this first part provides a historical perspective that serves as a primer to the second part— the potential use of BCG or other vaccines as protective agents against dementias such as AD [9].
BCG: THE ANTI-TUBERCULOSIS VACCINE
BCG began as a stand-alone vaccine for tuberculosis (TB, Box 1), and recently passed its 100th anniversary, as celebrated in detailed reviews [10–13]. Albert Calmette’s (Fig. 1A) rendering of the BCG story in an article in 1931 began by his description of the many earlier attempts to obtain attenuated pathogens for vaccination [14]. He was certain that ‘only living viruses are able to cause solid and durable immunity, as does the disease itself’. Here Calmette uses the term ‘virus’ to designate any noxious microorganism, following Villemin who was the first to demonstrate that TB is transmissible (Box 1). Thus in 1901, following his observations and those of his colleague Camille Guérin (Fig. 1A) that tubercular infections in wildlife were by ingestion, they infected by oral intubation young calves with an emulsified bacterial preparation, first in their food and later in egg yolk and in sterilized bile. They evolved a method of culturing the organism in glass tubes on potatoes cooked in 5% ox-bile in glycerine, their so-called bile–potato medium. The inoculum was of Mycobacterium bovis isolated in 1902 from a tuberculous udder. After 30 passages, it was no longer virulent to guinea-pigs, and after 60 passages was still ‘slightly virulent’ to rabbits and horses, but not at all virulent to guinea-pigs, monkeys, or calves [15, 16]. WWI interrupted planned large-scale trials in calves, but a sentinel event changed the direction of their research.
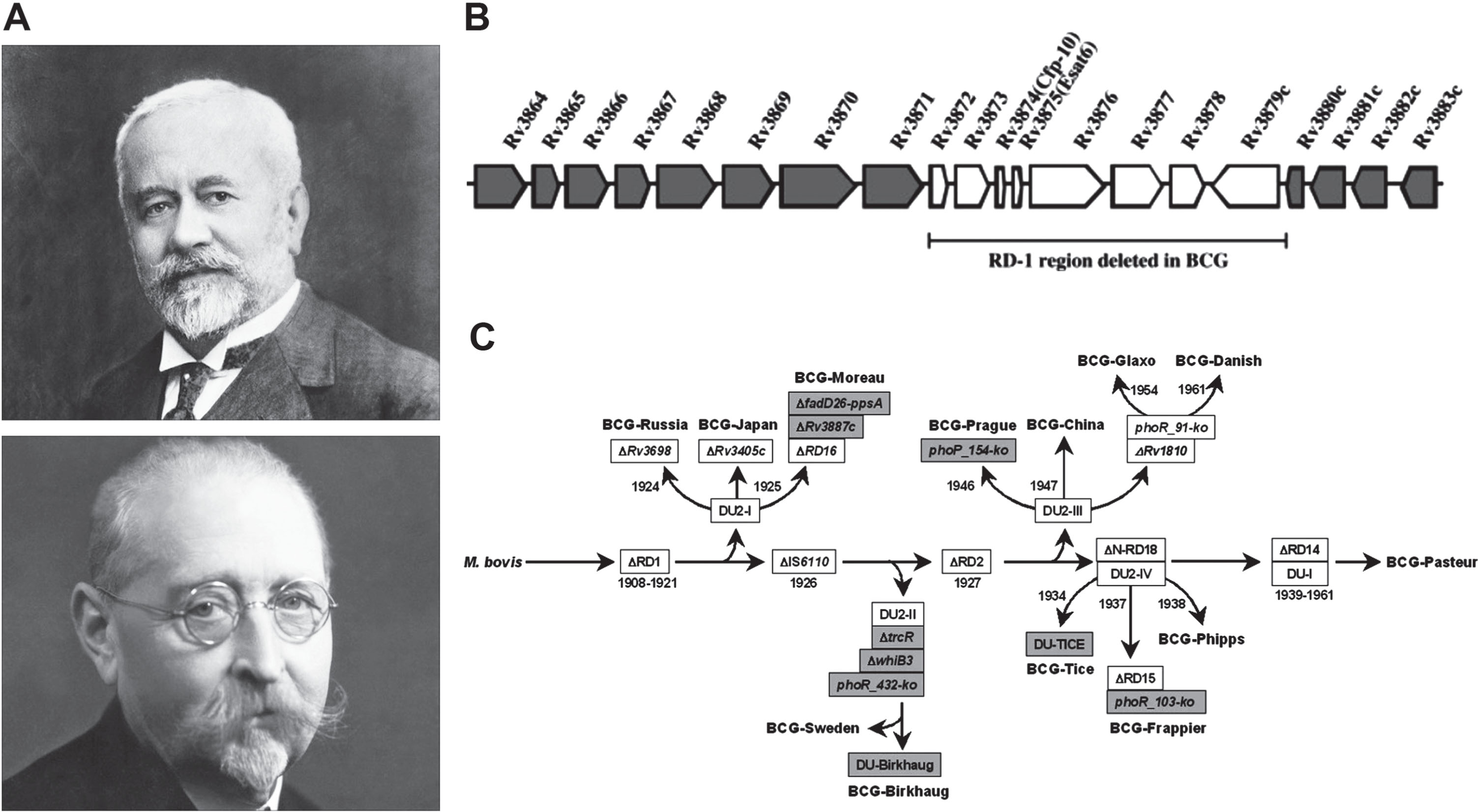
Evolution of Bacille Calmette–Guérin (BCG). A) Léon Charles Albert Calmette (1863–1933; above) and Jean-Marie Camille Guérin (1872–1961; below) who first developed BCG at the Institute Pasteur de Lille, France. B) Deletion of the RD-1 region of the Mycobacterium bovis genome during passage by Calmette and Guérin. C) Further genomic polymorphisms including four deletions and two duplications during further passage of BCG vaccine strains, highlighting that different BCG strains may differ in their properties; additional detailed studies on the phylogeny of BCG strains can be found in [151]. Panel (A) reprinted, with permission, from [152]; panel (B) reprinted, with permission, from [153]; panel (C) reprinted, with permission, from [154].
