
Abstract
Background:
Traumatic brain injury (TBI) has been linked to multiple pathophysiological processes that could increase risk for Alzheimer’s disease and related dementias (ADRD). However, the impact of prior TBI on blood biomarkers for ADRD remains unknown.
Objective:
Using cross-sectional data, we assessed whether a history of TBI influences serum biomarkers in a diverse cohort (approximately 50% Hispanic) with normal cognition, mild cognitive impairment, or dementia.
Methods:
Levels of glial fibrillary acidic protein (GFAP), neurofilament light (NFL), total tau (T-tau), and ubiquitin carboxy-terminal hydrolase-L1 (UCHL1) were measured for participants across the cognitive spectrum. Participants were categorized based on presence and absence of a history of TBI with loss of consciousness, and study samples were derived through case-control matching. Multivariable general linear models compared concentrations of biomarkers in relation to a history of TBI and smoothing splines modelled biomarkers non-linearly in the cognitively impaired groups as a function of time since symptom onset.
Results:
Each biomarker was higher across stages of cognitive impairment, characterized by clinical diagnosis and Mini-Mental State Examination performance, but these associations were not influenced by a history of TBI. However, modelling biomarkers in relation to duration of cognitive symptoms for ADRD showed differences by history of TBI, with only GFAP and UCHL1 being elevated.
Conclusions:
Serum GFAP, NFL, T-tau, and UCHL1 were higher across stages of cognitive impairment in this diverse clinical cohort, regardless of TBI history, though longitudinal investigation of the timing, order, and trajectory of the biomarkers in relation to prior TBI is warranted.
INTRODUCTION
Traumatic brain injury (TBI) has been identified as a risk factor for Alzheimer’s disease and related dementias (ADRD). For instance, a nationwide Danish study involving 2.8 million people found that the risk of ADRD was increased in those with prior TBI, increasing with a higher number of injuries [1]. Though the risk is relatively small (0.6% increased incidence) and there are mixed findings in the literature [2, 3], several other studies have also found an association between TBI history and later development of dementia [4–6]. Importantly, the timing and severity of injury may be moderating factors [7]. The onset of ADRD, and not just calculated risk, also appears to be influenced by a history of TBI, as individuals with a prior TBI have been found to present with earlier onset of cognitive symptoms in the context of mild cognitive impairment as well as dementia [8]. However, the underlying mechanisms for TBI being a risk factor for an earlier onset of ADRD are unclear and represent a critical area of research.
Non-human animal studies have identified that TBI in transgenic mice leads to increased deposition of tau pathology, axonopathy, neuroinflammation, and neurodegeneration as soon as 2 weeks after the event [9, 10]. TBI has also been linked with greater cognitive deficits in mice, including slowed learning and progressive behavioral impairment in some mouse models [11, 12]. In clinical studies with humans, glial fibrillary acidic protein (GFAP), neurofilament light (NFL), ubiquitin carboxy-terminal hydrolase-L1 (UCHL1), and total tau (T-tau) have all been found to be increased in cerebrospinal fluid as well as blood in the acute stage after TBI compared to control individuals [13–15]. Although the reasons for these markers being detected in biofluids such as blood remain to be identified, the current interpretation is that these are soluble markers of neuronal (T-tau, UCHL1), axonal (NFL), and astrocyte injury (GFAP) [16–18]. Along these lines, an increase of all four markers after TBI correlates with severity of injury and poorer clinical outcomes [19–22], and T-tau and NFL specifically have also shown to be related to cortical and/or white matter neurodegeneration months after injury in moderate-severe TBI [13, 23]. In contrast to mice models, there has been normalization of each of the markers (GFAP, NFL, T-tau, and UCHL1) over time (within a year) in most of the longitudinal human studies, and only a small subset of individuals appear to develop cognitive decline long after injury [8]. NFL, however, has been reported to remain elevated in blood for years after injury in a proportion of individuals with moderate-severe TBI [13, 19]. All four markers have also been shown to increase in neurodegenerative diseases, including ADRD [24, 25]. Thus, it has been posited that the neuronal, axonal, and astrocyte injury induced by TBI might predispose individuals to have a higher level of pathophysiologic markers and risk for ADRD.
