
Abstract
Background:
Increased blood-brain barrier (BBB) permeability and amyloid-β (Aβ) peptides (especially Aβ1–42) (Aβ42) have been linked to Alzheimer’s disease (AD) pathogenesis, but the nature of their involvement in AD-related neuropathological changes leading to cognitive changes remains poorly understood.
Objective:
To test the hypothesis that chronic extravasation of bloodborne Aβ42 peptide and brain-reactive autoantibodies and their entry into the brain parenchyma via a permeable BBB contribute to AD-related pathological changes and cognitive changes in a mouse model.
Methods:
The BBB was rendered chronically permeable through repeated injections of Pertussis toxin (PT), and soluble monomeric, fluorescein isothiocyanate (FITC)-labeled or unlabeled Aβ42 was injected into the tail-vein of 10-month-old male CD1 mice at designated intervals spanning ∼3 months. Acquisition of learned behaviors and long-term retention were assessed via a battery of cognitive and behavioral tests and linked to neuropathological changes.
Results:
Mice injected with both PT and Aβ42 demonstrated a preferential deficit in the capacity for long-term retention and an increased susceptibility to interference in selective attention compared to mice exposed to PT or saline only. Immunohistochemical analyses revealed increased BBB permeability and entry of bloodborne Aβ42 and immunoglobulin G (IgG) into the brain parenchyma, selective neuronal binding of IgG and neuronal accumulation of Aβ42 in animals injected with both PT and Aβ42 compared to controls.
Conclusion:
Results highlight the potential synergistic role of BBB compromise and the influx of bloodborne Aβ42 into the brain in both the initiation and progression of neuropathologic and cognitive changes associated with AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive destruction and loss of neurons and their synapses in the brain, gradually escalating cognitive decline and, ultimately, dementia and death [1–6]. Currently, treatment options are limited and, at best, only temporarily alleviate some symptoms with no significant effect on disease progression [7–9]. Seeking a better understanding of events and conditions that trigger and propagate AD pathogenesis is critical before effective disease-modifying therapies can be proposed and tested in clinical trials. Despite progress in our understanding of some key aspects of AD-related neuropathological and cognitive deficits, a complete picture of sequential stages during AD pathogenesis and its relationship with neuropsychological phenotypic outcome remains elusive [3, 5]. AD has been associated with a number of pathological features, including reactive gliosis, inflammation, and the appearance of hallmark amyloid plaques (APs) and neurofibrillary tangles (NFTs). These and other changes in the brain have been temporally and spatially linked to a progressive and severe decline in cognitive function [10–12].
To identify potential early triggers of AD, determine its pathogenetic course, and ascertain which pathological features play a central role in driving the observed cognitive, memory and behavioral changes, much research has focused on APs, NFTs, neuroinflammation, and vascular pathology [13]. While the exact nature and magnitude of the contribution of each of these pathological features to disease progression continue to be debated, there is little doubt that the appearance of APs and NFTs in the brain parenchyma is directly linked to AD pathogenesis. It is generally thought that brain amyloid deposition in the form of APs precedes the appearance of NFTs [14–18]. APs principally contain the amyloid-β1–42 (Aβ42) peptide, a self-assembling amyloidogenic peptide composed of 42 amino acid residues derived from cleavage of the amyloid-β protein precursor (AβPP) [19–25]. Increased production of Aβ42 and its deposition in the brains of patients with early onset, familial AD, as well as in certain transgenic mice, have confirmed correlations between Aβ42 levels and the formation of APs, loss of synapses, and cognitive deficits [15, 26–29]. Numerous studies have described Aβ42 deposition in the form of APs and intraneuronal deposits, the latter primarily in the perikaryon of pyramidal cells in humans, transgenic rodents, and in vitro models [15, 30–51]. Mechanisms associated with intraneuronal Aβ42 deposition at each of these sites and their relationship to neuronal dysfunction and loss are being investigated [52, 53]. Determining the source(s) of the Aβ42 peptide that eventually deposits within neurons and APs in the brain and the underlying reason for the requirement of advanced age remain key questions.
Aging-associated changes in the structural and functional integrity of blood vessels in the human brain have been extensively studied. Among these changes, a chronic increase in blood-brain barrier (BBB) permeability, resulting in a disruption of brain homeostasis, has long been suspected of playing a role in the triggering and/or progression of AD as well as in other neurodegenerative diseases, post-operative delirium, post-operative cognitive decline, Sundowner’s syndrome and the short- and long-term effects of traumatic brain injury [31, 54–64]. There is some evidence that increased BBB permeability allows greater access of bloodborne Aβ peptides to neurons residing in the brain parenchyma [31]. Aβ peptides are detected in human blood and cerebrospinal fluid (CSF) [65–70]. Since Aβ42 peptide is detected in the blood (plasma) of both cognitively normal and AD subjects, it is not unreasonable to suspect that, in the context of BBB compromise and a prolonged pre-symptomatic phase, the blood could serve as a chronic source of the Aβ42 peptide that is deposited in AD brains [31, 65–70]. This scenario is supported by our earlier reports demonstrating the extravasation of various vascular components into the brain parenchyma in both AD and age-match non-demented controls [31, 71]. Our previous studies of human, porcine, rat and mouse brain have demonstrated a close spatial association between BBB leak sites and the locations of Aβ42-burdened neurons [31, 73]. For example, in a study on mice with an acutely compromised BBB, we tracked the fate of bloodborne, soluble Aβ42 peptide and demonstrated that it readily entered the brain, was selectively bound to pyramidal neurons and was internalized by these cells, the same neuronal subtype that is typically overburdened with Aβ42-rich deposits in human AD brains [31].
In the present study, we used the middle-aged (10 months old) mouse as a model system to investigate the long-term neuropathological effects of a chronically induced increase in BBB permeability and the resulting influx of soluble bloodborne fluorescein isothiocyanate (FITC)-labeled and unlabeled Aβ42 into the brain parenchyma on the animals’ ability to learn (acquisition) and retain (long-term memory) information, as well as on selective attention. To induce chronic BBB dysfunction, Pertussis toxin (PT) was repeatedly injected into the tail vein of 10-month-old male CD1 mice for a little over three months. Mice also received tail vein injections of purified monomeric FITC-labeled or unlabeled Aβ42 peptide at regular intervals over the same time period. During the last month of treatment, animals underwent a battery of cognitive and behavioral assessments. Mice injected with PT plus Aβ42 demonstrated a deficit in long-term retention and increased susceptibility to interference in selective attention compared to controls exposed to either PT or saline only. Immunohistochemical analyses of brain samples revealed an increased influx of Aβ42 and IgG into the brain parenchyma, selective neuronal binding of IgG, and intraneuronal accumulation of exogenous Aβ42 in mice injected with PT plus Aβ42 compared to controls. These findings highlight the potential contribution of an impaired BBB and the resulting influx of soluble, bloodborne Aβ42 influx into the brain parenchyma in triggering the earliest cognitive and neuropathological changes associated with AD-related pathogenesis.
MATERIALS AND METHODS
Animals
All procedures involving animals were performed following the guidelines of the institutional animal care and use committee (IACUC) in accordance with NIH guidelines. Ten-month-old male CD-1 mice (Harlan) were randomly separated into three treatment groups: PT-Aβ42 (n = 20) (animals injected with both PT and Aβ42), saline (Sal) only controls (i.e., where PT and Aβ42 injections were each replaced with saline) (n = 12), and PT-Sal controls (i.e., animals injected with PT but with Aβ42 injections replaced with saline) (n = 12). Animals were acclimated to the laboratory for 14 days prior to the initiation of any treatments and were handled (held and manipulated by an experimenter) for 90 s/day during this period. This handling ensured that differential stress responses to the experimenters and any associated effects on learning were minimized. Animals were individually housed in clear boxes with floors lined with wood shavings in a humidity- and temperature-controlled vivarium adjacent to testing rooms. A 12/12-h light/dark cycle was maintained.
Preparation of Aβ42 peptides and PT
Aβ peptides (FITC-Aβ42, Anaspec, San Jose, CA; unlabeled Aβ peptides, Biosource, Camarillo, CA) were solubilized to the monomeric form according to the method described by Zagorski et al. (1999) [74]. Briefly, Aβ peptide was solubilized (1 mg/ml) in trifluoroacetic acid (TFA), followed by removal of TFA under a slow, steady stream of nitrogen gas. This was followed by three sequential solubilizations in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) followed by removal of HFIP under nitrogen gas each time. Stock solutions (50.0μM) were prepared in 0.5×PBS, 250.0 mM HEPES buffer (pH 8.5). Working solutions were diluted in 0.9% NaCl to 6.9μM and used for tail-vein injection. Peptide concentrations were confirmed using the Pierce Micro BCA Assay (Rockford, IL). Working solutions of PT (Sigma-Aldrich, St. Louis, MO) were diluted in 0.9% saline to 3.0 × 10−3 μg/μl prior to use in tail vein injection. Concentrations were confirmed using the Micro BCA Assay (Pierce; Rockford, IL).
Animal treatments
Tail vein injections of saline only (Sal), PT plus saline (PT-Sal), or PT plus Aβ42 peptide (PT-Aβ42) were administered according to the following time intervals and concentrations. To effectively achieve a permeable BBB, PT-Aβ42 and PT-Sal mice were treated with PT (300 ng in 100μl 0.9% saline) for 3 months via tail vein injection, which has previously been shown to reliably induce increased BBB permeability in adult mice [31]. PT injections occurred on days 0, 3, 17, 33, 47, 61, and 75 of the 3-month injection period. To augment plasma levels of Aβ42 in mice and track its fate, soluble Aβ42 peptide was administered via tail vein injection (100 microliters of 6.9 micromolar in 0.9% saline) on days 7, 11, 14, 18, 21, 25, 28, 32, 35, 39, 42, 46, 49, 53, 56, 60, 63, 67, 70, 74, 77, 81, 84, 88, 91, and 95 during the same 3 month period. Given expected inherent levels of endogenous Aβ42 peptide present in mice which, under conditions of increased BBB permeability, can also potentially enter the brain parenchyma, and to facilitate tracking the fate of tail vein-injected, bloodborne Aβ42, a subset of PT-Aβ42 treated mice (n = 12) were injected with FITC-labeled Aβ42. In the remaining subset of PT-Aβ42 mice (n = 8), animals were administered unlabeled Aβ42 (U-Aβ42) peptide to confirm observations with FITC-labeled Aβ42 and to address the possibility that addition of the FITC moiety to the Aβ42 peptide might somehow modify its behavior and/or fate in the animal. Controls for Aβ42 and PT were injected with similar volumes of saline (100μl of 0.9% saline) with or without Aβ peptides following the same schedule described above. Following completion of one month of treatments, a battery of behavioral training and testing was initiated and continued for around five weeks post last injection on D-95.