Jean Antoine Villemin (1827–1892) was the first to demonstrate that TB is transmissible by inoculating human tuberculous extracts into rabbits [140]. Although he and others at the time used the loose term ‘virus’ to designate pathogens in general (‘La tuberculose est l’effect d’un agent causal spécifique, d’un virus en un mot’, p. 216), he specifically likened TB to glanders (morve équine), a severe infectious respiratory disease of horses caused by a bacillus (now Burkholderia mallei). The causal bacterium of TB, Mycobacterium tuberculosis, a Gram-positive rod-shaped aerobe, was identified by Robert Koch in 1882 [141].∥TB as a disease was recorded by early Greek physicians including Galen and Hippocrates ([142], see also [143]). However, the disease has been around since much earlier. M. bovis sequences were detected in bone samples from the remains of two individuals in the Eastern Mediterranean dated to 9000 years ago [144], signs of tubercular infections have been found in Egyptian mummies dating to 2000–3000 BC (https://en.wikipedia.org/wiki/Tuberculosis), and traces of TB infection have been found in an extinct bison dating to 17,000 years ago [145]. Phylogenetic analysis of 259 strains of the M. tuberculosis complex (MTBC) suggests that MTBC first arose about 70,000 years ago, and accompanied human migration out of Africa [146].
M. tuberculosis is classified as being Gram-positive, but DNA sequencing reveals that is more closely related to Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa than it is to other Gram-positive bacteria [147]. Although mycobacteria are often regarded as being intracellular pathogens (e.g.., [148, 149]), there is direct evidence for persistence as free forms in the extracellular milieu [150].∥Principally affecting the lung, TB can also affect other tissues including bone and the nervous system. TB can be treated with antibiotics but can be life-threatening if untreated. Infections are commonplace in cattle and other animals including nonhuman primates, and mankind has probably been exposed to TB for eons: this could explain why over 90% of people infected are asymptomatic, suggesting that robust immune defenses have emerged through Darwinian selection. Nevertheless, perhaps a quarter of the world population presently harbors latent TB, and the disease remains the second leading cause of death worldwide from infectious disease after COVID-19.
On July 1, 1921, a physician from the children’s department of the Charité Hospital in Paris consulted Calmette and Guerin about a child whose mother had died of TB shortly after birth. Not only was the mother tuberculous, but also the grandmother who was caring for the infant. At that time, an infant born under these conditions would have a 7–25% chance of dying before reaching the age of 1 year, whereas this was under 2% in a normal family. Calmette, then in Paris, agreed to treat the child with BCG, which at that time had been passaged 230 times. The child was given three doses of 6 mg of BCG orally. No ill effects were observed, and over the next 3 years 317 infants were vaccinated. For reasons not specified, the amount per dose given three times was raised to 10 mg. Of the vaccinees, 21% were born into tuberculous families and the remainder into unaffected families.
From a historical perspective, Calmette’s decision to administer the vaccine was perhaps influenced by the precedent of his eminent colleague, Louis Pasteur, who in 1885 (also in Paris) performed the first human anti-rabies vaccination [17].
In his 1931 article Calmette recounts the experience of the next 7 years in which nearly a million children were vaccinated in France, Romania, Sweden, Poland, and Bulgaria [14]. Although widespread vaccination was not practiced in the USA or Europe, it was widely deployed in a few large cities including New York, Montreal, Montevideo, Barcelona, and Amsterdam. Calmette gives a smattering of statistical data, at the same time noting the fact that they called in chief statisticians to certify their results. In Romania he noted that general mortality was reduced by 50%, whereas in Sweden general mortality was reduced from 9.5% to 2.3% . In a New York study in the first year of life, 8% of the children died of TB, but only 0.9% of the vaccinated, whereas in the second year the numbers were 3.8% for the non-vaccinated and 0% for the vaccinated. Later studies in Africa confirmed these results [18].
Other than saving these many children, Calmette was energetic in shooting down reports claiming that they had either observed or caused BCG to revert to a virulent state, an issue of great importance. Today we know that a portion of the genome of Mycobacterium bovis was deleted in the attenuation process. Mahairas et al. [19] identified the RD1 region which contains the genes for the antigens ESAT-6 and CFP-10 which relate to tuberculosis virulence (Fig. 1B). When RD1 was deleted from M. tuberculosis, infectivity was lost for isolated cells and mice, and the action of BCG was mimicked [20].
Non-specific immune boosting
Vaccines are paradigmatically deemed to protect specifically against the organism from which they are derived. For example, cowpox infection against smallpox, as observed by Edward Jenner in 1796, is because cowpox and smallpox are related viruses that share common antigens. However, this is only part of the story, and there are multiple reports of protection against distinct infectious agents. For example, E. Gildemeister and Kurt Herzberg reported in 1924 overlapping protection in rabbits between smallpox and the unrelated virus, herpes [21]. Paul Forster in Los Angeles and Brooks Abshier in New York observed in 1926 that some individuals receiving smallpox vaccine (vaccinia virus) displayed regression of lesions caused by herpes simplex virus [22]. They stated: ‘The effect of smallpox vaccination was first called to our attention in 1926, during an epidemic of smallpox in Los Angeles, when one of us (P. D. F.) had had attacks of herpes labialis so frequently that a white scar still remains. During the epidemic he was vaccinated approximately six times, and since that time no further attacks have occurred.’ Prompted by this observation, they carried out more extensive studies. Thirty-five patients with recurrent herpes simplex lesions were treated with ∼4 smallpox vaccinations. The lesions were markedly decreased all patients, and over a 2-year follow-up there was no recurrence in 86% [22]. Although the protective finding has been queried, it has been confirmed elsewhere [23, 24]. Importantly, this is not a peculiar interaction restricted to poxviruses and herpes. A different vaccine, attenuated poliovirus-based vaccine, can also have therapeutic effects against herpes [25, 26]. Vaccination with oral poliovirus has been reported to decrease postnatal influenza morbidity by threefold [27], and similar findings have been reported for enteroviruses [28, 29]. From such observations emerged the concept of adjuvants –agents that generically stimulate the immune system (Box 2).
