
Abstract
Background:
Vitamin D has neuroprotective and immunomodulating functions that may impact glial cell function in the brain. Previously, we reported molecular and behavioral changes caused by deficiency and supplementation of vitamin D in an Alzheimer’s disease (AD) mouse model. Recent studies have highlighted reactive astrocytes as a new therapeutic target for AD treatment. However, the mechanisms underlying the therapeutic effects of vitamin D on the glial cells of AD remain unclear.
Objective:
To investigate the potential association between vitamin D deficiency/supplementation and the pathological progression of AD, including amyloid-β (Aβ) pathology and reactive astrogliosis.
Methods:
Transgenic hemizygous 5XFAD male mice were subjected to different dietary interventions and intraperitoneal vitamin D injections to examine the effects of vitamin D deficiency and supplementation on AD. Brain tissue was then analyzed using immunohistochemistry for Aβ plaques, microglia, and astrocytes, with quantifications performed via ImageJ software.
Results:
Our results demonstrated that vitamin D deficiency exacerbated Aβ plaque formation and increased GABA-positive reactive astrocytes in AD model mice, while vitamin D supplementation ameliorated these effects, leading to a reduction in Aβ plaques and GABA-positive astrocytes.
Conclusions:
Our findings highlight the significant impact of vitamin D status on Aβ pathology and reactive astrogliosis, underscoring its potential role in the prevention and treatment of AD. This study provides the first in vivo evidence of the association between vitamin D and reactive astrogliosis in AD model mice, indicating the potential for targeting vitamin D levels as a novel therapeutic approach for AD.
Keywords
INTRODUCTION
Vitamin D deficiency is a pervasive nutritional issue in contemporary society, characterized by its high prevalence [1]. It may contribute to significant health problems, including weakened immunity, impaired bone health, and an increased risk of chronic diseases [2]. Recent studies have indicated a potential link between vitamin D deficiency and the exacerbation of Alzheimer’s disease (AD) progression [3]. AD is pathologically characterized by increased amyloid-β (Aβ) plaques, neurofibrillary tangles, and neuroinflammation [4]. Insufficient vitamin D levels contribute to the pathogenesis of AD by increasing the accumulation of Aβ load and promoting neuroinflammation, thereby leading to cognitive decline [5]. However, this relationship between vitamin D and AD is controversial, and the exact mechanisms remain unclear [3, 6]; therefore, the findings emphasize the importance of maintaining adequate vitamin D levels as a promising preventive measure against cognitive decline and AD [7].
We demonstrated that vitamin D deficiency could increase the production of Aβ in an AD mouse model, while upregulating genes involved in Aβ synthesis and downregulating genes involved in Aβ degradation [8]. In addition, it has been associated with alterations in microglia- and astrocyte-related genes, which may contribute to neuroinflammation and synaptic dysfunction, potentially exacerbating the progression of AD [8, 9]. Prolonged astrocyte activation can generate a neurotoxic environment and exacerbate the pathological progression of AD [10, 11]. Reactive astrocytes can also cause an inflammatory response in neurodegenerative disorders, and the administration of vitamin D suppresses astrocyte activation [12, 13].
In recent years, there has been growing interest in the role of astrocyte changes in the context of AD, particularly regarding the increase in astrocytic gamma-aminobutyric acid (GABA) releases [14]. Astrocytes exhibit a reactive response surrounding Aβ plaques in AD, characterized by hypertrophy and tonic release of GABA [14–16]. The GABA synthesis in reactive astrocytes is linked to elevated monoamine oxidase B (MAO-B) gene expression, which has been implicated in the pathophysiology of AD [17]. The relationship between astrocytic GABA and MAO-B gene expression may contribute to developing novel therapeutic strategies that target these pathways to mitigate the progression of AD [18].
However, despite growing interest in the role of astrocytic GABA and its potential implications in AD, a direct connection between vitamin D deficiency and reactive astrocytes remains unestablished. Consequently, our study aims to investigate the changes in reactive astrocytes in vitamin D deficiency and supplementation in the AD model mice to examine the causal relationship between vitamin D and glial responses.
METHODS
Animals
Transgenic hemizygous 5XFAD male mice (Jackson Laboratory, Bar Harbor, ME, USA) were used in this study, overexpressing both the human PS1 gene and the human APP gene. All experimental procedures involving animals adhered to the Institutional Animal Care and Use Committee guidelines and were approved by the ethics committee at Gwangju Institute of Science and Technology.
The experimental methodology used here has been described in detail in our earlier publication [8]. The 5XFAD mice were randomly assigned to six groups (n = 6 per group): 1) Early-Control (Early-Con) group received a control diet (RodFeed, DBL, Eumseong, Korea) containing 60% carbohydrate, 20% protein, 4% fat, and 8 IU/kg vitamin D, in accordance with NIH-41 Open Formula. 2) Early-Deficiency (Early-Def) group was given a vitamin D-deficient diet (89123, ENVIGO, Indianapolis, IA, USA) devoid of vitamin D (59% carbohydrate, 16% protein, 10% fat, and 0 IU/kg vitamin D) starting at 1 month of age. 3) Late-Control (Late-Con) group consistently received the control diet. 4) Late-Deficiency (Late-Def) group consumed a vitamin D-deficient diet starting at 3 months of age. 5) Vitamin D-supplemented (Inj) group was fed the control diet and received weekly intraperitoneal injections of 410 ng/g vitamin D3 (PHR1237, Merck, Rahway, NJ, USA) in 1% ethanol saline solution beginning at 4.5 months old. 6) Vehicle-injected (Veh) group was fed the control diet and intraperitoneally injected with saline containing 1% ethanol (injection volume: individual body weight (g)/100 mL saline) from 4.5 months old. All mice were housed under constant temperature and humidity conditions with unrestricted access to food and water.