Behavioral training and testing procedures
All animals were tested on five cognitive tasks (odor discrimination, passive avoidance, spatial water maze, Lashley Maze, and selective attention/disengagement) as described in detail previously (see the Supplementary Material for additional details) [75–77]. These five tasks were chosen based on their suggested sensitivity to AD-related pathologies and/or their conceptual relevance to cognitive impairments associated with AD [78, 79]. Furthermore, while AD pathology is associated with learning impairments, tests of retention are sometimes more sensitive to mild or early impairments, and several of the tests employed here are amenable to tests of long-term retention [78]. The order in which tasks were administered was designed to separate tasks based on similar motivations, processes, or motor requirements (e.g., mazes of a similar nature, activity versus passivity) to minimize any potential transfer between tasks.
For the learning tasks requiring food deprivation, ad lib food was removed from the animals’ home cages at the end of the light cycle approximately 40 h prior to the start of training (and thus encompassing the “rest” day between successive tasks). During this deprivation period, animals were provided food in their home cages for 90 min/day during the last 2 h of the light cycle, and thus were approximately food-deprived for 16 h at the time of training or testing. To manage the large number of animals being tested, we split the animals into two batches and ran one behavioral test at a time. Each batch of animals included mice from all treatment groups. The sequential order of the behavioral tests run was as followed: Water Maze, Lashley’s III Maze, Passive Avoidance, Odor Discrimination, Water Maze Retention, Lashley’s III Maze Long-term, and Selective Attention.
Odor discrimination and choice
Rodents rapidly learn to use odors to guide appetitively reinforced behaviors [75]. Here, mice learned to navigate a square field in which unique odor-marked (e.g., almond, lemon, mint) food cups were in three corners. Although food was present in each cup, it was accessible in only one cup (e.g., that marked by a mint odor). Each animal was placed in the empty corner, after which it explored the field and eventually retrieved the single piece of available food. In subsequent trials, the location of the accessible food cup was changed, but was consistently marked by the mint odor. On successive trials, animals required less time to retrieve the food and made fewer approaches (i.e., “errors”) to food cups lacking food. In this task, poor learning performance is indicated by an increased number of errors (i.e., contacting a cup marked by an odor not associated with available food). Using this procedure, errorless performance is typically observed within 3–4 training trials.
One-trial passive avoidance
Animals learn to suppress movement to avoid contact with aversive stimuli [75]. This “passive avoidance” response is exemplified in “step-down” avoidance procedures, where commonly, an animal is placed on a platform, whereupon stepping off the platform it encounters an aversive stimulus [75]. Following only a single trial, animals are subsequently reluctant to step off a safe platform. In the present study, upon stepping off the platform, animals were exposed to a combination of bright light and a loud oscillating noise. The ratio of post-training to pre-training step-down latencies was calculated for each animal and served to index learning. It has been determined that asymptotic performance is apparent in group averages following 1–3 training trials; thus, performance after a single trial reflects (in most instances) sub-asymptotic learning and is sensitive to variations in the rate of learning between animals. Thus, higher Post/Pre-step-down latencies indicate better learning, i.e., acquired fear of leaving the safe platform.
Spatial Water Maze
For this task, animals are immersed in a round pool of opaque water from which they could escape onto a hidden (i.e., submerged) platform. The latency for animals to find the platform decreases across successive trials [80]. The performance of animals can improve across trials despite the animals beginning each trial from a new start location (i.e., efficient navigation requires the implementation of a “cognitive map”, rather than simply following a fixed route). Such a procedure mitigates against egocentric navigation (as could determine performance in the Lashley Maze) or fixed motor patterns and promotes the animals’ dependence on extra maze spatial landmarks and is said to reflect the animal’s representation of its environment as a “cognitive map”.
During training, shorter latencies to locate the escape platform across trials is indicative of more rapid acquisition of a stable cognitive map. To estimate capacity for long-term retention in this task, animals were tested with a “probe” trial following a 30-day retention interval from completion of acquisition training. On the probe test, the escape platform was removed from the pool, and all animals were started from the same start location. A 60-s test was conducted in which the animals’ time searching in the target quadrant (that in which the escape platform was previously located) and non-target quadrants were recorded. This measure served to index long-term retention in this task, i.e., an animal with good retention should spend significantly more time in the target quadrant (previous location of the escape platform) than non-target quadrants. Conversely, an animal with poor retention would not express any preference for the target quadrant.
Lashley III Maze
Mice can learn a pattern of egocentric navigation to locate food in a maze [75]. The Lashley III Maze consists of a start box, four interconnected alleys, and a goal box containing a food reward [75]. Over trials, the latency of rats to locate the goal box decreases, as do their errors (i.e., wrong turns or retracing). Here, mice were first acclimated to the Lashley Maze, then received five training trials during which errors were recorded. Using our parameters, mice reach asymptotic levels of performance (i.e., errors decrease to a stable level) in 4–6 training trials, thus five trials is sensitive to differences in rate of learning across animals. To estimate capacity for long-term retention in this task, following a 30-day retention interval from completion of training, animals were placed in the start box and allowed to traverse the maze again for two trials. To index the long-term retention using this task the difference between the first trial after the retention interval (T7) and the average of the last two trials in the acquisition period (T5 and T6) was used for each animal.
Selective attention/attentional disengagement
In the human Stroop Interference Test Condition, subjects are instructed to name the color of the ink that is used to spell a word rather than read the word. Subjects respond more quickly and make fewer errors when the written word and the color of ink are congruent (e.g., the word RED is spelled with red ink) relative to when the ink color and word disagree (e.g., the word RED is spelled with green ink). The increased latency and tendency to generate errors in the incongruent condition are believed to reflect a failure of selective attention, i.e., the subject is distracted from the task by the written word [81]. We have developed an analog of the Stroop Test for mice, wherein the attention directed toward an odor (or visual) cue is impaired when the animal is tested in the presence of a task-relevant visual (or odor) distractor [77, 82].
In the present task, mice were trained to perform an odor discrimination or a visual discrimination, each in a distinct context. In this manner, the context came to “instruct” the animal which discrimination to perform. After reaching asymptotic (near errorless) performance in each task, a test of selective attention was performed. For this test, the visual cues were added to the odor discrimination box. Given the previously learned instructions, the animal should ignore the visual cues and approach only the relevant odor cues. An increase in errors was deemed as a failure of the ability to selectively attend to the odor cues. After the completion of this test, the animals were subjected to a test of their capacity to disengage from the previous tendency to approach the odor cues. For this test, animals were placed in the visual discrimination box with both visual cues (the relevant targets) and odor cues (the task-relevant distracters) present. To perform effectively in the task, animals were required to inhibit their approach to recently reinforced odor cues and now attend only to visual cues to find food. Animals then alternated between boxes and were tested in this manner until they had been tested for performance in each box two times (for a total of four test trials). The number of errors accumulated over the test trials served as the index of selective attention and the animals’ ability to shift between relevant cues (i.e., to disengage from one type of cue and focus attention on the alternate type of cue). Errors were scored in the same manner described above for the Odor Discrimination task. A detailed description of this task is provided in the Supplementary Material.
Tissue preparation for neuropathological studies
Animals were euthanized at ∼14.5 months of age using pentobarbital (200 mg/kg) administered intraperitoneally. Brains were removed rapidly and fixed in 4% paraformaldehyde in PBS at 4°C for 7 days. From each animal brain, tissue blocks were generated for immunohistochemistry (IHC), including the cerebral cortex (from multiple locations), hippocampus and cerebellum. Following fixation, tissue was either paraffin-embedded or frozen. Paraffin-embedded tissues were sectioned at a thickness of 5.0μm, and sections were placed on Superfrost® Plus glass slides (48311-703, VWR, Bridgeport, NJ) previously coated with Poly-L-Lysine (P8920, Sigma-Aldrich, St. Louis, MO). Slides were baked at ∼60°C for 1 h to ensure tissue was fully adherent. Following fixation, tissue to be used for frozen sectioning was infiltrated with 10% and then 30% sucrose in PBS also containing 1.0% paraformaldehyde, pH 7.4) at 4°C under constant gentle agitation. Specimens were placed atop a solid 4.0% gelatin matrix and the resulting tissue/gelatin blocks were snap-frozen in liquid nitrogen and stored at −80°C. Cryosectioning was performed using a Leica CM1850 Cryostat (Deerfield, IL). Cryosections (12.0μm thick) were placed on Superfrost® Plus glass slides and stored in a dry container at 4°C until use.
Brightfield immunohistochemistry
Paraffin-embedded tissues were processed for brightfield IHC as described previously [30, 84]. Briefly, after removal of paraffin with xylene and rehydration through a graded series of decreasing concentrations of ethanol, protein antigenicity was enhanced by microwaving sections in Target Buffer (Dako, Carpenturia, CA) for 2 min. Following a 30-min incubation in 0.3% H2O2, sections were treated for 30 min in normal blocking serum and then incubated with primary antibodies (anti-Aβ42 antibodies polyclonal, 1:50 dilution, AB5078P, Chemicon International, Temecula, CA; anti-mouse IgG, dilution 1:100, Millipore, Temecula, CA) for 1 h at room temperature. After a thorough rinse in PBS, a secondary biotin-labeled antibody was applied for 30 min at room temperature. Immunoreactions were treated with the avidin–peroxidase-labeled biotin complex (ABC, Vector Labs, Foster City, CA) and visualized with 3-3-diaminobenzidine-4 HCl (DAB)/H2O2 (Biomeda, Foster City, CA). Sections were lightly counterstained with hematoxylin, dehydrated using increasing concentrations of ethanol, cleared in xylene and mounted in Permount. The specificity of immunoreactivity was confirmed by incorporating various antibody controls during each IHC run. Since the specificity of anti-Aβ42 antibodies are controversial, we used one of the most widely used and tested anti-Aβ42 antibodies. All specimens were examined and photographed with a Nikon FXA microscope, and digital images were recorded using a Nikon DXM1200F digital camera and processed using Image Pro Plus (Phase 3 Imaging, Glen Mills, PA) imaging software.
Fluorescent imaging
A subset of frozen brain sections from mice treated with PT and FITC-labeled Aβ42 were washed in PBS, mounted using 4,6-diamidino-2-phenylindole, or DAPI (H-1500, Vector Laboratories, Burlingame, CA), and examined via fluorescence microscopy to assess the localization of FITC-conjugated peptides.