The finding that exposure to one agent can enhance non-specific immune responses against a unrelated agent (main text) led to the concept of immune ‘adjuvants’, from Latin adiuvare, to help or aid (reviewed in [155]). Multiple different substances were investigated in the 1920 s, including toxins, silver salts, turpentine, and other noxious agents. Whereas Paul Lewis and Dorothy Loomis [30] used live tubercle bacillus to boost the immune response, Jules Freund (Figure A) and Katherine McDermott used a combination of killed tubercle bacteria in a lanolin/oil suspension in their classic paper [40] (main text). The most powerful adjuvant known to date, Freund’s complete adjuvant (FCA), is thus based on heat-killed Mycobacterium tuberculosis in mineral oil [44]. Nevertheless, the potency of FCA in inducing an immune reaction is so high that it can induce lesions at the site of inoculation and is therefore not approved for human use [44].∥The most widely used adjuvant in humans consists of aluminum salts, as first used by Alexander Glenny in 1926 (reviewed in [156]). Other molecules known to stimulate vaccine efficacy include bacterial lipopolysaccharide (LPS) and muramyl dipeptides (MDPs, main text). However, only four adjuvant molecules are currently licensed for human use. The most common is aluminum hydroxide and related aluminum salts, known as ‘alum’; more recently licensed adjuvants include lipid-based molecules (monophosphoryl lipid A, MF59/squalene), a triterpene glycoside (QS-21), and CpG 1018 (a synthetic bacterial DNA mimetic) (https://www.cdc.gov/vaccinesafety/concerns/adjuvants.html; [156]). The combination of monophosphoryl lipid A and QS-21 is known as AS01 [157]. However, alum remains the key adjuvant molecule in > 90% of vaccines, and there is an urgent need to develop new immunopotentiating agents [158].∥ In contrast to FCA, the anti-TB vaccine based on attenuated Mycobacterium bovis, BCG, is approved for human use and is the only vaccine for TB. It is most effective for childhood M. tuberculosis infections and variably effective for adult pulmonary TB [159]. Importantly, BCG shares many of the non-specific immunostimulatory effects of FCA (e.g., against bladder cancer; main text). Hence, two of the most powerful adjuvants known, FCA and BCG, are both based on Mycobacterium species.

Mycobacteria as immunostimulators: TB versus cancer
At the same time as Calmette and Guérin were developing a prophylaxis for TB, other researchers were increasingly intrigued by these cross-agent adjuvant effects. In 1924, Paul Lewis and Dorothy Loomis at the Rockefeller Institute in Princeton discovered that the titer of hemolytic antibody against sheep red blood cells could be increased by up to 20-fold by experimental infection of guinea pigs with live pathogenic ‘tubercle bacillus bovine C2’ (Mycobacterium sp., exact species uncertain) [30] (Fig. 2). This was a remarkable finding because it began for the first time to indicate a mechanism of action of cross-agent protection— infection with a pathogen could boost a specific arm of the immune system.
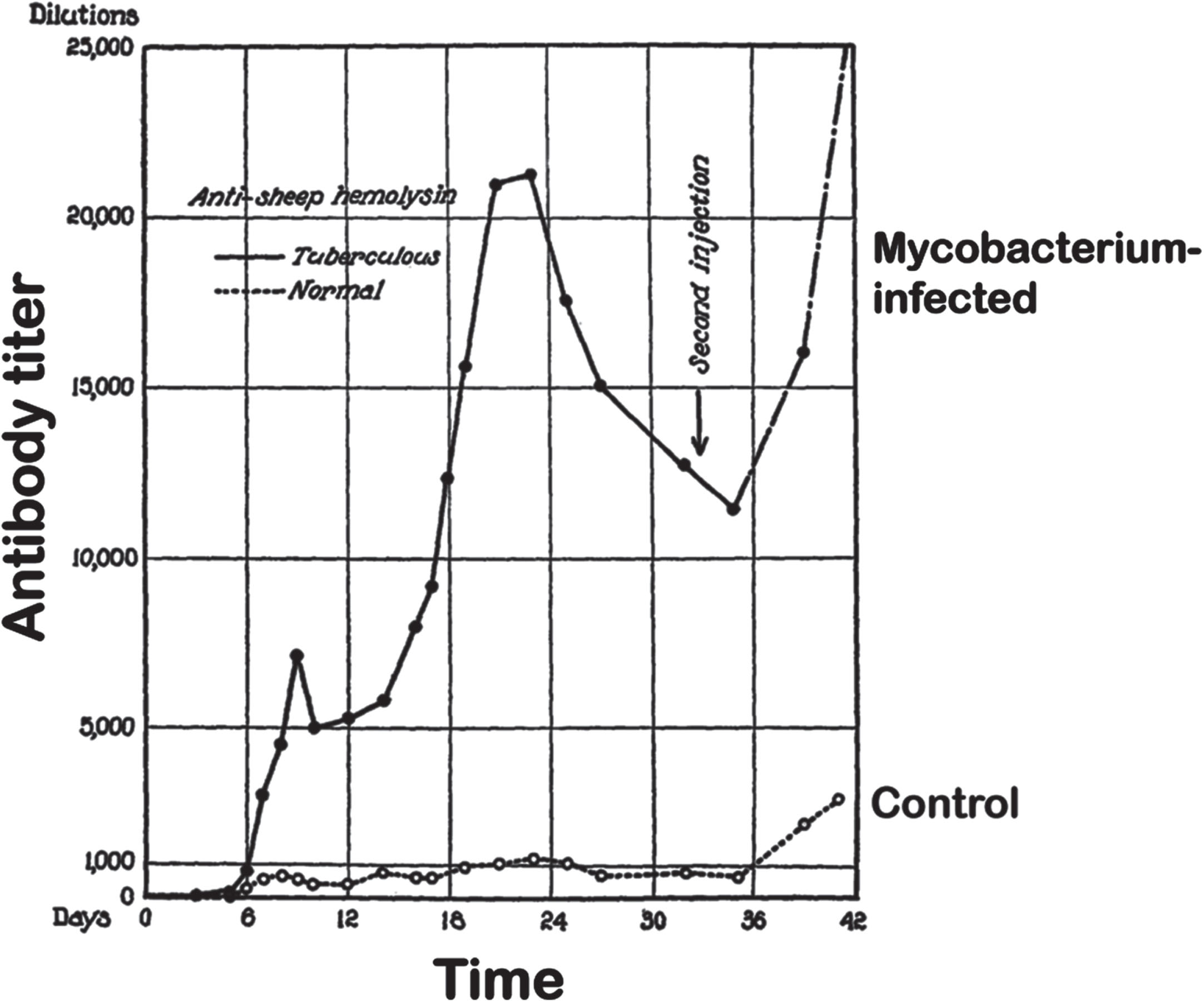
Titers of anti-sheep red blood cell antibodies in control and tuberculous guinea pigs. Animals were infected 21 days before the start of the experiment. At day zero animals were inoculated with a suspension of sheep red blood cells, followed by a second injection on day 33. Figure adapted, with permission, from [30].