Tissue collection
The vitamin D-deficient and control groups (Early-Def, Early-Con, Late-Def, and Late-Con) were sacrificed at ∼3.5 months after the administration of a vitamin D-deficient diet, while the vitamin D-supplemented and vehicle-injected groups (Inj and Veh) were sacrificed at 2 months after starting the injection. Mice were deeply anesthetized using isoflurane and transcardially perfused with 1X phosphate-buffered saline (PBS). Two hemispheres of the brain were dissected following brain harvesting. Before use, the left hemisphere was stored in a 4% paraformaldehyde (PFA) solution in PBS at 4°C.
Immunohistochemistry
The left hemisphere of the brain was fixed in a 4% PFA solution in PBS and submerged in a 30% sucrose solution in PBS at 4°C until it sank. Brain coronal sections of 40μm thickness were obtained using a cryotome (Leica Biosystems, Buffalo Grove, IL, USA). Sections were washed three times with PBS containing 1% Triton X-100 (PBST) for 10 min at room temperature with shaking at 100 rpm. Blocking was performed for 2 h at room temperature in a solution containing 3% normal donkey serum (D9633, Sigma Aldrich, St. Louis, MO, USA) with shaking at 100 rpm. Microglia were visualized using primary antibody rabbit anti-Iba1 (1 : 1000 dilution with 3% normal donkey serum, 019-19741, Wako, Japan) and secondary antibody Alexa FluorTM 594 donkey anti-rabbit IgG (1 : 500 dilution with 3% normal donkey serum, A21207, Invitrogen, Waltham, MA, USA). Astrocytes were stained using primary antibodies rabbit anti-GFAP (1 : 1000 dilution with 3% normal donkey serum, 019–19741, Wako, Japan) and anti-GABA antibody (1 : 200 dilution with 3% normal donkey serum, AB175, Merck, Rahway, NJ, USA), and secondary antibodies Alexa FluorTM 555 donkey anti-rabbit IgG (1 : 500 dilution with 3% normal donkey serum, A31572, Invitrogen, Waltham, MA, USA) and Alexa FluorTM 647 AffiniPure donkey anti-guinea pig IgG (1 : 500 dilution with 3% normal donkey serum, 706-605-148, Jackson Immuno Research Labs, West Grove, PA, USA). Aβ plaques were stained using 1 mM Thioflavin S (T1892, Sigma Aldrich, St Louis, MO, USA) in 50% ethanol. Finally, the sections were mounted on saline-coated slide glass (5116-20F, Muto Pure Chemicals, Tokyo, Japan).
Quantification of Aβ plaques, microglia, and astrocyte
Histological images from a coronal section of each brain region, including cortex (at AP +1.3 mm from bregma), hippocampus (at AP –1.6 mm from bregma), and brainstem (at AP –6.3 mm from bregma) were collected by a research slide scanner (VS200, Olympus, Tokyo, Japan), and Z-stack of 30μm in 5μm steps were captured with a 20x objective lens in extended focal imaging (EFI) and deblur modes. Regions of interest for quantification were identified according to Paxinos and Franklin’s The Mouse Brain [19] (Supplementary Figs. 1 and 2). Quantification of Aβ plaques, microglia, and astrocytes in each brain section was quantified using ‘Analyze Particle’ tool installed in the Image J software (NIH, Bethesda, MD, USA). Also, the GABA-positive reactive astrocytes were quantifying the co-labeling of GFAP and GABA, using the ‘Colocalization Threshold’ plugin in Image J software.
Quantification and statistical analysis
GraphPad Prism version 9 (GraphPad Software, La Jolla, CA, USA) was used to analyze all data and presented as the means±SEM (*p < 0.05). For simple comparisons, the Student’s t-test and Mann-Whitney test were used.
RESULTS
No substantial changes in Aβ plaques and microglia by vitamin D deficiency or supplementation
We investigated the effects of vitamin D deficiency on AD pathology by quantifying Aβ plaques and microglia in the brain, using thioflavin S staining and immunohistochemistry against Iba1 (Figs. 1 and 2). The Early-Def group exhibited a significantly higher number of Aβ plaques in the hippocampus than the Early-Con group (p < 0.05; Fig. 1D-F). In addition, vitamin D deficiency groups showed lower mRNA levels of the Interleukin-10 (IL-10) gene, which is an inflammatory cytokine (Supplementary Fig. 3). However, no significant differences between the Late-Def and Late-Con groups were found between Aβ plaques and microglia (Fig. 1G-I).