Counting the number of IgG or Aβ42 positive neurons
The number of Aβ42-positive cells was determined from sections of brain tissue from four animals in each of the treatment groups. Sections of paraffin-embedded tissue were immunostained with anti-Aβ42 antibodies under identical conditions. Five random images at 20X magnification from each animals’ cortical region were recorded under identical illumination, magnification and camera settings using a Nikon FXA microscope equipped with a Nikon CCD camera. Similarly, Aβ42-positive cells were visualized in the five random images at 20X magnification of the frozen brain cortical region using fluorescence settings of Nikon FXA microscope and Nikon Immunofluorescence camera. The total number of Aβ42-positive cells in each 20X viewing field was counted. Various statistical tools were used to test the significance of differences among the different treatment groups.
Statistical analyses
After data collection, descriptive statistics for each group was examined along with the normality of the distribution. Multiple tests were used to ensure that the data followed a normal distribution, in which case an ANOVA analysis was carried out. A mixed effects analysis was preferred when there were missing data points in animal behavioral studies. If the distribution pattern of the acquired data were not normal, unpaired, non-parametric Mann-Whitney t-tests were carried out. Analyses were carried out using SPSS (v28) and GraphPad Prism (v10) while GraphPad Prism (v10) was used to generate graphs.
RESULTS
Overall, on several cognitive tasks and acquisition tests, mice treated with PT plus Aβ42 (PT-Aβ42) showed deficits compared to mice treated with PT plus saline (in place of Aβ42) (PT-Sal) or saline only (Sal). By contrast, in most tests, PT-Sal mice generally did not exhibit decrements in performance compared to Sal mice. A similar pattern was observed in tests of long-term retention, where PT-Aβ42 mice demonstrated a marked inability to recall trained activities relative to PT-Sal and Sal mice. Results from specific tests are described below.
Odor discrimination
No significant difference in the number of errors was observed between groups in Trial 1 (before learning could have occurred; Fig. 1A). However, closer inspection of the acquisition curves suggests that the rate of acquisition was slower in PT-Aβ42 mice relative to the other groups, an effect that became increasingly apparent as trials progressed. This pattern was reflected in a significantly greater number of errors (p < 0.01) in the final two trials (Trials 4 and 5) for PT-Sal and Sal mice. These observations suggest a specific deficit in the rate of acquisition in the PT-Aβ42 group.
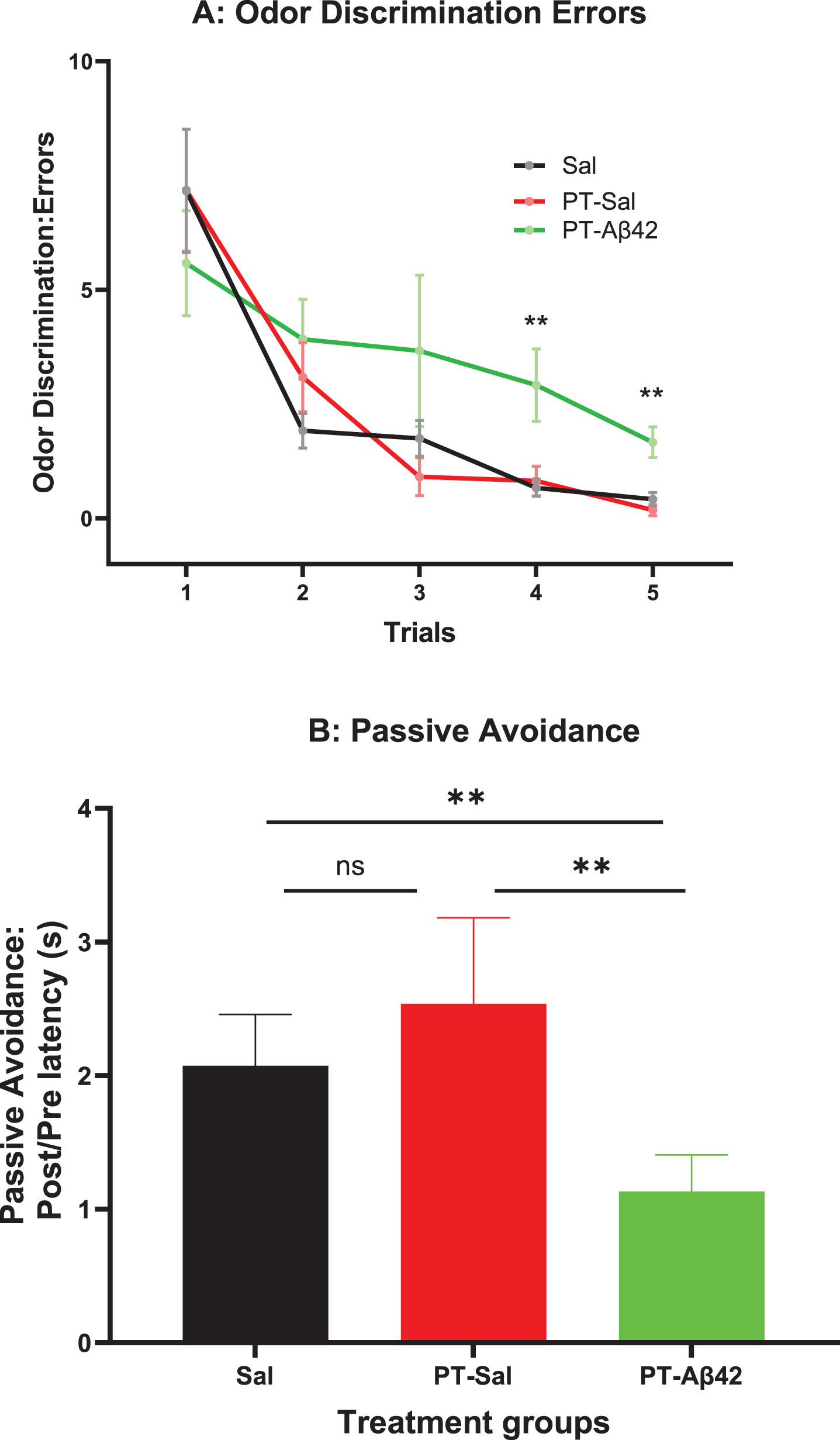
Mice treated with PT-Aβ42 (PT-Aβ42) showed deficits compared to PT-Sal and Sal-only mice. A) Odor Discrimination. Animals were trained to locate food in randomly assorted locations by using an odor cue. Mean errors to locate the target food cup for animals in each treatment group are shown. Brackets indicate standard error. Using a mixed effects analysis, we did not find any significant difference between the treatment groups across trials 1 through 3; however, when trials 4 and 5 were compared, a statistically significant difference (p < 0.01) was observed in the PT-Aβ42-treated mice compared to the other groups. B) Passive Avoidance: Post/Pre-Latency(s). Upon stepping from a safe platform, animals were exposed to a bright light, loud noise, and vibration. The mean ratio of post-training to pre-training step-down latencies was calculated for each animal and served to index learning. Brackets indicate standard errors, and the values were analyzed using the Mann-Whitney test. Animals treated with PT-Aβ42 showed a significantly lower step-down latency compared to the Sal group (PT-Sal, p < 0.01). Likewise, the PT-Aβ42 group demonstrated significantly lower step-down latency compared to PT-Sal group (p < 0.01). The PT-Sal and Sal-only groups failed to demonstrate any significant differences.
Passive avoidance: post/pre-latency(s)
PT-Aβ42 mice exhibited inferior performance on the passive avoidance task, but this difference did not achieve statistical significance in the overall ANOVA (Fig. 1B). However, comparisons between the PT-Aβ42 and Sal groups or the PT-Aβ42 and PT-Sal groups indicated that the performance of the PT-Aβ42-treated group was significantly worse than in animals treated with PT-Sal or Sal as these comparisons reached statistical significance (p < 0.01) (Fig. 1B). On the contrary, paired comparison between mice treated with PT-Sal and Sal were not statistically different from each other (Fig. 1B).
Spatial Water Maze
We investigated the animals’ ability to learn a task (spatial navigation, Fig. 2A) and retain over a long retention interval (Fig. 2B) the learned response for all treatment groups. All groups were found to exhibit equivalent performance in their ability to learn the task across trials, i.e., the groups did not differ in their rate of acquisition (Fig. 2A). The latency to locate a submerged platform gradually declined in later trials (Fig. 2A), suggesting that PT-Aβ42 mice also learned spatial navigation at a normal rate (Fig. 2A). On mixed effect analysis, we observed statistical difference between the trials (p < 0.0001) without any difference between three treatment groups in each trial (Fig. 2A).