This was followed by the fascinating discovery, by Raymond Pearl at Johns Hopkins University Hospital in 1929, that TB was not all bad— indeed it seemed to lower the risk of developing cancer [31]. In over 800 patients with malignancies, 6.6% had active TB whereas a control group of similar size matched for race, sex, and age, with no evidence of malignancies, 16.3% were tuberculous. Pearl had earlier worked with the famous English statistician Karl Pearson, and this method of statistical analysis colored much of his work. His range of interests was enormous, and he published on population biology, longevity, fertility, and growth in populations as varied as poultry, corn, fruit flies, and humans [32]. Pearl concluded that active TB was an antagonist to cancer, and in his report, there is mention of ‘Some experiments are now being carried out treating malignant tumour in human beings with some form of tuberculin and although the results so far are satisfactory no definite statement can be made at this early stage’ [31]. More than 20 years passed before, in 1959, Lloyd Old and colleagues in New York demonstrated that BCG could retard or prevent tumor development in mice inoculated with cancer cells [33], and BCG (not tuberculin) became a major player in cancer immunotherapy (reviewed in [34]).
This aspect has a long history: records from early Egypt suggest a crude treatment for tumors involving deliberate infection of the tumor site [35], and William Coley in the 1890 s explored the use of extracts of bacteria (Streptococcus pyogenes and Serratia marcescens) in inducing tumor regression, often with considerable success, but this appeared to require direct inoculation into the tumor site, although it is likely that remission of distant metastases was also achieved in some cases (reviewed in [36]). These findings are reminiscent of reports in the 1960 s that inoculation of tumors with wild-type vaccinia or cowpox virus could lead to tumor regression [37, 38]: although the general interpretation at that time was that the virus directly infected and killed the tumor cells, regression was sometimes also seen in uninjected lesions (reviewed in [39]).
Freund develops a very effective adjuvant, the mycobacteria step in
In 1942, Jules Freund and Katherine McDermott working in the Department of Health of New York City were determined to enhance the production of antibodies in animals [40]. We know something of Freund in that he had a successful career in the Austro-Hungarian army, and after serving in WWI he returned to the University of Budapest, rising to Commissioner for Hygiene in the army. After becoming an assistant professor in preventive medicine, he moved to Hamburg Medical School in Germany, and then in 1922 to Harvard University. In the US, he became a major figure in the immunological community, ending his career at the National Institutes of Health (https://www.aai.org/About/History/Past-Presidents-and-Officers/JulesFreund). Regarding McDermott, a 2016 article by Hilda Bastian remarks “another woman on the immunology timeline who doesn’t have a Wikipedia page. Katherine McDermott is credited for adjuvants in 1942. Who is she? What’s her story?” [41]. Bastian also makes note of McDermott in a more recent article on women in vaccine science where she writes: ‘I cannot find anything online about McDermott’ [42].
Freund with McDermott developed his adjuvant based in part on the observation by Lewis and Loomis that tuberculous guinea pigs produced more antibodies to antigens than non-infected animals (Fig. 2 and [30]). Dienes showed it was possible to substitute living tubercle bacilli with dead ones by first sensitizing a lymph node with the bacteria and subsequently injecting the antigen— in this case egg white [43]. Several variations of these injections were also successful in producing abundant antibodies. Paraffin oil was found to enhance the effect of bacilli and so it was used with the bacteria in presensitizing the animals. The next step involved mixing the antigen with a lanolin-like substance (aquaphor) in an aqueous phase, then this was suspended in twice the amount of killed bacilli in paraffin oil. When guinea pigs were sensitized by injecting this mix into the back of their neck by three injections in the first month, then at 6 months and 1 year, the adjuvanted antigen gave much stronger skin sensitization and precipitin reactions than the antigen in saline. Thus, Freund’s complete adjuvant (FCA) was born.