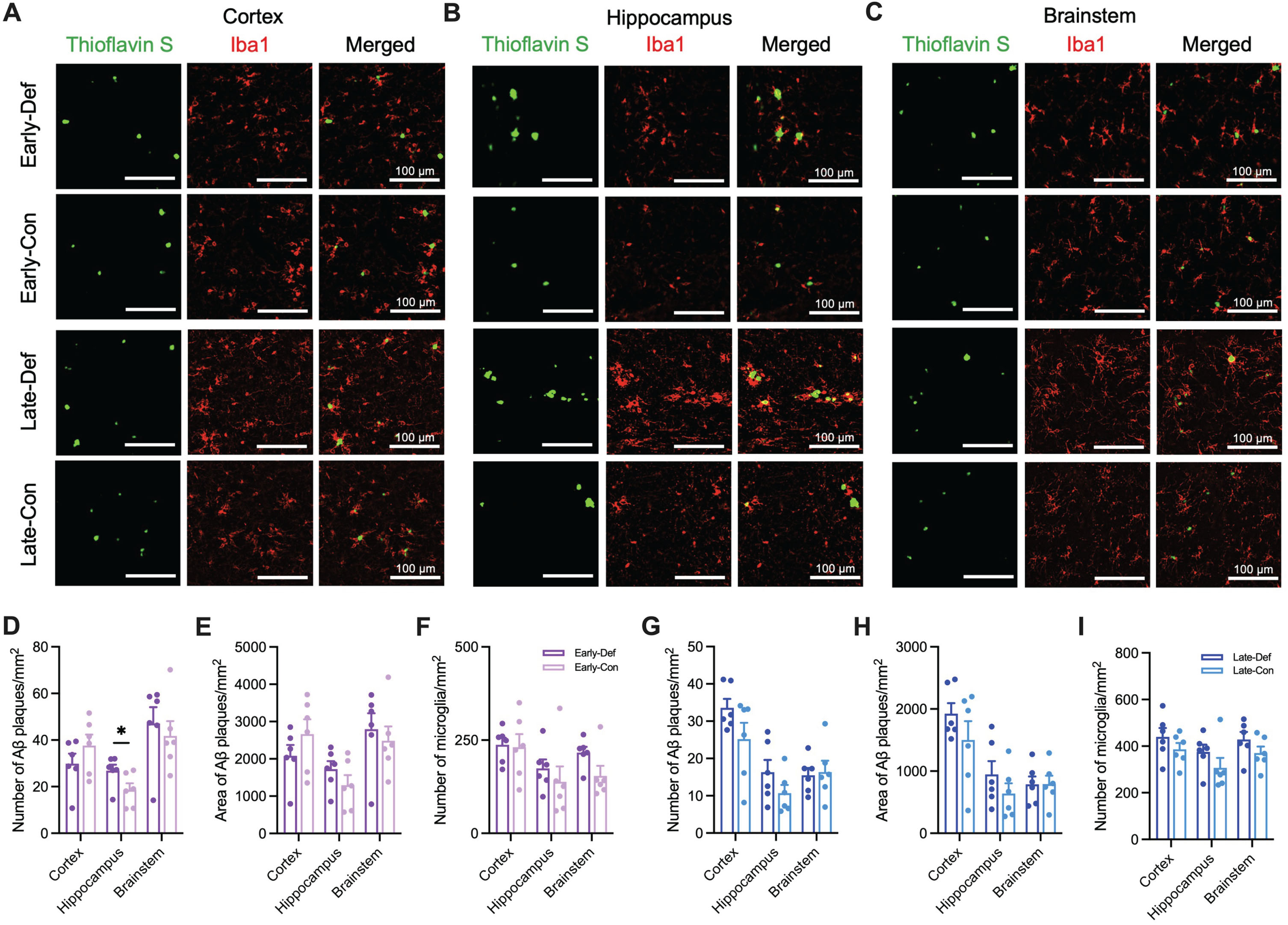
Vitamin D deficiency increased Aβ load and microglia in the brain. A) Representative image of cortical Aβ plaques (Thioflavin S; Green) and microglia (Iba1; Red) in the primary motor cortex (Scale bar, 100μm) of Early-Def, Early-Con, Late-Def, and Late-Con groups. B) Representative images of Aβ plaques and microglia in the hippocampus of four groups. C) Representative images of Aβ plaques and microglia in the nucleus tractus solitarius of four groups. D) The number of Aβ plaques per unit area in the cortex, hippocampus, and brainstem of Early-Def and Early-Con groups. E) Quantification of the area of Aβ plaques per unit area in the cortex, hippocampus, and brainstem of Early-Def and Early-Con groups. F) Quantification of the number of microglia (Iba1 + cells) per unit area in the cortex, hippocampus, and brainstem of Early-Def and Early-Con groups. G) The number of Aβ plaques per unit area in the cortex, hippocampus, and brainstem of Late-Def and Late-Con groups. H) Quantification of the area of Aβ plaques per unit area in the cortex, hippocampus, and brainstem of Late-Def and Late-Con groups. I) Quantification of the number of microglia (Iba1 + cells) per unit area in the cortex, hippocampus, and brainstem of Late-Def and Late-Con groups. Data are presented as mean±SEM. *p < 0.05 by Student’s t-test or Mann-Whitney test. Aβ, amyloid-β; Iba1, ionized calcium-binding adaptor molecule 1.
Similarly, in the vitamin D-supplemented group, we observed a significantly reduced number and area of Aβ plaques in the cortex compared to the vehicle group, reaching statistical significance (p < 0.05; Fig. 2D-F). However, the number of microglia was not significantly different between Veh and Inj groups (Fig. 2G-I).
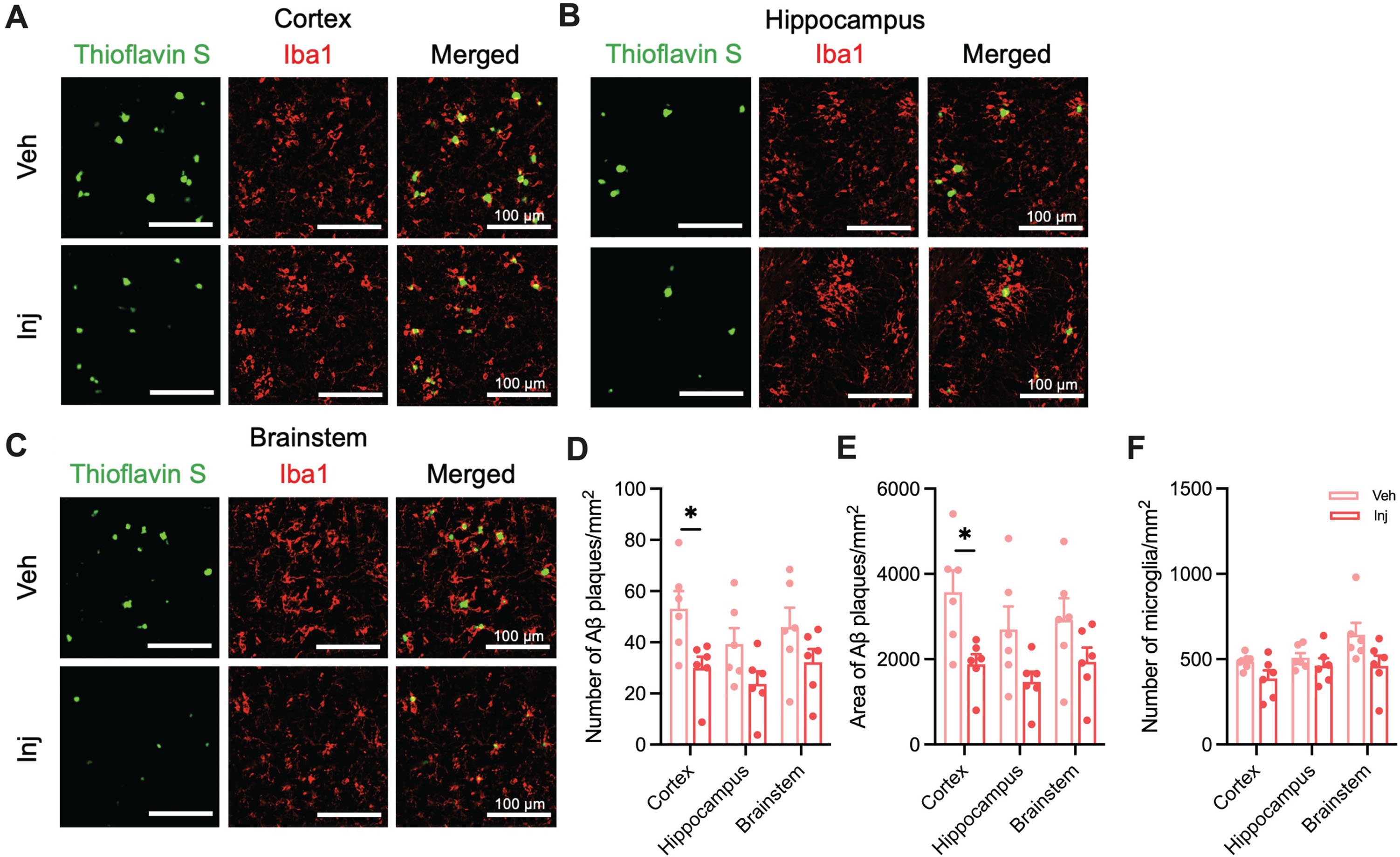
Vitamin D supplementation reduced the number and area of Aβ plaques in the brain. A) Representative image of cortical Aβ plaques (Thioflavin S; Green) and microglia (Iba1; Red) in the primary motor cortex (Scale bar, 100μm) of Veh and Inj groups. B) Representative images of Aβ plaques and microglia in the hippocampus of two groups. C) Representative images of Aβ plaques and microglia in the nucleus tractus