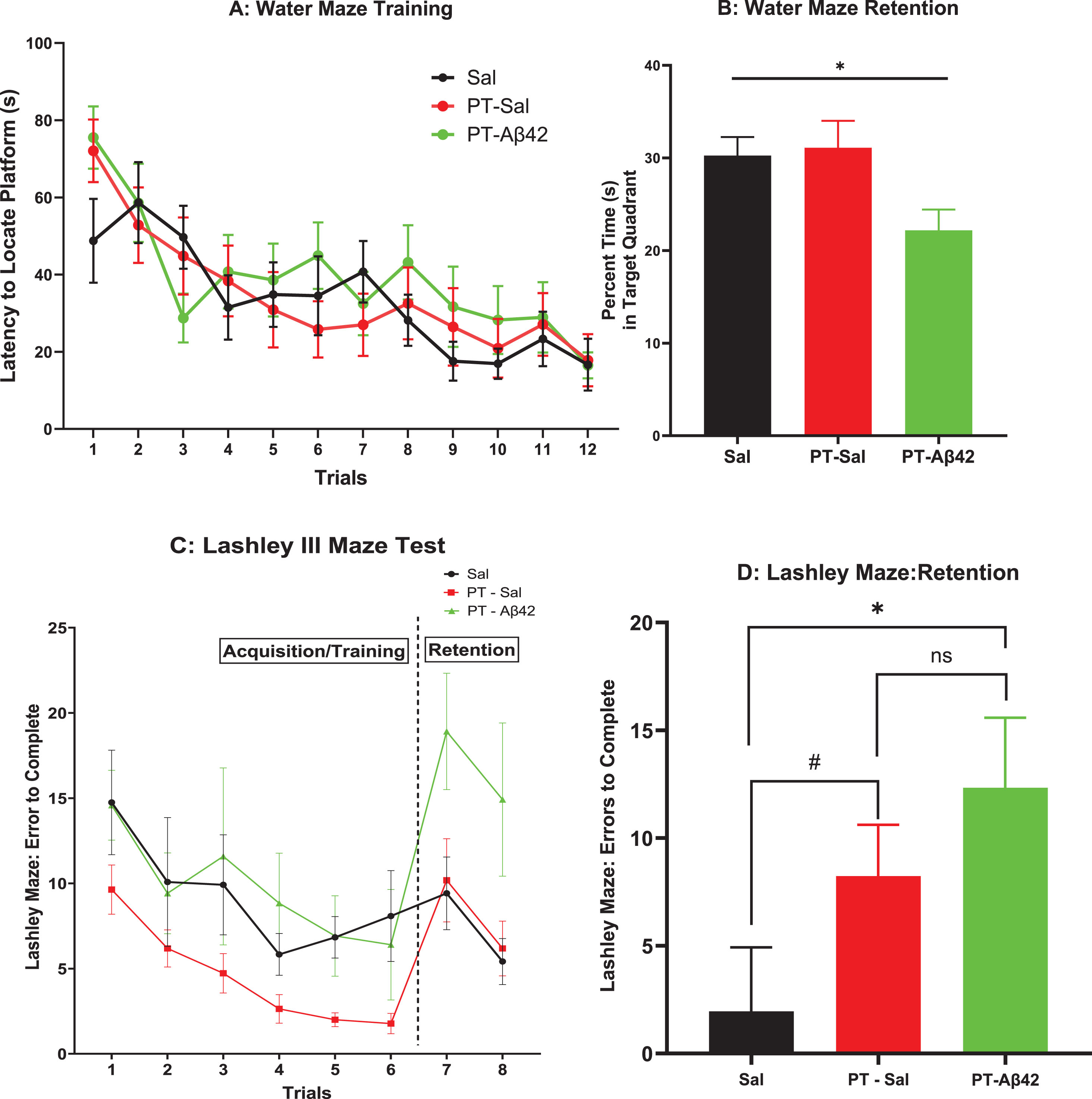
A) Spatial Water Maze: Latency to Locate Platform. Animals were trained to locate a hidden platform submerged below the surface in a tank of water. The latency to locate the hidden platform was recorded across each of 12 training trials for animals in each treatment group. Brackets indicate standard error. The mice in all treatment groups demonstrated steady progress in locating submerged platform as revealed by the declined period from first to twelfth trial. Mice in all treatment groups also demonstrated comparable abilities to learn a task. B) Spatial Water Maze Retention: Percent Time(s) in Target Quadrant. Following a 30-day retention interval, animals were administered a 60-s “probe” trial in which the escape platform was removed from the maze and the percentage of time spent searching in the target quadrant was recorded. Mean time in the target quadrant is depicted. Brackets indicate standard error. For comparison across the three treatment groups, we used the Brown-Forsythe ANOVA test and found significant difference (p < 0.05) across the treatment groups. Overall, the PT-Aβ42 group spent less time in the target quadrant compared to both PT-Sal and Sal groups. C) Lashley Maze: Errors to Complete: Long-term Retention. Mice were trained to locate food in the Lashley Maze over 6 trials (T1–T6) and re-tested on 2 trials (T7–T8) following a 30-day retention interval. Mean errors to reach the goal box across trials are depicted for each treatment group. Brackets indicate standard error. As the training progressed, T1–T6, the number of errors was consistently reduced for mice treated with PT-Sal and Sal. Sal and PT-Aβ42 groups demonstrated inconsistent results as their number of errors fluctuated considerably. At T7, PT-Aβ42 mice demonstrated a significant increase in errors compared to both PT-Sal and Sal groups. The number of errors made by mice from the PT-Aβ42 group at T7 was higher than during T1. The number of errors made by mice in the PT-Sal and Sal groups at T7 was similar and lower than T1. D) A measure of retention was computed for each animal by obtaining a difference score between the errors committed on the first post-time lapse trial (i.e., T7) and the average of the last two acquisition trials (T5–6). Brackets indicate standard error. A comparison of the errors between the Sal and PT-Aβ42 groups attained statistical significance (*p = 0.0409). Interestingly, between the PT-Aβ42 and PT-Sal groups, the PT-Aβ42 group demonstrated more errors and the numbers approached a similar trend (#p = 0.0571).
Thirty days after completion of training, the same mice were tested for long-term retention of the learned response in the Spatial Water Maze. Mice treated with PT-Aβ42 exhibited significant impairments of long-term retention relative to the other groups. This difference reached statistical significance in the overall Brown-Forsythe ANOVA (p < 0.05) (Fig. 2B). Paired comparison between PT-Aβ42-treated mice to PT-Sal mice as well as PT-Aβ42-treated mice to Sal mice demonstrated a reduced tendency in PT-Aβ42-treated mice to retain the learned task (Fig. 2B). This pattern is particularly notable given the intact acquisition observed on this task in PT-Aβ42-treated animals (Fig. 2A).
Lashley III Maze
In the Lashley III Maze, PT-Aβ42-treated mice learned how to navigate the maze in a manner comparable to mice in the other groups, as overall rates of acquisition were largely similar in this task (Fig. 2 C). These values did not reflect the subjects’ failure to learn, as they were evident on the first trial prior to any learning having occurred. As in the Spatial Water Maze, mice were tested for long-term retention in the Lashley III Maze at a 30-day interval. In contrast to their varying pattern of performance on tasks assessing acquisition abilities, mice treated with PT-Aβ42 again exhibited a performance deficit compared to the other groups (Fig. 2D). This impaired retention in PT-Aβ42 animals reached significance relative to mice in the Sal group (p < 0.05, Fig. 2D). A similar trend of difference in this regard was noted between the PT-Sal and Sal groups (p = 0.0571, Fig. 2D). Interestingly, paired comparison between the PT-Aβ42-treated and PT-Sal-treated groups failed to attain statistical significance (Fig. 2D). Most importantly, the observed decline in retention in PT-Aβ42 animals was especially notable in the context of the fact that animals in all groups demonstrated comparable acquisition rates during initial training on this task (Trials 1–6, Fig. 2C).
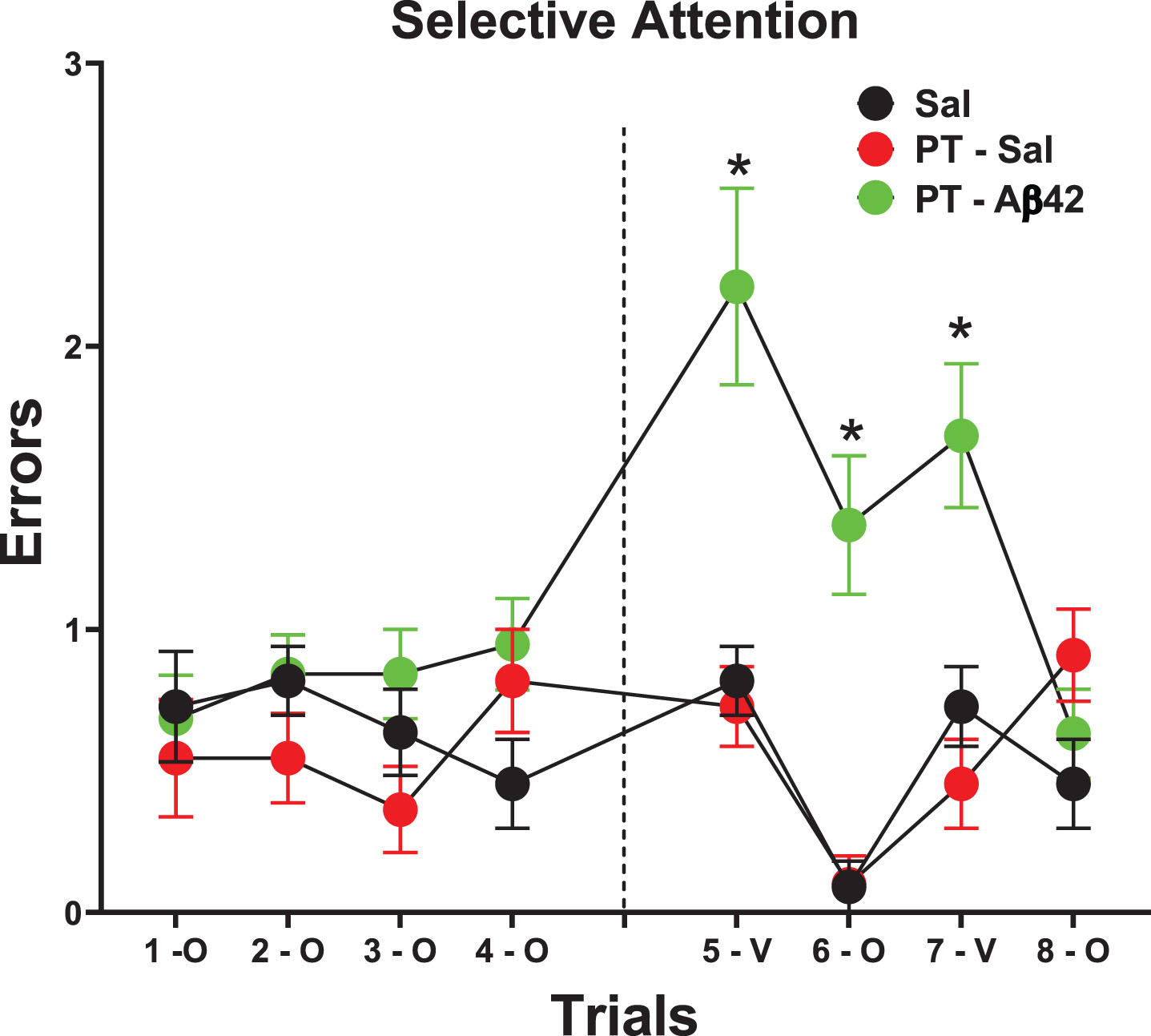
Selective attention
The selective attention task is a test that is distinct from the other cognitive tasks used here. It occurs after asymptotic acquisition and makes no nominal demands on learning or long-term retention. On this test, a severe deficit was expressed by PT-Aβ42 mice relative to all other groups as determined by a trials×Group interaction (p < 0.001, Fig. 3). This deficiency appeared to be related to an interference effect that likely accumulated across successive trials given its specific emergence on later trials when the odor cue served as the task-relevant distractor (after previously serving as the target cue; Fig. 1A). This was evidenced by significant increases in errors relative to each other group on trials 5–7, as determined by planned comparisons (p < 0.05, Fig. 3). This pattern of results suggests that PT-Aβ42 mice were cognitively “rigid” and could not adapt to the changing conditions required for effective directed attention (i.e., when odor cues changed from target to distractor).