Most studies that have informed the pathophysiology of ADRD and associated biomarkers have been largely composed of non-Hispanic White participants [26] despite dementia rates being considerably lower compared to persons from Black or Hispanic backgrounds [27]. An underrepresentation of persons from minority backgrounds in dementia research is problematic, as it limits the potential generalizability of biomarker findings for diagnostic, prognostic, and/or treatment purposes across diverse populations, particularly in those with higher risk for ADRD [28, 29]. Along these lines, Hispanic individuals carrying an apolipoprotein ɛ4 (APOE4) allele, the largest genetic risk factor identified for developing ADRD, showed lower Alzheimer’s pathologic changes on position emission topography compared to White individuals [30]. Weaker associations between APOE4 and risk for cognitive decline have also been found for Hispanic individuals [31, 32]. Taken together, there may be differences in the level of biomarkers for common syndromes among different racial and ethnic groups due to medical comorbidities and prevalence of exposure to risk factors. Pathways implicated in ADRD, including alterations in blood brain barrier integrity, axonal myelination, synaptic architecture, lipid metabolism, mitochondrial processes, and inflammatory reactions have been shown to be modulated by social determinants of health, cardiovascular illness, and TBI, among others [33, 34].
While studies have begun to examine ADRD biomarker differences within more diverse groups, including Hispanic individuals [35], such studies have not yet examined the role TBI might play in the accumulation and presence of pathophysiologic markers. Recently, cerebrospinal fluid markers of amyloid and tau deposition as well as cognitive function were compared in well-matched samples of patients with Alzheimer’s type dementia having no history of TBI and those having a prior mild TBI [36]. Cognition and amyloid deposition did not differ between the groups, but there was a link between higher total tau (T-tau) in cerebrospinal fluid among individuals having a history of TBI, in whom most had a single injury and were on average nearly 3 decades out from injury. However, similar to prior biomarker studies, the samples were comprised mostly of non-Hispanic White participants. Furthermore, there have been no studies to our knowledge on the influence of a history of TBI on biomarkers within blood along the cognitive spectrum of normal cognition (NC), mild cognitive impairment (MCI), and dementia. In light of these gaps, there is a pressing need for research that examines TBI-related effects on biomarkers within diverse communities to better comprehend whether a link between TBI and ADRD involves overlapping or unique processes to predispose some individuals after TBI to dementia later in life. The current study leveraged the Texas Alzheimer’s Research and Care Consortium (TARCC), which is a multicenter project involving 10 institutions across Texas that have enrolled a large cohort of participants identifying as Hispanic as well as non-Hispanic ethnicity for aging and neurodegeneration research [37]. As a result, TARCC provides an opportunity to examine whether a history of TBI influences the concentrations of blood-derived T-Tau, GFAP, NFL, and UCHL1 across the cognitive spectrum in a diverse cohort.
MATERIALS AND METHODS
Dataset
TARCC collects sociodemographic, medical, and clinical data, in addition to blood from participants during in-person visits across multiple sites. Participants are diagnosed as having NC, MCI, or dementia per established clinical criteria [38, 39] during a consensus conference in which TARCC clinicians review information from a clinical interview, neurological exam, and neuropsychological testing. For individuals diagnosed with MCI and dementia, a suspected etiology is determined by consensus, and the timing of when cognitive decline began is estimated by a study partner and/or TARCC clinician. Data obtained for this study were from participants aged 50+ across the cognitive spectrum (NC, MCI, dementia) who had serum stored from their initial TARCC visit. Mini-Mental State Examination (MMSE) scores were obtained as a global measure of cognitive staging and classified as being Normal to Mildly Low (scores = 30–28), Moderately Low (scores = 27–24), and Severely Low (scores <24) for analyses similar to prior work [40, 41].