solitarius of two groups. D) The number of Aβ plaques per unit area in the cortex, hippocampus, and brainstem. E) Quantification of the area of Aβ plaques per unit area in the cortex, hippocampus, and brainstem. F) Quantification of the number of microglia (Iba1 + cells) per unit area in the cortex, hippocampus, and brainstem. Data are presented as mean±SEM. *p < 0.05 by Student’s t-test or Mann-Whitney test. Aβ, amyloid-β; Iba1, ionized calcium-binding adaptor molecule 1; Veh, vehicle-injected 5XFAD mice (n = 6); Inj, vitamin D-injected 5XFAD mice (n = 6).
Decreased Aβ plaques in vitamin D supplementation
To explore the therapeutic effect of vitamin D on AD pathology, we administered intraperitoneal injections of vitamin D to induce a vitamin D-supplemented state in the AD mouse model using thioflavin S staining and Immunohistochemistry against Iba1 (Fig. 2A-C). The Inj group showed significantly reduced numbers and area of Aβ plaques in the cortex compared to the Veh group. The number of Aβ plaques in the Inj group displayed a decreasing trend in comparison to the Veh group, reaching statistical significance in the cortex (p < 0.05; Fig. 2D) and a trend towards significance in the hippocampus (p = 0.0806; Fig. 2D). The area of Aβ plaques in the Inj group also showed a significant decrease in the cortex (p < 0.05; Fig. 2E) and a decreased trend in the hippocampus (p = 0.0619; Fig. 2E). In addition, trend level differences were observed in the number of microglia between the Veh and Inj groups in the cortex and brainstem, though these did not reach statistical significance (p = 0.0791, cortex; p = 0.0649, hippocampus; Fig. 2F).
Increased GABA-positive reactive astrocytes in vitamin D deficiency
We evaluated the potential impact of vitamin D deficiency on reactive astrogliosis in the brains of AD model mice by examining the glial response through immunohistochemistry for GFAP for astrocytes staining and GABA staining (Fig. 3A-C). Although no significant differences in astrocyte count were found between the two groups (Fig. 3D), the Early-Def group showed a significant rise in GABA-positive astrocytes in the hippocampus and brainstem compared to the Early-Con group (p < 0.05; Fig. 3E).