Repetitive Pertussis toxin (PT) treatments facilitate chronic compromise of BBB integrity and leak of plasma components, including IgG antibodies, into the cerebral cortex
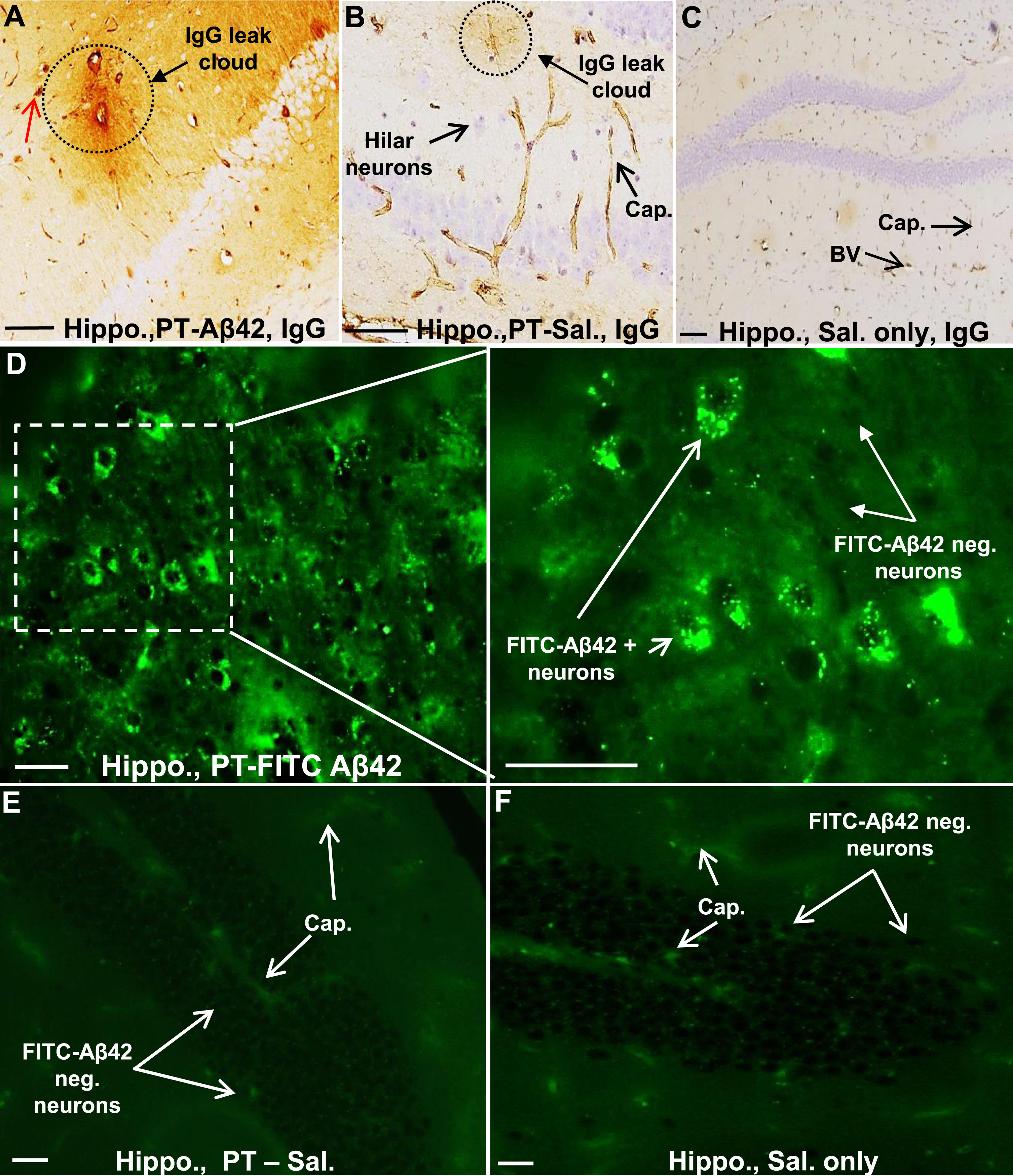
To chronically increase BBB permeability in mice, PT was repeatedly injected (seven times over three months) into the tail vein as described previously [31]. Leak of IgG from the brain vasculature was used as a marker of PT-induced BBB breach in the cerebral cortex and hippocampus (Fig. 4A–D). Such leaks were observed in all PT-treated mice. Interestingly, IgG leaks did not occur uniformly along the length of brain blood vessels, but rather were variable in location and extent, and were not limited to blood vessels of a single hierarchical type. Leaks ranged from small focal plumes of material emerging from a single site (Fig. 4A, B) to more extensive leaks occurring along a greater length of the vessel and its tributaries. In general, larger vessels (especially arterioles) displayed more extensive perivascular leak clouds than smaller vessels. Areas lacking discernible leaks often exhibited a sharp interface between IgG-positive and -negative regions (Fig. 4C, D). By contrast, IgG was mostly confined to the brain vasculature in Sal mice (Fig. 4E). Thus, repetitive PT treatments facilitate chronic compromise of BBB integrity and leak of plasma components, including IgG antibodies, into the cerebral cortex.
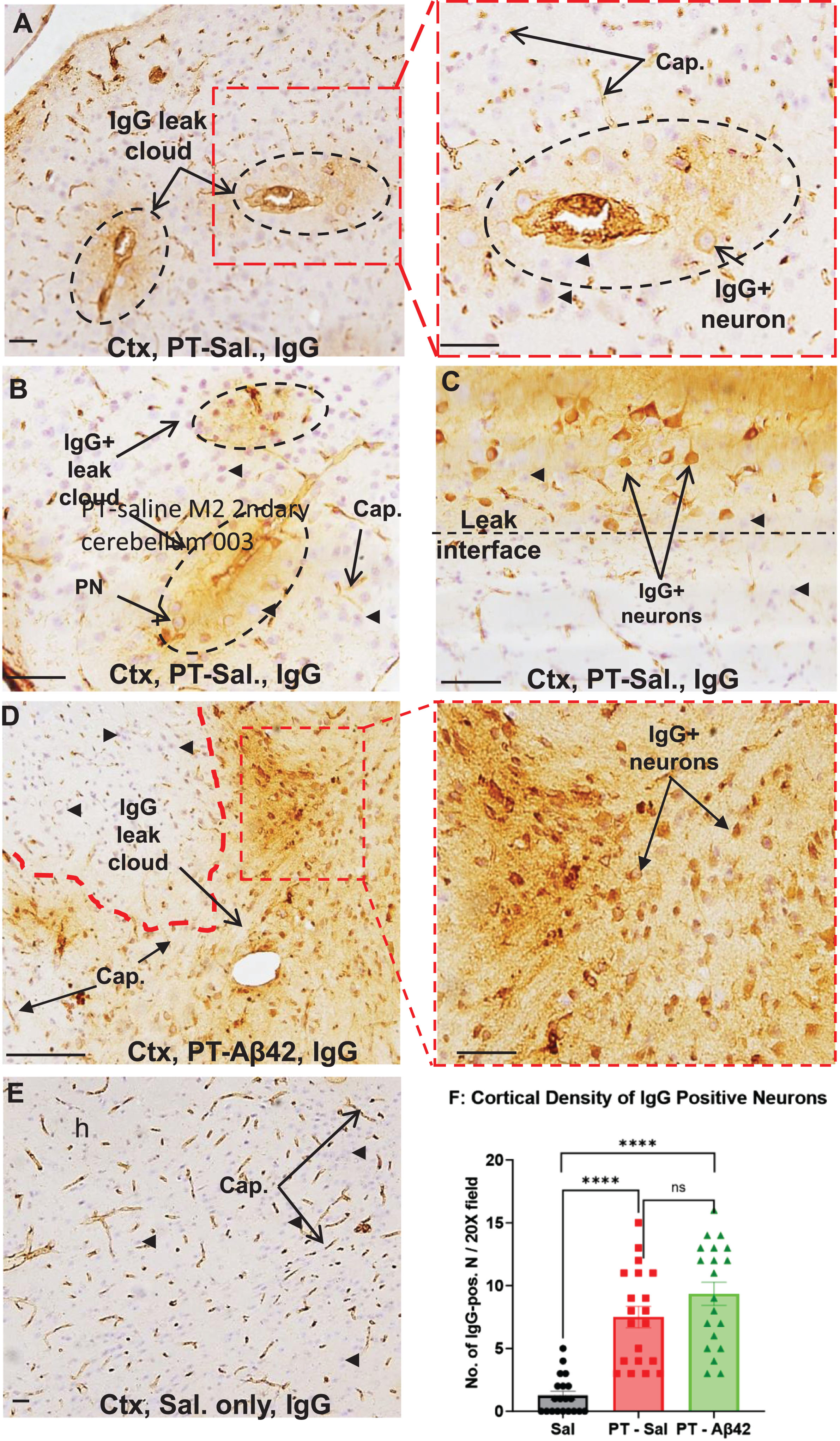
Chronically increased BBB permeability allows IgG antibodies to enter the brain parenchyma and bind selectively to neurons, especially pyramidal cells in the cerebral cortex
IgG entering the brain interstitial space was found to bind preferentially to certain neuronal subtypes, especially pyramidal neurons positioned within or near perivascular leak clouds (Fig. 4A–D). The intensity of neuronal IgG immunoreactivity decreased rapidly with increasing distance from the vessel leak site, often forming a well-defined leak interface (Fig. 4A–D). While increased permeability of the BBB to IgG was evident in the cerebral cortex of PT-Aβ42 and PT-Sal mice relative to Sal controls, vascular leaks were most prevalent in the brains of PT-Aβ42 mice, leading to an increased cortical density of IgG-positive neurons (Fig. 4D, F). In contrast to PT-Aβ42 and PT-Sal animals, Sal animals exhibited a generally intact BBB within the cortex (Fig. 4E, F). We also found a significant difference in the density of IgG-positive cortical neurons across treatment groups (Fig. 4F). Paired comparison between the PT-Sal and Sal groups reached statistical significance, as did comparison of Sal mice with PT-Aβ42 mice (p < 0.0001) (Fig. 4F). Although we observed a greater density of IgG-positive cortical neurons in PT-Aβ42 mice compared to PT-Sal mice, this difference failed to achieve statistical significance (Fig. 4F).
Chronic BBB compromise allows bloodborne Aβ42 to enter the brain parenchyma and accumulate within neurons in the cerebral cortex
The fate of exogenous Aβ42 in animals with and without chronic PT-induced BBB compromise was tracked over a three-month period (final age ∼14.5 months) using FITC-labeled Aβ42 peptide administered via tail vein injection. A group of PT-injected mice also received FITC-Aβ42 injections throughout this period (26 times over 95 days), and the fluorescent signal was used to localize FITC-Aβ42 in tissue sections of the cerebral cortex (Fig. 5). In regions exhibiting vascular leak, FITC-Aβ42 was observed emerging from the walls of blood vessels and entering the surrounding brain interstitial space (Fig. 5A). Many (but not all) neurons within or in the vicinity of perivascular leak clouds exhibited intense FITC-Aβ42 fluorescence in their cytoplasm, but not in nuclei, indicating a selective affinity of exogenous FITC-Aβ42 for these neurons and its localization in the cytoplasmic compartment (Fig. 5A–D). As for IgG, neurons showing the greatest intensity of neuronal FITC-Aβ42-positive fluorescence were those positioned closest to the source vessel (Fig. 5A, B). FITC-Aβ42 was also localized within numerous discrete granules in neuronal perikarya, suggesting that exogenous, cell surface-bound FITC-Aβ42 can be internalized and most likely makes its way into elements of the lysosomal compartment (Fig. 5C, D). Intraneuronal deposition of FITC-labeled Aβ42 was clearly selective for certain types of neurons, especially pyramidal cells (Fig. 5A), with the most prominent labeling associated with larger pyramidal neurons that populate the lower layers of the cerebral cortex. Interestingly, great variations in the relative content of intracellular Aβ42 were observed among neurons (e.g., compare Fig. 5A–D). FITC-Aβ42-positive neurons were lacking in the cerebral cortex of PT-Sal and Sal mice (Fig. 5E, F).