To this day, in experimental antibody production, to our knowledge FCA has not been surpassed by any other adjuvant. However, its use is not without various serious side effects, including pain, and has therefore not been used in human or veterinary medicine. Stills Jr has described the gradual steps by which the original FCA has been modified while maintaining its basic oil in water emulsion character [44]. These have included the introduction of more purified oil and surfactant components, and with time the virulent Mycobacterium tuberculosis was replaced with non-virulent tubercle bacilli (M. tuberculosis H37Ra) or with M. butyricum. Nevertheless, it remains the case that the two most powerful immunostimulants known, FCA and BCG, are both based on Mycobacterium spp. In the next section we explore how BCG has grown to replace M. tuberculosis as the mycobacterial component.
How BCG went from a stand-alone vaccine to become a general immune booster
How did BCG come into the adjuvant picture? Although reviews generally trace its role to Pearl’s observation that in tuberculous individuals there was a reduction in the incidence of cancer [31], the antibody boosting seen in guinea pigs [30] was undoubtedly influential. The active use of BCG in oncology rather than a ‘form of tuberculin’ began in 1959, first based on animal models (see earlier), but soon extended to bladder cancer, leukemia, lung cancer, and melanoma [34].
In addition to cancers, successful use of BCG has been reported for the treatment of warts caused by human papillomaviruses [45, 46], infections by respiratory syncitial virus [47], clinical cases of herpesvirus infection [48, 49] (although this has been queried [50]), and recurrent pneumonia in the elderly patients [7]. BCG inoculation has been observed to confer protection against unrelated infectious agents including viruses [51, 52], malaria [53], and respiratory infections [54, 55]. This has prompted growing appreciation that vaccines, notably BCG, can induce protective non-specific ‘trained’ immunity against tumors, viruses, and bacteria [56–60].
Trained immunity: a brief primer
Innate immunity describes the ability of the body to resist microbial infection independently of antigen-specific immunoglobulins and cell-mediated immunity. Innate defenses include diverse antimicrobial proteins, interferons, NO-mediated pathogen toxicity, extracellular traps (NETosis), and autophagy of infected cells, among others. ‘Trained immunity’ is a specific branch of innate immunity in which the immune system is primed to respond more aggressively to non-specific infections. As developed by key protagonists such as Mihai Netea, Peter Aaby, and others, exposure to specific microbial agents such as BCG, and/or the molecules they release, provokes changes in the immune system that can last for months to years or more (reviewed in [13, 62]).
Epigenetic reprogramming of monocytes and macrophages (effects on immune progenitor cells in bone marrow are discussed below) is central to trained immunity; key changes involve alterations in cellular energy-generating pathways and potentiation of immune signaling and defense pathways. Transcriptomic studies in monocytes from individuals inoculated with BCG have revealed long-lasting (more than 3 months) changes in the expression of cytokine (IL1B, IL6, TNF) and chemokine (CXCL9, CCL2, CCL3, CCL4) genes, among many others [63, 64], that are inferred to allow the body to react more swiftly to new infections. In normal human volunteers given BCG before an attenuated yellow fever virus vaccine strain, the key correlate of the rise in antiviral titers was IL-1β [52], whereas heightened IFN-γ production was implicated in BCG-induced protection against SARS-CoV-2 [65].
BCG AT WORK: SITE OF ADMINISTRATION, TIMING, AND MODE OF ENTRY
BCG has been administered orally, by injection, by prick in the skin, intravenously, by aerosol, into the urinary bladder, rectally, directly into tumors, and probably in other ways as well. The difference in dosage has sometimes been more than a thousand-fold [3, 67].
As an anti-TB vaccine, BCG was originally administered orally to infants. This approach described by Calmette emerged from their observations: ‘As early as 1901 certain experiments on roebucks and young bovines had shown Guérin and myself that the absorption of virulent bacilli by the mucous membranes of the digestive tract plays a predominant part in tuberculous infection of the lungs, just as it does in pulmonary glanders in horses. In studying this mode of infection we tried to imitate as closely as possible the condition of natural infection. The animals absorbed carefully prepared suspensions of bacilli, mixed with their food, or by means of an esophageal tube’ [14].
Thus, BCG in its early stages was introduced to infants orally. This method was generally followed until the disastrous Lübeck disaster when, in the late 1920 s, 72 infants died following a mistake when they were given orally BCG contaminated with a virulent TB strain [68]. Following this vaccination debacle, most nations abandoned oral administration and developed various intradermal (ID) injections. Aside from local lesions at the inoculated site, which can be successfully treated, few adverse effects have been reported, although immunodeficient and HIV-infected children can be at risk of disseminated infection [69].
How mode of entry affects immune output
As noted, oral BCG vaccine was generally replaced by injected forms, although Brazil was an exception [66, 70]. Long-term follow-ups of 50 years of percutaneous administration revealed that newborn infants retained at least a partial immune memory of their TB resistance [71, 72]. To understand the action of BCG when given orally (PO) versus ID injection. Hoft et al. examined differences in mucosal and systemic responses [67]. ID BCG administration produced a stronger systemic Th1 and IFN-γ response than PO BCG. By contrast, PO BCG elicited stronger mucosal TB-specific secretory IgA and bronchoalveolar lavage T cells. In their studies, CD4 + T cells from PO or ID vaccinees that were then stimulated with BCG-infected dendritic cells gave distinct gene expression patterns. Nevertheless, both PO and ID administration are associated with immunopotentiation.
Do mycobacterial infections persist life-long?