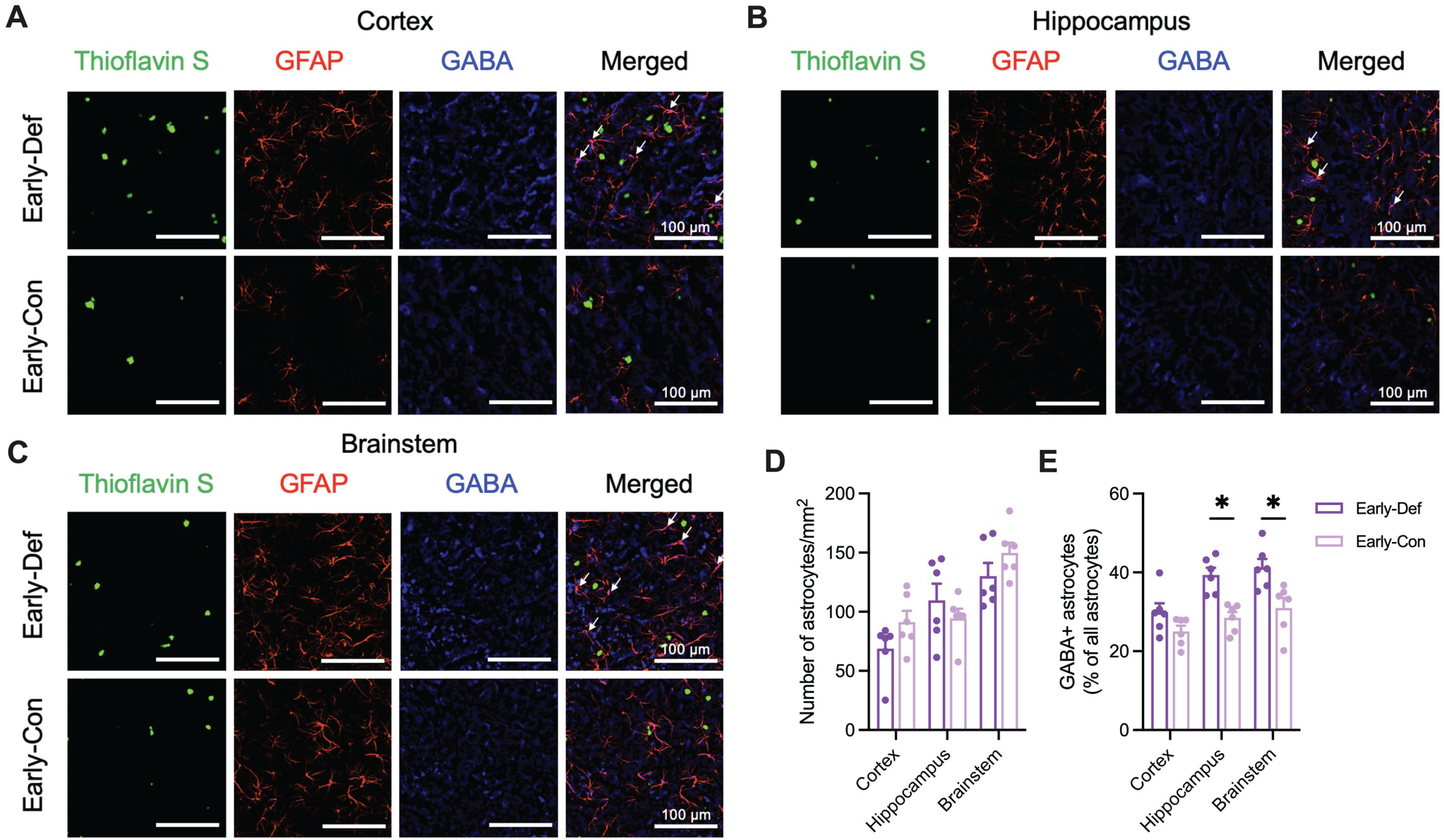
Vitamin D deficiency showed increased GABA-positive reactive astrocytes in the early phase of 5XFAD mice. A) Representative image of cortical Aβ plaques (Thioflavin S; Green), astrocytes (GFAP; Red), and GABA (Blue) in the primary motor cortex (Scale bar, 100μm) of Early-Def and Early-Def groups. There is colocalization between GFAP and GABA (white arrows). B) Representative images of Aβ plaques, astrocytes, and GABA in the hippocampus of two groups. There is colocalization between GFAP and GABA (white arrows). C) Representative images of Aβ plaques, astrocytes, and GABA in the nucleus tractus solitarius of two groups. There is colocalization between GFAP and GABA (white arrows). D) The number of astrocytes (GFAP + cells) per unit area in the cortex, hippocampus, and brainstem. E) Colocalization of GABA-positive astrocytes in the cortex, hippocampus, and brainstem. Data are presented as mean±SEM. *p < 0.05 by Student’s t-test or Mann-Whitney test. Aβ, amyloid-β; GFAP, glial fibrillary acidic protein; GABA, gamma-aminobutyric acid.
Representative images indicated that the Late-Def group also exhibited an elevated number of GABA-positive astrocytes in the hippocampus compared to the Late-Con group (Fig. 4A-C). While no significant differences in astrocyte count were observed between the two groups (Fig. 4D), the Late-Def group displayed a significant increase in GABA-positive astrocytes in the hippocampus compared to the Late-Con group (p < 0.05; Fig. 3E).
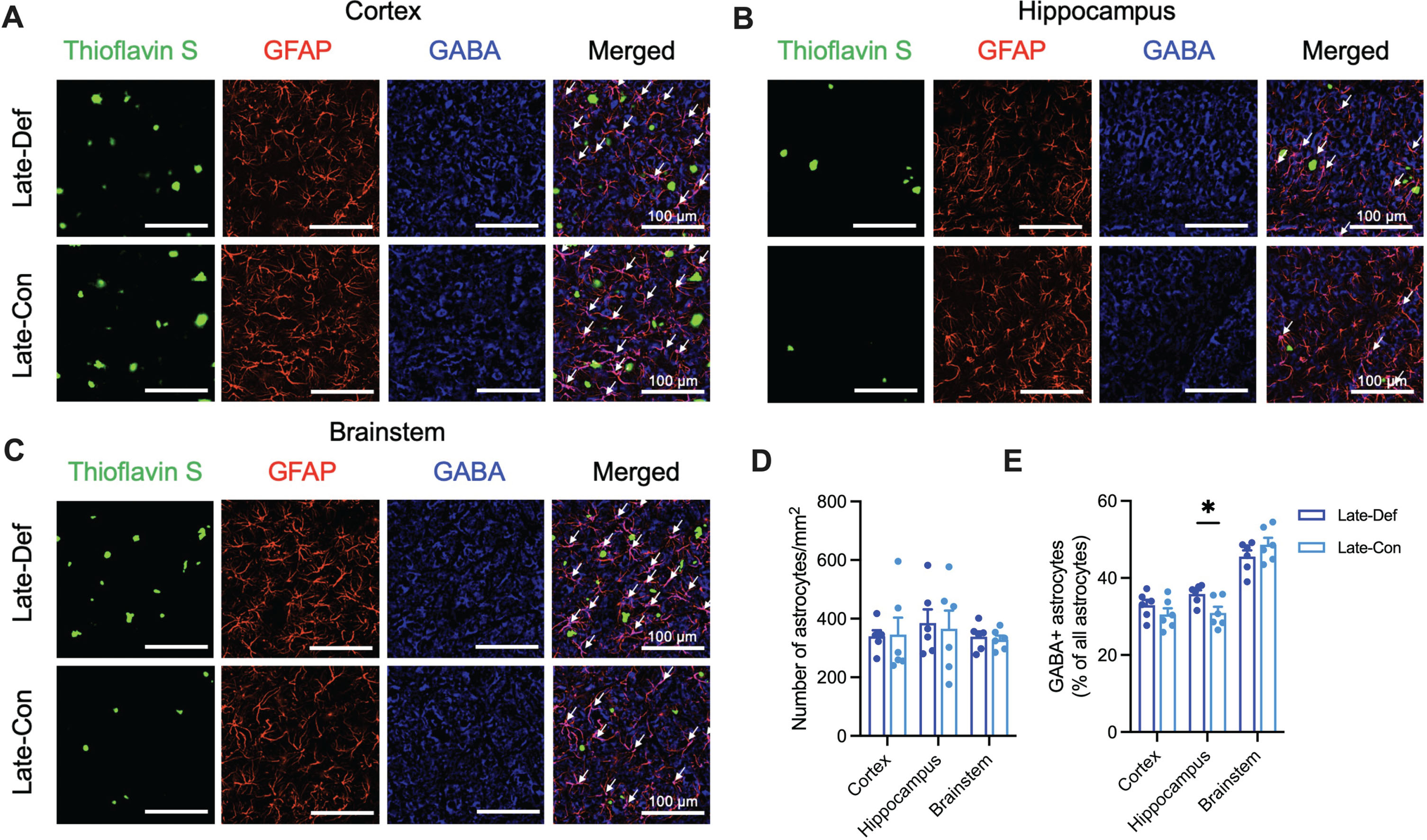
Vitamin D deficiency showed increased GABA-positive reactive astrocytes in the late phase of 5XFAD mice. A) Representative image of cortical Aβ plaques (Thioflavin S; Green), astrocytes (GFAP; Red), and GABA (Blue) in the primary motor cortex (Scale bar, 100μm) of Late-Def and Late-Def groups. There is colocalization between GFAP and GABA (white arrows). B) Representative images of Aβ plaques, astrocytes, and GABA in the hippocampus of two groups. There is colocalization between GFAP and GABA (white arrows). C) Representative images of Aβ plaques, astrocytes, and GABA in the nucleus tractus solitarius of two groups. There is colocalization between GFAP and GABA (white arrows). D) The number of astrocytes (GFAP + cells) per unit area in the cortex, hippocampus, and brainstem. E) Colocalization of GABA-positive astrocytes in the cortex, hippocampus, and brainstem. Data are presented as mean±SEM. *p < 0.05 by Student’s t-test or Mann-Whitney test. Aβ, amyloid-β; GFAP, glial fibrillary acidic protein; GABA, gamma-aminobutyric acid.
Reduced GABA-positive reactive astrocytes in vitamin D supplementation
Representative images revealed that the vitamin D-injected group had lower GABA-positive astrocytes in hippocampus, and brainstem compared to vehicle-injected group (Fig. 5A-C). The vitamin D-injected groups had significantly decreased GABA-positive astrocytes in the hippocampus and brainstem compared to the vehicle-injected group (p < 0.05; Fig. 5D, E).