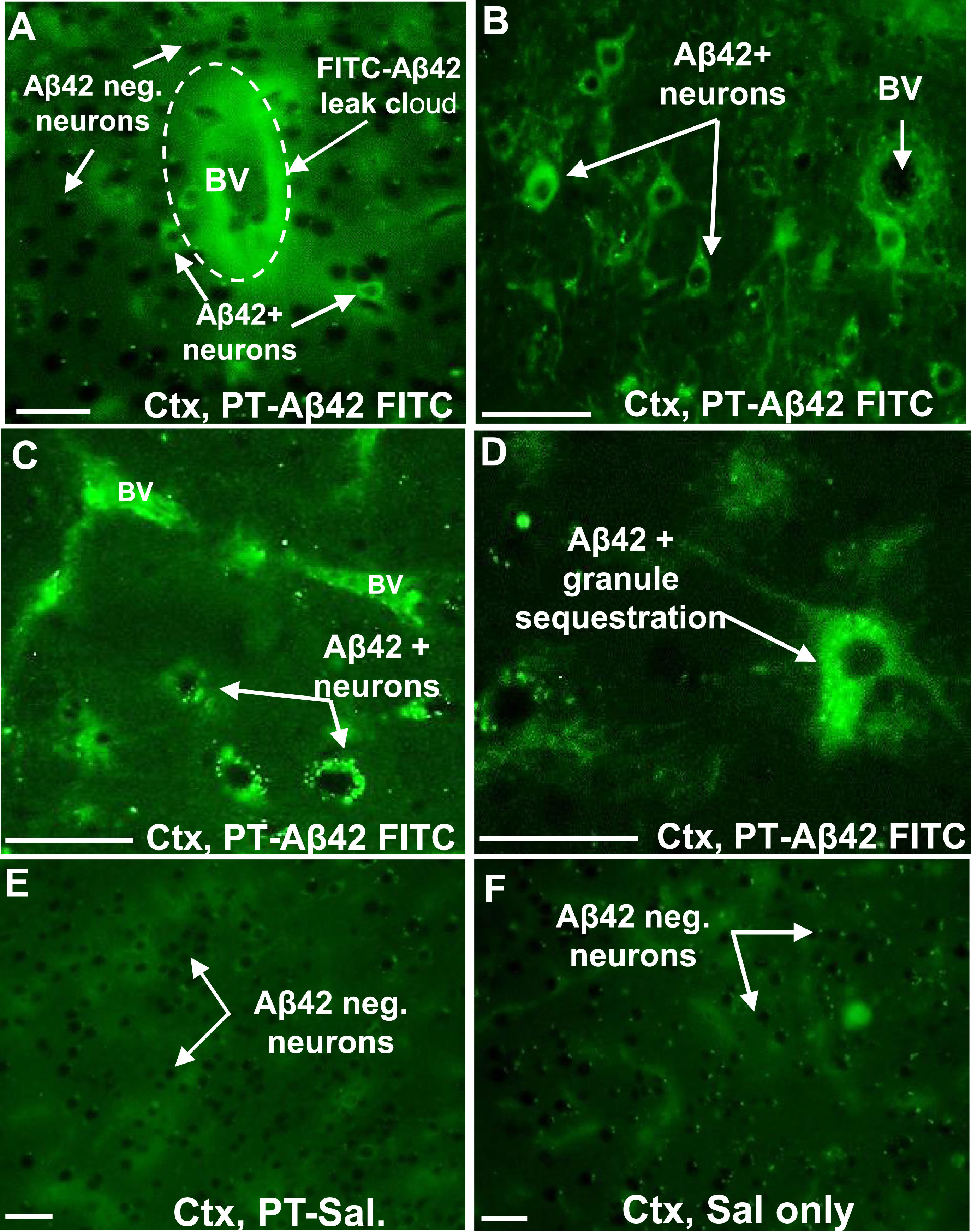
In PT-treated animals injected with unlabeled Aβ42 (U-Aβ42) peptide instead of the FITC-labeled peptide, the distribution of U-Aβ42 in the brain following the long-term treatment course was investigated by bright field immunohistochemistry (red arrows, Fig. 6A, B). As expected, PT-U-Aβ42 treated mice demonstrated a U-Aβ42 distribution of pattern within the brain parenchyma that closely resembled that of FITC-Aβ42-injected animals (red arrows, Fig. 6A, B). Controls injected with saline in place of U-Aβ42 showed no detectable U-Aβ42 in the brain parenchyma (Fig. 6C). Interestingly, PT-Sal mice (not injected with U-Aβ42) also showed increased intraneuronal Aβ42, although less than that of PT-U-Aβ42 treated mice, suggesting that endogenous mouse Aβ42 is also able to enter the brain parenchyma via a compromised BBB and preferentially associate and accumulate within local neurons (Fig. 6D).
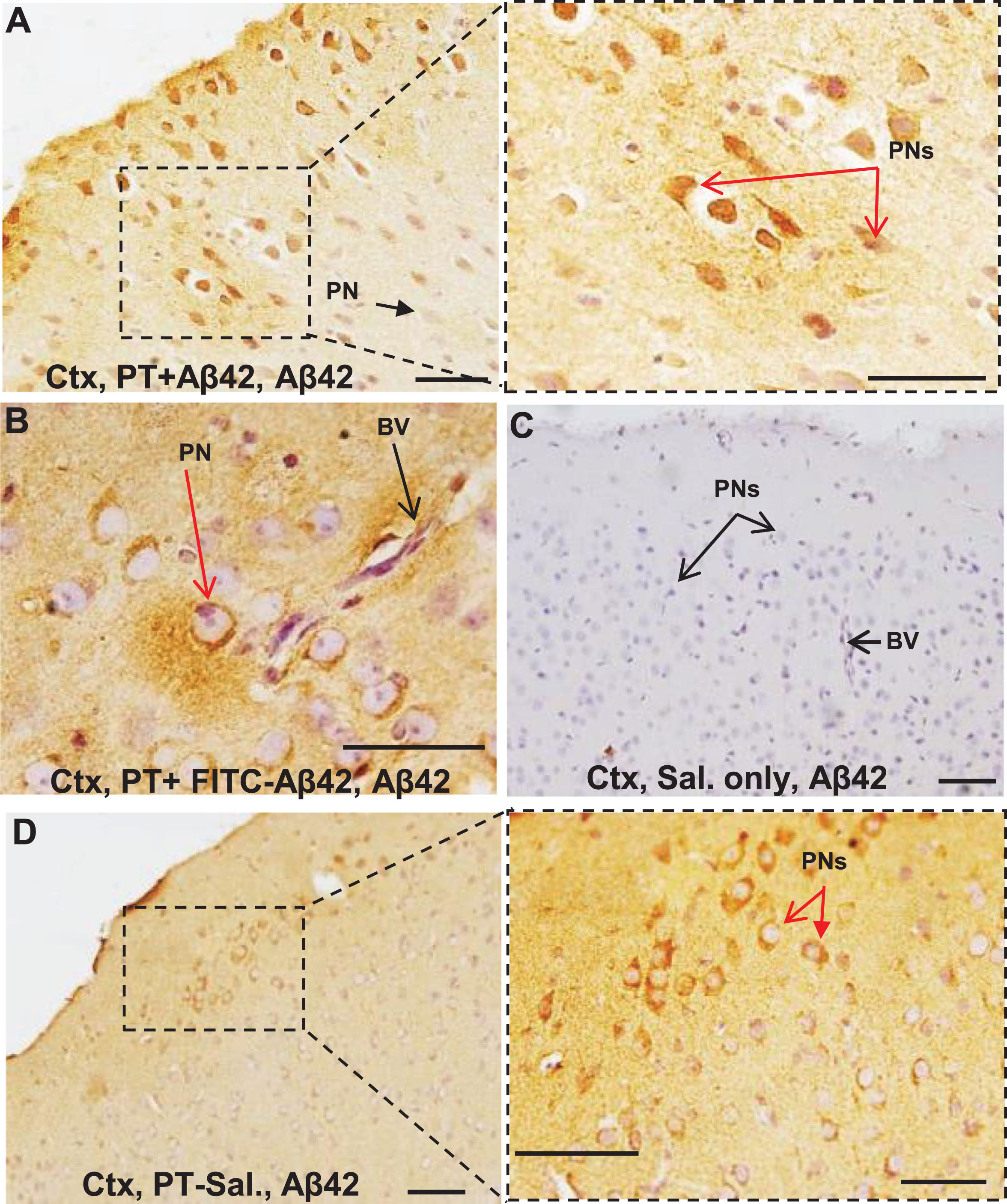
BBB compromise increases the density of Aβ42-positive neurons
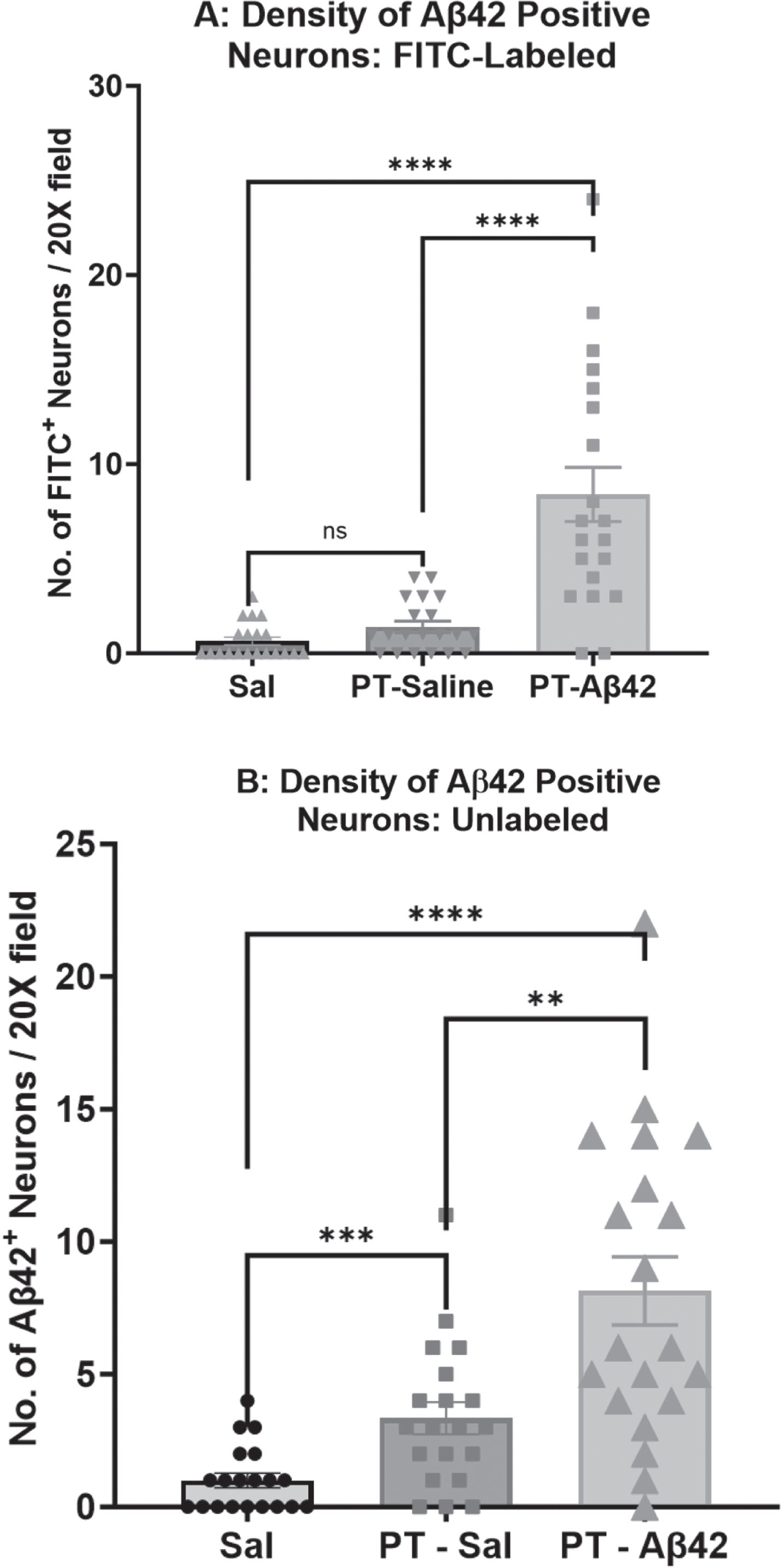
We measured the density of Aβ42-positive neurons (those exhibiting surface-bound and/or intracellular Aβ42) in sections of the mouse cerebral cortex processed for detection of FITC-Aβ42 using fluorescence microscopy (Fig. 7A) or U-Aβ42 using Brightfield IHC and purified anti-Aβ42 antibodies (Fig. 7B). Fluorescence data demonstrated a significantly higher density of FITC-Aβ42-positive neurons in mice injected with PT plus FITC-labeled Aβ42 (p < 0.0001) compared to Sal and PT-Sal groups (Fig. 7A). Similarly, mice exposed to PT plus U-Aβ42 showed an increased density of U-Aβ42-positive neurons within the cortex relative to PT-Sal (p < 0.01) and Sal (p < 0.0001) mice, (Fig. 7B). Brightfield IHC also showed a significantly higher number of Aβ42-positive neurons in the PT-Sal group compared to the Sal group (p < 0.001) (Fig. 7B).
Accumulation of bloodborne Aβ42 within hippocampal hilar neurons
In PT-FITC-Aβ42-treated animals, the hippocampus also showed blood vessels with prominent perivascular IgG-positive leak clouds extending out into the hippocampal parenchyma (Fig. 8A, D), which were not seen in PT-Sal mice (Fig. 8B, E) and Sal mice (Fig. 8C, F). As in the cerebral cortex, larger vessels in the hippocampus displayed the most extensive leak clouds (Fig. 8A). In mice injected with PT-FITC-Aβ42, the fate of FITC-Aβ42 in the hippocampus mirrored that in the cerebral cortex. A considerable number of the large hilar neurons exhibited intense FITC-Aβ42 fluorescence localized within discrete granules in the neuronal perikaryon, again suggesting that exogenous FITC-Aβ42 is selectively internalized by these neurons and most likely deposited within elements of the lysosomal compartment (Fig. 8D). No fluorescent signal similar to that from FITC-Aβ42 was seen in hippocampal neurons of animals PT-Sal and Sal control animals (Fig. 8E, F).
DISCUSSION
The goal of the present study was to use an aging mouse model to investigate the effects of long-term, chronic BBB compromise on the fate of exogenous bloodborne Aβ42 peptide and cognitive function. To achieve this, 10-month-old mice were treated repeatedly with Pertussis toxin (PT) to induce a chronic increase in BBB permeability over a period of three months. During the same interval, animals were also repeatedly administered purified FITC-labeled or unlabeled Aβ42 via tail vein injection. Controls were similarly injected with PT plus saline or saline only. FITC-labeled Aβ42 was used to track the fate of bloodborne Aβ42 in the brain tissue. During the last month of treatment, all mice underwent extensive behavioral testing. Animals were subjected to a wide range of learning, attentional, and long-term retention tests that relied on diverse motivational and neural systems. Following completion of behavioral testing, animals were sacrificed, and the brains removed and evaluated to determine the distribution of tail vein injected Aβ42 peptide (FITC-labeled and unlabeled) and bloodborne IgG, both of which are normally blocked from entry into the brain in animals with a functionally intact BBB.