The traditional view is that, once infected with M. tuberculosis, the infection lasts a lifetime. The Centers for Disease Control currently state ‘Many people who have latent TB infection never develop TB disease. In these people, the TB bacteria remain inactive for a lifetime without causing disease’ (https://www.cdc.gov/tb/topic/basics/tbinfectiondisease.htm). M. tuberculosis infection of the lung is associated with the formation of persistent fibrotic and often calcified lesions (known as Ghon and Ranke complexes) containing viable bacteria (e.g., [73]) that may evade the immune system by this means, although viable M. tuberculosis can be found in otherwise healthy lung tissue. However, the Tuberculosis Network (TBnet) stated in 2009 that it remains unclear whether latency, as assessed by tuberculin skin test and interferon release assays, depends on the presence of living mycobacteria [74]. Behr et al. argued that most people with ‘latent TB’ are no longer infected, and that immunoreactivity can outlast infection by many years [75]. In detailed analysis of lifetime curves, Emery et al. argued that 24% of individuals self-clear over 10 years following infection, and 73% over a lifetime [76]. Nevertheless, these authors point out that the analysis addresses lung infection in particular, and they raise the issue of whether M. tuberculosis might persist in other tissue reservoirs. Of note, the calculations suggest that 26% of individuals do not clear the bacterium, even over a lifetime. Moreover, the majority of individuals remain immunoreactive despite evidence of clearance, suggesting that some mycobacteria-induced innate immunity is likely to be maintained for decades if not longer.
A similar debate exists for BCG. In studies on young mice (8–10 weeks of age), titers of M. tuberculosis and M. avium administered by nebulizer or IV were found to rise in lung, spleen, and thymus for up to 25 weeks. BCG continued to grow in the thymus after a 2-week lag period, whereas colony numbers fell in the lungs and liver, and in the spleen were stable [77]. Thus, when BCG is given early (at least in a mouse), it can maintain its presence and persist as a living organism. In adult rabbits (∼12 weeks) intracisternal inoculation of BCG was followed by rapid dissemination in brain, liver, spleen, and lung by day 8 that persisted to day 21 [78]. In a non-human primate (Macaca mulatta) immunized (IV) with BCG, it appears that live bacteria could be cultured from 4 of 10 animals 9 months after BCG treatment [79]. In human, disseminated BCG infection was reported in a HIV-infected patient 30 years after BCG vaccination [80], and there are multiple reports of BCG persistence not only in immunocompromised patients (e.g., [81]) but also in healthy subjects. In one 62-year-old Japanese woman, > 50 years after ID inoculation, four lesions reappeared on the arm where she was injected, and another lesion on the other arm. BCG was cultured and identified [82].
Although live BCG may persist in some individuals over a lifetime, this remains uncertain. Nevertheless, BCG revaccination offers no apparent improvement in protection against TB [83, 84], although a more recent study was indicative of a small benefit [85], suggesting that immunity once present is long-lasting.
Tissue tropism
Despite wide dissemination, there appears to be some tropism for lymphoid tissue. In mice inoculated orally, 48 h later BCG was predominantly associated with lymphoid tissues in the alimentary tract [86]. In mice inoculated IV with BCG, the microbe was detected in bone marrow where it persisted for 7 months or longer [87]. However, when injected ID it did not appear to proliferate in the bone marrow [87], although ID administration did lead to long-term transcriptomic changes therein [88]. Moreover, in a bladder cancer patient given intravesicular BCG, infection of the bone marrow was reported 2 years after treatment [89]. Of note, the causative agent of TB, M. tuberculosis, can sometimes cause overt infections of the bone marrow, but such cases are generally rare [90]. In summary, multiple studies indicate that BCG may persist in the body for lengthy periods of time, particularly in lymphoid tissues. Here we include the brain given that the CNS controls the immune system. Indeed, mycobacteria have developed mechanisms to cross the blood–brain and/or blood/CSF barriers (e.g., [91, 92]). Neurons are targets for M. tuberculosis infection [93] and BCG is reported to ‘hide’ in the brain where it escapes immune recognition [94]. Long-term persistence of BCG in the CNS and other tissues warrants investigation. However, whether mycobacteria are ultimately cleared in a large proportion of individuals (as suggested for M. tuberculosis, above) remains to be determined.
Does BCG manipulate the immune stem cell niche?
In mice, BCG vaccination is associated with heightened responsiveness to mycobacteria, accompanied by epigenetic/transcriptional changes in bone marrow progenitors and cells derived from them [87]. Another potent immunogen, β-glucan, also induced expansion of bone marrow immune cell progenitors [95]. Comparable results were obtained in humans following inoculation with BCG, where persistent transcriptional changes in bone marrow progenitors were observed, suggesting that BCG somehow ‘rewired’ the bone marrow niche [88]. Does this require persistence of the live bacterium (see above)? In the study by Kaufmann et al. [87] the increased immune responsiveness persisted despite aggressive antibiotic therapy. However, there are caveats, because bacteria (particularly intracellular bacteria) in niche sites can be refractory to systemic antibiotics, and complete removal of BCG was not demonstrated.
Gene-level analyses specifically of bone marrow have highlighted two transcription factors, hepatocyte nuclear factors (HNFs) 1α and 1β, in this reprogramming [88], but the signals that activate their activity and expression remain unknown. Of note, BCG was not detected in the progenitor cells themselves [87]; however, other studies indicate that mycobacteria may enter and persist in other cells in the bone marrow. Local activation of progenitor/stem cells by diffusible signals from BCG (see below) within the bone marrow could contribute to persistent immune activation. It would certainly be of interest to investigate, in both animal models and in humans, the outcomes of bone marrow transplantation from BCG-positive versus -negative individuals to BCG-naive individuals to determine whether the beneficial effects of BCG are transplanted along with the bone marrow.