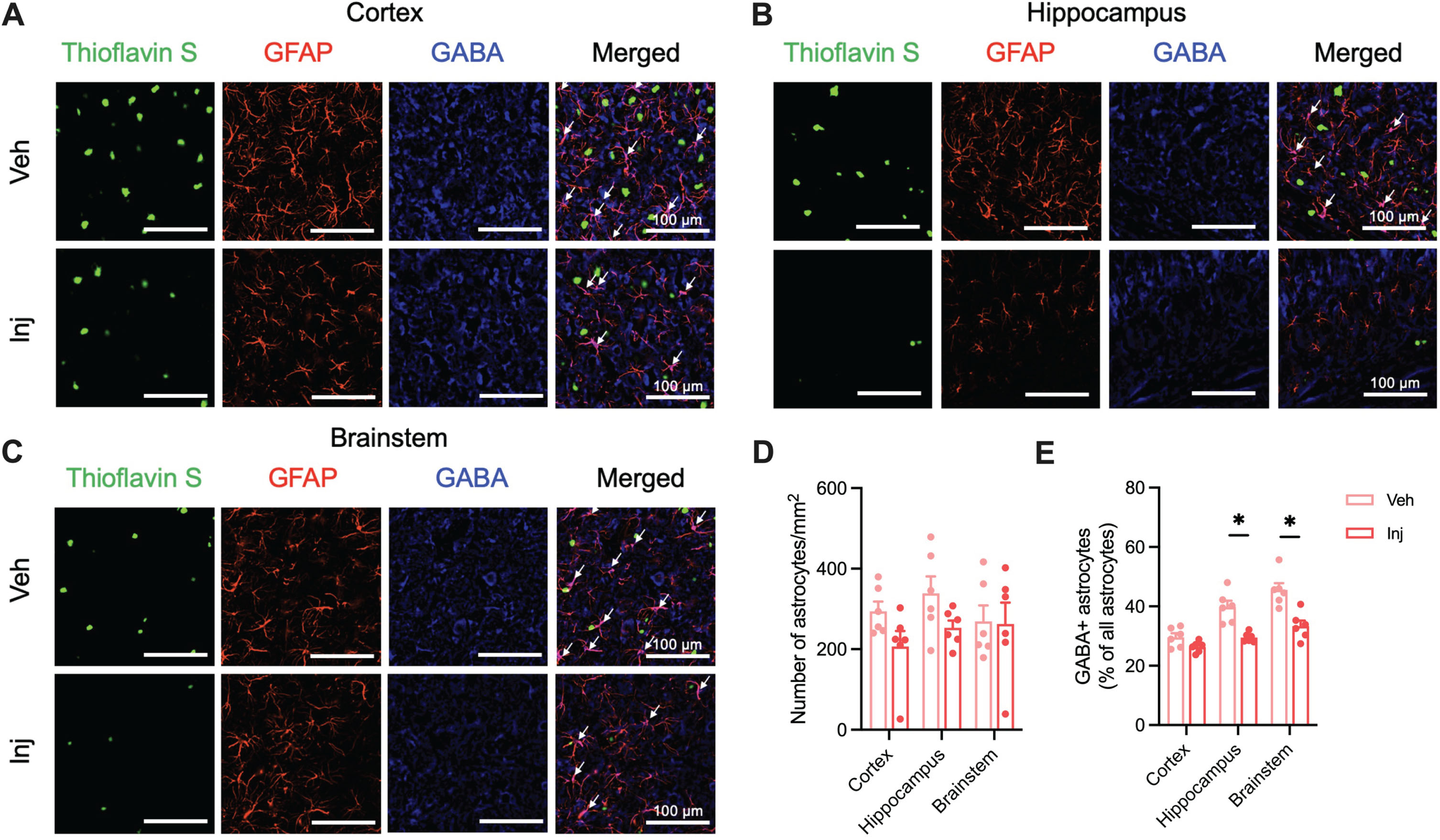
Vitamin D supplementation showed decreased GABA-positive reactive astrocytes in the AD model mice. A) Representative image of cortical Aβ plaques (Thioflavin S; Green), astrocytes (GFAP; Red), and GABA (Blue) in the primary motor cortex (Scale bar, 100μm) of Veh and Inj groups. There is colocalization between GFAP and GABA (white arrows). B) Representative images of Aβ plaques, astrocytes, and GABA in the hippocampus of two groups. There is colocalization between GFAP and GABA (white arrows). C) Representative images of Aβ plaques, astrocytes, and GABA in the nucleus tractus solitarius of two groups. There is colocalization between GFAP and GABA (white arrows). D) The number of astrocytes (GFAP + cells) per unit area in the cortex, hippocampus, and brainstem. E) Colocalization of GABA-positive astrocytes in the cortex, hippocampus, and brainstem. Data are presented as mean±SEM. *p < 0.05 by Student’s t-test or Mann-Whitney test. Aβ, amyloid-β; GFAP, glial fibrillary acidic protein; GABA, gamma-aminobutyric acid; Veh, vehicle-injected 5XFAD mice (n = 6); Inj, vitamin D-injected 5XFAD mice (n = 6).
DISCUSSION
Our study aimed to investigate the potential relationship between vitamin D deficiency/supplementation and the progression of AD pathology. We discovered that vitamin D deficiency was associated with increased GABA-positive reactive astrocytes, while vitamin D supplementation led to a decrease in GABA-positive astrocytes. These findings suggest the potential role of vitamin D on reactive astrogliosis in AD pathology. To our knowledge, this is the first in vivo report demonstrating the association between vitamin D and reactive astrogliosis in the brains of AD model mice, indicating the potential for targeting vitamin D levels as a novel therapeutic approach for AD.
Recent epidemiological reports have highlighted a potential association between vitamin D deficiency and AD [5, 18–22]. Moreover, vitamin D hypovitaminosis is relevant for increased risk of disease and development of cognitive impairment and AD and is associated with decreased volumes in the hippocampal regions in elderly patients with cognitive decline [3, 23–25]. These findings propose that insufficient vitamin D levels may contribute to the progression of AD, with possible implications for prevention and treatment strategies [26]. However, the effects of vitamin D on the pathological process of AD were controversial [6, 27–29]. We employed an AD mouse model to examine the impact of vitamin D on AD and determine whether vitamin D supplementation may help mitigate the pathological process of AD.