This study had four main findings. First, repetitive PT treatments induced a chronic increase in BBB permeability, leading to an influx of plasma components into the brain parenchyma, including IgG antibodies and FITC-labeled and unlabeled Aβ42 peptide. BBB leaks were evident as IgG- and Aβ42-positive perivascular leak clouds in PT-treated mice and were most prominent in mice treated with PT plus Aβ42. Second, after entering the brain parenchyma, both IgG and Aβ42 preferentially associated with certain types of neurons, mostly pyramidal neurons in the cerebral cortex and the large hilar neurons in the hippocampus. In these neurons, internalized FITC-labeled Aβ42 was most often localized within discrete cytoplasmic granules, presumably structures associated with the neuronal lysosomal compartment. We also found that endogenous Aβ42 was detectable in neurons in PT-treated mice (PT-Saline Control), but not in mice injected with saline-only (Saline-Saline Control), suggesting that disruption of the BBB in mice is required for entry of bloodborne Aβ42 into brain. Third, chronic BBB compromise increased the density of Aβ42-positive neurons and thus total brain Aβ42 load in animals injected with Aβ42 via the tail vein. Cells containing the greatest amounts of Aβ42 were positioned either within or in the vicinity of the leak, but no Aβ42-immunopositive amyloid plaques were found in any treatment group. Fourth, with respect to cognitive functioning, animals with chronic BBB compromise and injected Aβ42 displayed a pattern of variable disruption of the acquisition of learned behaviors, a more consistent and preferential deficit in the capacity for long-term retention, and increased susceptibility to interference in selective attention. Taken together, these results indicate that chronic disruption of the BBB and the resulting influx of exogenous Aβ42 and neuron-binding IgG antibodies (autoantibodies) from the blood into the brain in mice can mimic pathological events associated with early AD-related pathology in humans as well as cause comparable early changes in behavior and cognition. In addition, our data supports the notion that blood is a likely source of exogenous Aβ42 peptide that eventually deposits in the brains of people afflicted with AD, with autoantibody-induced endocytosis as a route of Aβ42 entry into neurons.
BBB compromise and AD
Results of the present study are in accord with previous work suggesting that cerebrovascular changes are key contributors to early AD pathogenesis and subsequent disease progression [31, 85–93]. Studies on humans and animal models have suggested that long-term, functional compromise of the BBB may contribute to initiation and/or progression of AD pathology, although the exact nature of its contribution is yet to be defined [30, 85–93]. Despite this, it is reasonable to expect that long-term increase in BBB permeability result in a chronic, gradually escalating disruption of brain homeostasis due to the influx of plasma components, including immunoglobulins (Igs) and Aβ peptides, into the brain [30, 92]. For obvious reasons, studies on the causal mechanisms of BBB compromise and long-term consequences in humans have been limited, necessitating the use of animal models. A missing feature that weakens the utility of mouse models for studies on the potential role of BBB compromise in AD pathogenesis is that, unlike in humans, mice lack comparable aging-associated changes in the structural/functional integrity of brain blood vessels, including those thought to lead to chronically increased BBB permeability in humans, and apparently do not live long enough to allow the full extent of AD-related pathological changes to evolve. In a previous study, we examined the fate of FITC-labeled Aβ42 that was introduced into mice via a single bolus tail vein injection, with the BBB rendered permeable by a single treatment with PT [31]. This study showed that FITC-Aβ42 readily penetrated the BBB and was localized in neurons. To test the requirement for disruption of the BBB to allow the influx of bloodborne FITC-Aβ42 into the brain tissue, mice were subjected to saline injections in place of PT prior to administration of FITC-Aβ42 by tail vein injection. Results showed that FITC-Aβ42 was detected in the brain but was confined to the lumen of blood vessels, and neurons throughout the brain were devoid of FITC-Aβ42 [31]. In the present study, we induced a chronic, increase in BBB permeability in mice via repetitive PT injections over a period of three-months, an approach confirmed to be successful by demonstrating a resulting influx of bloodborne IgG and FITC-labeled and unlabeled Aβ42 into the brain parenchyma. Although PT has been widely used in developing various animal models with impaired BBB, details on how PT mediates the observed increase in BBB permeability remains poorly understood [94]. The observation of rather sharp interfaces between IgG- and Aβ42-positive and -negative brain regions in these mice indicated that the site and extent of BBB compromise was not uniform in the cerebral cortex or hippocampus. A similar phenomenon has been reported in postmortem AD brains [30, 31], providing a plausible explanation for the often unpredictable pattern of symptoms that emerge early in the course of the disease, which later become more generalized as the disease progresses.