MYCOBACTERIA INDUCE TRAINED INNATE IMMUNITY, BUT SOME EFFECTS CAN BE REPLACED BY CELL-WALL COMPONENTS: MOLECULES AND MECHANISMS
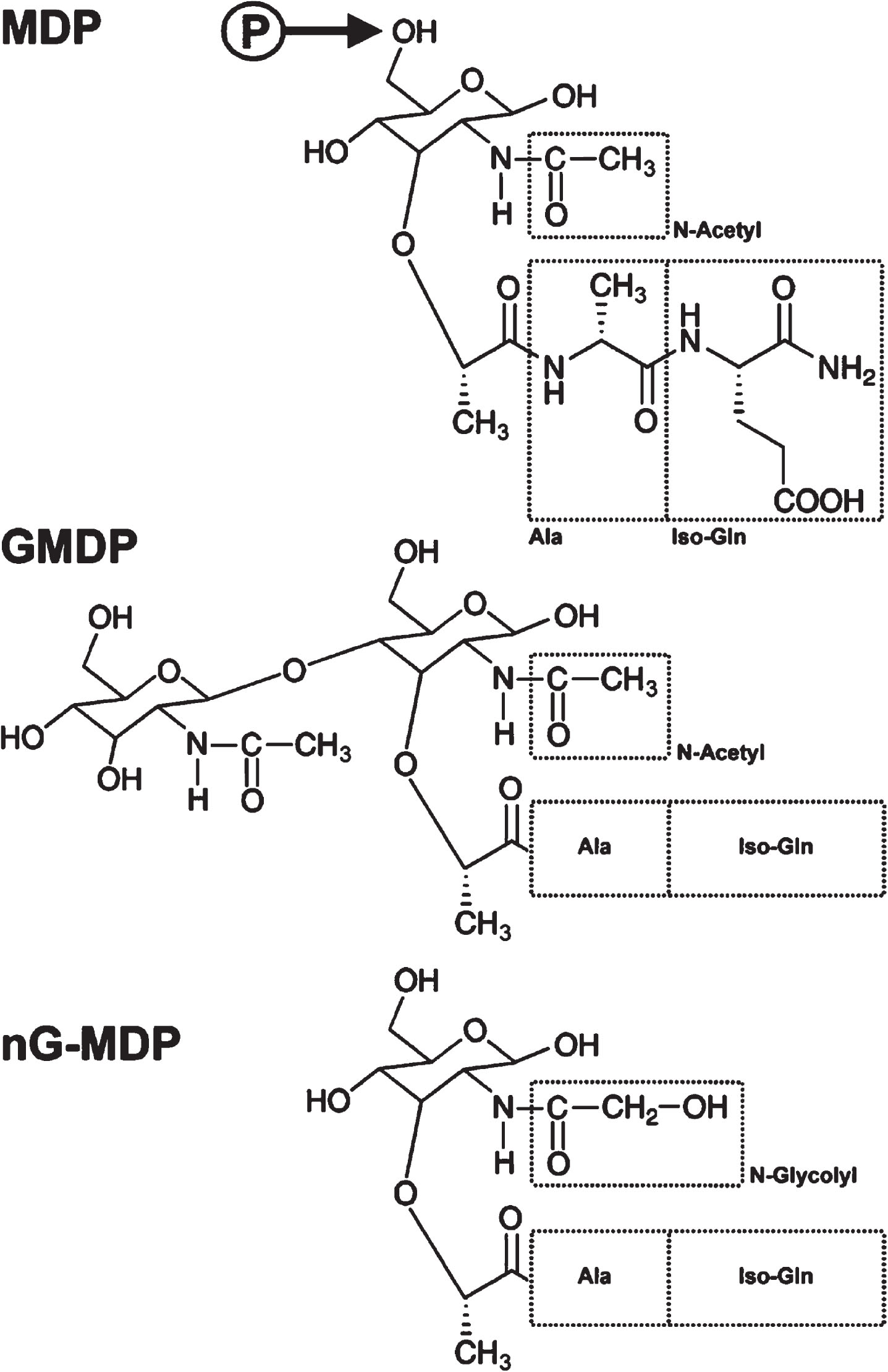
Freund established that the mycobacterial component of FCA could be inactivated without losing activity, but he did not determine what in the killed bacteria was the active agent. Because of the toxicity of FCA, investigators in the 1970 s sought to determine the key molecules (pathogen-associated molecular patterns, PAMPs; Box 3) involved in boosting the immune system, leading to the identification of muramyl dipeptide (MDP; N-acetyl muramic acid attached to a short L-Ala-D-isoGln amino acid chain) and its close relative, acetylglucosaminyl-MDP (GMDP; that contains an additional carbohydrate group) (Fig. 3) as major bacterial cell-wall peptidoglycan components that can replace mycobacterial cells in FCA ([96–98], reviewed in [99]).
Two groups of proteins have been specifically implicated in detecting and responding to microbial molecules. These are the Toll-like receptors (TLRs) and nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs). Evidence has accumulated over the years that these respond to specific molecules such as lipopolysaccharide (TLR4), CpG DNA (TLR9), bacterial lipoprotein (TLR2/1 and 2/6) and flagellin (TLR5), diaminopimelate/DAP peptidoglycan (NOD1), muramyl peptides (NOD2), flagellin (NLRC4), and bacterial/viral RNA/DNA (NLRP3) (reviewed in [160–162]). Many of these ligands have been shown to have adjuvant activity in that they broadly stimulate immune responses, including the production of proinflammatory cytokines (IL-1β, IL-6, TNF) and antimicrobial peptides (reviewed in [161]). Interestingly, there is emerging evidence that NLRP3, a potential alternative target for muramyl peptides (main text) may be a target for the adjuvant action of aluminum salts [163,164, 163,164]. However, Nlrp3 knockout mice were reported to respond normally to alum [165] and the role of NLRP3 in the adjuvant activity of alum remains uncertain [166]. Despite the focus on TLRs and NLRs, these are not the only receptors for PAMPS, exemplified by YB-1 that is targeted by muramyl dipeptides [118, 119].