As well as its neuroprotective and anti-inflammatory properties, vitamin D plays an important role in the development of the brain [30]. Vitamin D deficiency has been found to aggravate the pathological process of AD [31]. Insufficient vitamin D levels can exacerbate Aβ accumulation and promote neuroinflammation, thereby contributing to cognitive decline and the progression of AD [32]. Our previous study suggests that a vitamin D-deficient state showed more brain Aβ load, caused by increasing Aβ production and decreasing Aβ degradation (Supplementary Fig. 4) [8]. We also found increased Aβ plaques in the hippocampus of vitamin D-deficient 5XFAD mice. The hippocampus has an important role in visuospatial memory [33], and aggravated Aβ pathology by vitamin D deficiency results in memory impairment in 5XFAD mice (Supplementary Fig. 5) [8].
Vitamin D has a role in Aβ clearance in the brain of the elderly [34], induces a non-amyloidogenic process, and upregulates Aβ degrading enzymes [35]. Patients with daily oral vitamin D supplementation showed decreased biomarkers related to Aβ and improved cognitive function [36]. Our result also demonstrated reduced Aβ plaques in the cortex of vitamin D-injected 5XFAD mice. Vitamin D could protect against Aβ-driven cytotoxicity and oxidative stress by reducing Aβ pathology in the brain [37, 38]. Our findings suggest a potential modulation of Aβ plaques in vitamin D deficiency and supplementation. However, these observations warrant further validation.
In addition to Aβ pathology, AD is closely related to significant neuroinflammation and tau pathology (Supplementary Table 1), and 5XFAD mice are characterized by predominant inflammatory and immune responses [39, 40]. Vitamin D has been shown to be associated with glial cells, which play a crucial role in maintaining brain homeostasis and modulating neuronal function [30]. Recent studies have revealed that vitamin D may have a significant impact on glial cell activation and function, particularly in the context of neuroinflammation and neurodegenerative disorders such as AD [41]. The interaction between vitamin D and glial cells may provide insights into the molecular mechanisms underlying the neuroprotective effects of vitamin D, proposing a new therapeutic method for treating AD and other neurodegenerative disorders [42].
We conducted fluorescence staining of brain tissue to examine the effects of vitamin D deficiency/supplementation on glial cells. Although there were no changes in the number of microglia, vitamin D deficiency induced more GABA-positive reactive astrocytes in the hippocampus and brainstem, while vitamin D supplementation reduced GABA-positive reactive astrocytes in the hippocampus and brainstem. There are two possible interpretations for the decreased reactiveness: decreased Aβ led to decreased reactive astrogliosis; and vitamin D directly reduced the GABA synthesis by an unknown process. Interestingly, after vitamin D supplementation, the number of Aβ plaques remained comparable in the hippocampus and brainstem. In addition, our previous report showed decreased mRNA levels of the MAO-B gene after vitamin D supplementation [8]. These findings support the second interpretation that increased levels of vitamin D directly changed GABA synthesis in reactive astrocytes. The molecular mechanism of vitamin D’s modifying effects on reactive astrocytes should be elucidated in the future.