Bloodborne IgG and Aβ42 preferentially associate with pyramidal neurons
The present study shows that a chronic BBB compromise allows bloodborne IgG antibodies and FITC-labeled Aβ42 to gain access to neurons and glial within the brain parenchyma, suggesting that the blood can serve as a major source of the Aβ42 peptide that accumulates in AD brains under conditions of increased BBB permeability. Indeed, several studies have suggested that the blood can serve as a source of Aβ peptides that deposit in the brain of AD patients [30, 95–98]. Here, we show that, after entering into the brain parenchyma, bloodborne IgG and FITC-labeled Aβ42 introduced via tail vein injection preferentially associate with neurons, most notably pyramidal neurons in the cerebral cortex and large hilar neurons in the hippocampus, the same neurons that are IgG-immunopositive and have been shown to contain intracellular Aβ42 deposits in human AD brains [30, 31]. Of the three treatment groups, mice receiving PT + Aβ42 injections showed the highest number of cortical pyramidal neurons that were immunoreactive to IgG and Aβ42. Cells with the largest deposits of Aβ42 were most often positioned within a perivascular leak cloud or in the vicinity of a leaky blood vessel. In these cells, exogenous FITC-labeled Aβ42 was primarily localized within discrete granules, presumably structures associated with the neuronal lysosomal compartment. Little or no bloodborne FITC-Aβ42 entered the brains of mice with an intact BBB (saline controls), as has been reported previously [31]. The above findings suggest a causal relationship between chronic BBB breakdown, the resulting influx of soluble Aβ42 and IgG into the brain tissue, the selective association and internalization of Aβ42 in neurons and the pathogenesis of AD.
Evidence from a number of studies demonstrates that a substantial amount of Aβ42 accumulates intracellularly in neurons of AD brains and in elderly mutant transgenic mice overexpressing AD-relevant genes [15, 30–51]. Studies on AD transgenic mice have revealed that intraneuronal Aβ42 accumulation invariably precedes the appearance of plaques and temporally coincides with loss of synapses, the appearance of initial cognitive deficits and, in a triple transgenic mouse model characterized by overexpression of mutant tau, AβPP, and presenilin (i.e., 3xTg-AD), the development of neurofibrillary tangles [15, 99]. Collectively, this suggests that intraneuronal Aβ42 accumulation is an early event in a disease process that eventually leads, either directly or indirectly, to neuronal and synaptic loss, the appearance of amyloid plaques and neurofibrillary tangles and associated neurological sequalae [17, 100–102]. Based on this, it has been proposed that the gradual deposition of Aβ42 within vulnerable neurons might progressively impair the ability of these cells to structurally and functionally support their extensive dendrite arbors and synapses, thus precipitating a gradual dendrite collapse that ultimately leads to deterioration of cognition and memory [30, 34]. Nevertheless, very little has been conclusively ascertained thus far concerning how and why Aβ42 accumulates selectively in certain types of neurons, why aging predisposes these neurons to do so, and how this accumulation might induce its potential effects on neuron structure and function.
Possible relationship between the influx of brain-reactive antibodies and intraneuronal accumulation of Aβ42
The present study shows that the same neuronal subtypes in mouse brain that exhibit intracellular deposits of Aβ42 are also IgG-immunopositive, raising the possibility that the selective binding of IgG antibodies to the surfaces of these cells may somehow facilitate Aβ42 internalization and accumulation [30–32]. In a previous study, we tested the effects of human sera on the extent of neuronal IgG binding and intraneuronal deposition of soluble Aβ42 in adult mouse neurons in organotypic brain slice cultures [30]. This study showed that binding of human serum IgG to the surfaces of mouse neurons was accompanied by a dramatic increase the rate and extent of Aβ42 (both exogenous and endogenous) accumulation in neurons of the cerebral cortex and hippocampal region. Furthermore, replacement of human sera with specific antibodies targeting abundant neuronal surface proteins (e.g., the alpha7 nicotinic acetylcholine and glutamate R2 receptors) resulted in a comparable dramatic enhancement of exogenous Aβ42 internalization and accumulation in mouse cortical and hippocampal neurons, compared to neurons treated with Aβ42 alone [30, 103]. Taken together with the results of the present study, this suggests that increased BBB permeability allows bloodborne, brain-reactive IgG autoantibodies to access the brain interstitium and bind selectively to cognate targets on the surfaces of certain neuronal subtypes, which enhances intraneuronal deposition and accumulation of exogenous Aβ42 peptide, presumably via the natural tendency of neurons to clear surface-bound antibodies via endocytosis [19, 103].
Chronic, long-term BBB compromise and treatment with Aβ42 peptide caused deficits in cognitive functioning
With respect to cognitive functioning, animals subjected to PT-induced, long-term BBB compromise and repeated injections of unlabeled or FITC-labeled Aβ42 displayed a pattern of variable disruption of the acquisition of learned behaviors, a more consistent and preferential deficit in the capacity for long-term retention, and an increased susceptibility to interference in selective attention. We used a battery of behavioral and cognitive tests aimed at assessing animals’ learning (acquisition) and retention (long-term memory). Except for the odor discrimination task (where significant learning deficits were observed), experimental manipulations such as the PT-induced increase in BBB permeability and injection of Aβ42 peptide had only a small and inconsistent effect on initial learning. However, mice in the PT-Aβ42 group demonstrated significant deficits in long-term retention as revealed by the water maze and Lashley Maze tests. The specificity of the latter effect was particularly salient in light of intact acquisition on these same tasks.
In a test of selective attention that made no explicit demands (at the time of testing) on either learning or long-term retention, mice in the PT-Aβ42 group expressed profound deficits. Selective attention evidenced a pattern of performance consistent with increased susceptibility to interference, manifested in specific impairment on later test trials, a segment most sensitive to accrual of interference. Since attentional abilities broadly influence cognitive processing [104, 105] this suggests that BBB breakdown and exogenous Aβ42 may influence general cognitive abilities that extend beyond the tasks reported here.
The aggregate pattern of functional repercussions of experimental manipulation of BBB integrity and plasma Aβ42 levels is particularly illuminating in view of implications for AD pathogenesis. Taken together, this profile is most consistent with a prodromal or emergent phase of AD [i.e., subtle cognitive impairment/mild cognitive impairment (MCI)], both with respect to absolute degree and distribution of impairment across domains of cognition or behavior. Individuals suffering from the amnestic variant of MCI, a subtype of MCI generally considered a transitional state for AD, typically display a specific profile that includes preferential deficits in long-term retention as compared to initial acquisition, coupled with variability in other domains of cognitive functioning. Behavioral disturbance is also common, including mild to moderate neuropsychiatric problems and mood changes such as anxiety, irritability, and apathy [106, 107]. Furthermore, the variable pattern of cognitive and behavioral deficits demonstrated here mirrors the unpredictable behavioral presentation and lack of consistent characterization of cognitive impairment associated with MCI. However, the most compelling parallels concern the specific profile of cognitive and behavioral impairment exhibited by animals treated with PT-Aβ42, most prominently including a preferential deficit in long-term retention with respect to acquisition capacity, in addition to changes in selective attention. Remarkably, the pattern of results in the present model dovetails very closely with the staging observed in longitudinal studies assessing the progression of pathology in the 3xTg-AD model, a model which surpasses the present model in progression of pathology [36, 108]. In this case, studies have demonstrated that the emergence of a specific and preferential deficit in long-term retention (i.e., with respect to acquisition as well as other cognitive functions) coincides temporally with the initial appearance of intracellular accumulation of Aβ42, and both can be traced to initial phases of pathological progression, corresponding to early disease stages (i.e., MCI).
Limitations of the study
This study has some inherent limitations. First is the usage of PT for compromising cerebrovasculature and BBB. Though used extensively for this purpose, PT is a toxin that can lead to inflammatory changes. Secondly, this model does not show AD-associated terminal pathological changes such as amyloid plaques, neurofibrillary tangles, and significant neuronal loss, all of which may require longer study intervals. Another limitation concerns the age of the mice. Given the consistent and robust link between advanced age and development of AD (i.e., in sporadic, non-hereditary variants of AD), this is a core feature of AD that is likely to be important in attempts to replicate pathology and determine causal mechanisms. While the age of the mice used in the present model is sufficiently advanced to allow activation of some AD-related pathological processes, their relative youth may have attenuated key mechanisms implicated in AD pathogenesis and thus curbed emergence of more dramatic pathology. Such flaws are endemic to all mouse models that attempt to translate from rodent to human pathology. Lastly, these animals lack the larger arteries found in humans, blood vessels that are most associated with susceptibility to BBB dysfunction and ensuing vascular leak given their increased hydrostatic pressure. Nevertheless, despite these potential critiques, this model is comparable to other animal models in the reproduction of early pathological and behavioral alterations associated with AD, with the advantage of exhibiting pathogenic mechanisms in the absence of forced expression of various AD-relevant genes and their gene products.
Conclusions
Results presented here indicate that plasma derived Aβ42 peptide and anti-neuronal autoantibodies can penetrate a chronically defective BBB, bind selectively to the surfaces of certain types of neurons and accumulate within these cells in the characteristic pattern of AD. These findings demonstrate that chronic BBB compromise and the ensuing influx of exogenous Aβ42 and anti-neuronal autoantibodies from the blood over time can initiate or at least contribute to a cascade of neurological events and associated cognitive and behavioral changes that resemble those apparent in emerging AD. Taken in the context of research indicating widespread BBB deficiency in AD brains, these findings provide compelling evidence that the blood may serve as a chronic source of Aβ42 peptides as well as neuron-binding autoantibodies in AD as they gain access to the brain tissue through a compromised BBB, and that the influx of soluble, exogenous Aβ42 into the brain may be the origin of the intracellular Aβ42 that accumulates within neurons early in AD pathogenesis.
AUTHOR CONTRIBUTIONS
Robert G Nagele (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Writing – original draft; Writing – review & editing); Nimish K. Acharya (Formal analysis; Funding acquisition; Investigation; Methodology; Visualization; Writing – original draft; Writing – review & editing); Henya C. Grossman (Data curation); Peter M. Clifford (Data curation; Investigation; Methodology); Eli C. Levin (Data curation; Methodology); Kenneth R. Light (Data curation; Methodology); Hana Choi (Methodology); Randel L. Swanson (Formal analysis); Mary C. Kosciuk (Data curation; Methodology; Writing – review & editing); Venkat Venkataraman (Formal analysis; Investigation; Methodology); David J. Libon (Conceptualization; Writing – review & editing); Louis D. Matzel (Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
This manuscript includes a portion of the PhD thesis/dissertation work completed by Henya C Grossman (mentors, L Matzel and R Nagele) and Kenneth R Light (mentor, L Matzel), all of whom are co-authors.
The contents of this work do not represent the views of the Department of the Veterans Affairs or the US Government.
FUNDING
This work was supported by funds from the Osteopathic Heritage Foundation Endowment Intramural Funding Program and Rowan School of Osteopathic Medicine (to NKA and RGN), National Institute of Aging (NIA48387-06 to LDM), Alzheimer’s Association (to RGN) and Foundation of UMDNJ (RGN). RLS was supported, in part, by the US Department of Veterans Affairs Rehabilitation Research and Development Service under award number IK2 RX003651.
CONFLICT OF INTEREST
RGN and DJL are Editorial Board Members of this journal but were not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
The authors have no other conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.