MDP/GMDP are certainly not the only PAMPs that contribute to immune boosting by mycobacteria. Indeed, MDPs themselves include a wider group of cell wall-derived peptidoglycan molecules extending from MDP and GMDP to N-acetyl-d-glucosaminyl-(β1⟶4)-N-acetylmuramyl-L-alanyl-D-glutamic acid (GM-DPA), among others. The carboxyl functions can also be modified by amidation, methylation, or other changes. Of note, mycobacterial cell wall MDPs including those of BCG are unusual because they contain a proportion of N-glycolyl-muramic acid (nG-MDP) [100–103], a conversion mediated by the mycobacterial enzyme NamH [104], but this is not necessary for growth or virulence (e.g., [105]) and some mycobacteria such as M. leprae [103] lack the enzyme. Moreover, despite the primary focus on MDP, other mycobacterial molecules such as the glycolipid trehalose dimycolate are likely to be involved in the unusual immunopotentiating properties of mycobacteria [106,107, 106,107].
The adjuvant effects of MDPs in protecting against challenge infection with different bacteria (e.g., [108,109, 108,109]) and viruses (e.g., [110–112]) have been confirmed (reviewed in [113]). Because GMDP is considered to be less toxic than MDP [114], many studies have focused on this molecule; however, it remains uncertain whether GMDP and MDP are equally active in vivo, noting that mycobacteria produce a range of muramyl peptides. Indeed, nG-MDP may be particularly effective in immunopotentiation [115].
Two host molecules have been identified as receptor targets for muramyl peptide PAMPs –NOD2 and YB-1 [116–119]. NOD2 (and its relative NOD1 that responds to D-glutamyl-meso-diaminopimelic acid [120]) are generally held to be intracellular receptors that activate innate immunity. MDP is the essential core component, but diverse muramyl peptide derivatives are reported to activate the NOD2 pathway (e.g., [121, 122]). Nevertheless, unmodified muramyl peptides have low affinity for NOD2 [123, 124] and require further modification (phosphorylation by the principally intracellular N-acetylglucosamine kinase, NAGK) before they can bind to NOD2 [125] (Fig. 3). A detailed review of the clinical importance of NOD signaling has recently appeared [126].
In contrast to NOD2, YB-1 is a diffusible multifunctional transcription factor [127, 128] that is present not only in the nucleus but also in the cytosolic and extracellular compartments [129]. In addition, whereas muramyl peptides require phosphorylation of the molecule, YB-1 is targeted by unmodified GMDP in vitro [118, 119]. Blood levels of YB-1 are remarkably elevated in patients with sepsis [130], suggesting that YB-1 upregulation may be a protective response to infection. Indeed, treatment of mice with recombinant YB-1 protein was reported to act as an adjuvant to increase the immune response to an unrelated antigen (Yersinia pestis V antigen) and significantly promoted survival against lethal bacterial infection (Escherichia coli) [131], although this requires confirmation, and is active in a mouse model of AD [132].
The interplay between YB-1 and NOD2 is complex, and YB-1 might represent a sensor for extracellular MDP/GMDP whereas NOD2 detects muramyl peptides released by intracellular microbes. However, although intrinsically intracellular, membrane-bound versions of NOD2 are also reported at the cell surface [133–135] where it may interact with extracellular muramyl dipeptides and/or YB-1. In addition, experiments with NOD2 have majorly employed MDP, whereas YB-1 studies have principally employed GMDP, and it is possible that they differ in their recognition specificity.
Activation of both YB-1 and NOD2 induces NF-κB expression, and with it innate immunity. Although YB-1 can act alone, together with NOD2 is perhaps the more usual interaction, leading to the formation of higher molecular mass complexes containing both YB-1 and NOD2. A major recombinant fragment of NOD2 (NACHT–LRR), stimulated by GMDP, binds to YB-1. There is also subcellular colocalization of NOD2 and YB-1. The authors argue that maximal innate immunity activation by the muramyl peptides is mediated via an interaction between YB-1 and NOD2 [119].
In addition to NOD2 and YB-1, MDP may exert its effects in part by activating another PAMP receptor, NLRP3 (e.g., [136, 137]), although molecular modeling argues that MDP may be unable to activate NLRP3/NALP3 directly, and an interaction with another PAMP receptor such as NOD2 or YB-1 may be necessary for NLRP3 activation [138].
However, why do MDPs and other PAMPs released by the enteric microbiome not serve as a long-term immune booster? One answer may lie in the fact that MDP release and entry into the circulation remain low under normal conditions. By contrast, BCG vaccination by injection leads to a rise in circulating MDP concentrations for up to 3 months, and this correlated with inflammatory markers in the circulation [139]. Nevertheless, there was a wide range of MDP levels in both vaccinated and unvaccinated individuals, and the increase in MDP levels upon vaccination was numerically small [139], challenging the interpretation that increased circulating MDP levels are responsible for the adjuvant effects of BCG. Another possibility is suggested by findings that nG-MDP produced by some mycobacteria has particularly robust activity in activating human NOD2 [115, 122], and this particular molecule may be less well represented in the gut microbiome. In addition, continued production of MDP and its derivatives (and perhaps other molecules) in select Mycobacterium niches such as bone marrow and other immunological tissues could potentially maintain a state of heightened immune responsiveness in the longer term.
CONCLUSIONS
We have reviewed evidence that multiple infectious agents can have broad-spectrum immunopotentiating effects, leading to the development of BCG in particular as a treatment for some cancers and as a prophylactic against diseases of the elderly such as pneumonia. In Part II of this work [9], we address emerging reports that BCG and other vaccines can protect against the development of dementias including AD. Although we focus primarily on BCG, we emphasize that multiple different microbes and molecules can markedly boost immunity, but mycobacteria and BCG in particular do appear to be special in this regard, and the reasons why will require detailed attention in the future.
AUTHOR CONTRIBUTIONS
Charles Leonard Greenblatt (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing – original draft; Writing – review & editing); Richard Lathe (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Software; Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
We thank Janet Janbek, Shoshana Frankenburg, Tom Dow, and Brian Balin for helpful comments on the manuscript. Cure Alzheimer’s Fund is thanked for encouraging C.L.G. to think about how BCG works.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.