Reactive astrocytes have been increasingly recognized for their association with dementia and AD [43]. Astrocytes, a subtype of glial cells, undergo morphological and functional changes in response to neuroinflammation and neuronal injury, transforming reactive astrocytes [44]. Emerging evidence suggests that the neurotoxicity of reactive astrocytes may contribute to the progression of AD by promoting neuroinflammation, disrupting synaptic function, and impairing neuronal survival [45]. Moreover, these cells have been implicated in the formation and deposition of Aβ plaques, a hallmark of AD pathology [46]. Reactive astrocytes have been implicated in generating more tonic GABA release by increased MAO-B gene expression in AD models [14, 47]. GABA has been found to be elevated in reactive astrocytes, contributing to cognitive decline and neuronal dysfunction in AD [14, 48]. GABA released from reactive astrocytes activates neuronal GABAA receptors, which inhibit synaptic release. Tonic GABA release from reactive astrocytes reduces the spike probability at the synapse of the hippocampus, impairing synaptic plasticity and memory [14]. Vitamin D supplementation indicates decreased mRNA levels of MAO-B and improved memory functions in AD model mice, suggesting the therapeutic effects of vitamin D by reducing neurotoxic reactive astrogliosis [8]. In the hippocampus, we observed an increase in GABA-positive reactive astrocytes in vitamin D-deficient mice and a decrease in GABA-positive reactive astrocytes in vitamin D-supplemented mice. These findings suggested that vitamin D levels have a crucial role in the hippocampal-dependent memory function of AD model mice.
Recent studies highlight reactive astrocytes as a new target for AD treatment [49, 50], with MAO-B inhibitors proposed as potential therapeutic agents targeting these cells [51, 52]. We discovered that vitamin D, as a nutritional supplement, may also suppress reactive astrogliosis in AD, functioning similarly to MAO-B inhibitors. This finding could advocate for nutritional interventions using vitamin D in AD treatment. However, the optimal dose of vitamin D in AD has not yet been established [53]. Although vitamin D levels are recommended to be maintained between the lowest level (10 ng/ml) and toxic level (150 ng/ml) [54], additional research and clinical trials are needed to determine the precise dosage of vitamin D that provides therapeutic benefits in AD. Provided that these further investigations are conducted, our study offers a rationale for pursuing clinical improvements in AD through vitamin D supplementation within the normal range.
Limitations
We provided novel evidence for the potential role of vitamin D on reactive astrogliosis in AD pathology (Supplementary Table 2). Despite the significant findings of this study, several limitations need to be acknowledged. Although direct evaluation of serum vitamin D levels in our experimental mice would have strengthened our results, we failed to measure serum vitamin D levels due to technical error and inadequate sample volume. We inferred vitamin D deficiency status based on well-established previous studies with similar dietary conditions and serum 25-hydroxyvitamin D levels [55–57]. Likewise, we calculated the dose of vitamin D supplementation based on previous clinical studies and dosage conversion formula for mice model [58, 59]. Despite the absence of direct measurement, we anticipate that we achieved high vitamin D levels by vitamin D supplementation within the normal range. However, this limitation should be taken into account for the interpretation of our data. In addition, while we provide significant insights into the role of vitamin D on the pathology of AD, it should be noted that the lack of a wild type-control group, which could have helped determine the specific effects of vitamin D on the pathological process of AD, may restrict the interpretations of our findings. Furthermore, the lack of direct assessment of markers related to synaptic transmission restricted the meaning of our findings.
Our study also had the limitation of lacking a direct measurement for extracellular GABA levels in brain tissue. Previous research has demonstrated increased extracellular GABA levels with overexpression of MAO-B in astrocytes, as observed through microdialysis [60]. Given that MAO-B plays a pivotal role in GABA synthesis in reactive astrocytes and its increased gene expression has been implicated in AD pathology [14], these findings suggest reliance of extracellular GABA levels on intracellular levels and synthesis, specifically MAO-B expression levels. Our previous research showed that vitamin D supplementation decreased MAO-B mRNA levels and restored memory function in an AD mouse model [8]. Consequently, we used the proportion of GABA-positive astrocytes among all astrocytes as a surrogate marker of extracellular GABA levels, but further studies using microdialysis should be warranted to validate these results.
Conclusions
Our study provides valuable insights into the association between vitamin D levels and the pathological progression of AD. Our findings demonstrate that vitamin D deficiency exacerbates Aβ pathology and increases GABA-positive reactive astrocytes, whereas vitamin D supplementation ameliorates these effects. These results highlight the importance of maintaining adequate vitamin D levels for the prevention and treatment of AD, emphasizing the potential of targeting vitamin D as a novel therapeutic approach. Furthermore, this study expands our understanding of the complex interactions between vitamin D and glial cells, particularly reactive astrocytes, which may contribute to the development of new treatment strategies for AD and other neurodegenerative disorders. Future research should continue to explore the molecular mechanisms underlying the neuroprotective effects of vitamin D, thereby enabling more effective interventions to combat the growing burden of AD.
Footnotes
ACKNOWLEDGMENTS
We would like to thank Dr. H. Kim (Central Research Facility, Gwangju Institute of Science and Technology) for excellent technical support in imaging using the research slide scanner.
FUNDING
This work was supported by the Ministry of Health & Welfare (HU22C0150), Ministry of Science and ICT (2022-DD-RD-0010), and Ministry of Science & ICT (NRF-2017R1A5A1014708).
CONFLICT OF INTEREST
All authors declare that they have no competing interests.
DATA AVAILABILITY
The authors confirm that the data supporting the findings of this study are available within the article. Raw data that support the findings of this study are available from the corresponding author upon reasonable request.