Biomarker collection and processing
At the baseline visit, blood was collected, processed, and stored in accordance with established guidelines [42]. Briefly, blood was collected by venipuncture in the morning after an overnight fast. Serum tubes were allowed to clot for 30–60 min and were then centrifuged for 10 min at room temperature. Serum was aliquoted into polypropylene tubes and stored in –80°C freezers within 2 h of collection. Apolipoprotein E (APOE) genotyping was completed using PCR as previously described [37]. Serum levels of GFAP, NFL, T-Tau, and UCHL1 were assayed using the Simoa Neurology 4-Plex Kit on a Simoa HD-1 Analyzer (Quanterix, Lexington, MA) with previously described analytical ranges and inter-assay coefficients of variance [35]. All assays were performed at the University of Vermont Laboratory for Clinical Biochemistry using a single batch of reagents by a certified laboratory-technician blinded to clinical and demographic information.
Study cohort
TARCC participants, in combination with a study partner, were asked three questions related to TBI involving a) whether subjects had ever sustained a TBI resulting in <5 min loss of consciousness (mLOC), b) ≥5 mLOC, and c) whether they were left with a chronic deficit. Each question is coded as absent, recent/active (defined as occurring within 1 year of visit or currently requiring treatment), remote/inactive (defined as occurring >1 year of visit and either having recovered from the injury or no treatment is currently underway), or unknown. Participants reporting a history of TBI leading to a chronic deficit were excluded in an attempt to examine the potential influence of TBI on serum-derived biomarkers when there was full recovery from symptoms/dysfunction. Timing of injury data were not collected, and thus, participants with or without a TBI resulting in LOC occurring >1 year prior to the initial visit (i.e., remote/inactive) were selected to characterize presence (TBI+) and absence (TBI–) of a history of remote TBI. To minimize the possibility of there being differences in serum markers between TBI+ and TBI–groups due to non-injury factors, an individual matching approach was completed to derive a control group of TBI–participants. Case-control matching with replacement was randomly performed using SPSS without knowledge of biomarker data to identify 3 TBI–controls for every 1 TBI+ case (3:1 ratio) from a cohort of over 1,884 individuals. Exact matching was completed for clinical classification (NC, MCI, dementia), sex, and APOE4 status. Age and education matching was done based on the closest age and education level to TBI+ participants, with a tolerance of±3 years for both.
Statistical analysis
All statistical tests were performed using SPSS and all figures were created in GraphPad Prism. Biomarker values were examined for outliers within each diagnostic group (NC, MCI, Dementia) and excluded if they were above or below 3 times the Median Absolute Deviation, as this is a robust method for detecting outliers due to it being resistant from distortion unlike thresholds identified from multiple standard deviations around the mean [43]. There were 15 GFAP outliers, 18 NFL outliers, 12 T-tau outliers, and 41 UCHL1 outliers that were excluded. Frequency distributions were visually inspected (using histograms), and given all of the biomarkers were skewed, each was log10 transformed. Multivariable general linear models were used to compare the mean log10 concentrations for each biomarker in relation to a history of TBI (TBI+, TBI–) and its interaction with clinical classification (NC, MCI, Dementia), MMSE scores (Normal to Mildly Low, Moderately Low, Severely Low), and APOE4 status (carrier, non-carrier). Since the differences in blood-derived biomarker levels have been small between individuals with dementia and those that are cognitively normal [44], any potential differences associated with a history of TBI with LOC were expected to be narrow. As such, cognitively impaired groups were combined (MCI + Dementia versus NC) in a follow-up analysis to maximize statistical power in assessing biomarker levels in relation to a history of TBI. Biomarker levels were transformed to z-scores using the raw (non-log) mean and standard deviation values from the NC group (TBI+ and TBI–) to place each biomarker on a standardized scale for comparisons, and linear regression was used to examine if z-scored biomarkers were linearly associated with the estimated duration of cognitive symptoms, stratified by presence or absence of a history of TBI. Since few individuals had onset of cognitive symptoms more than 10 years before data collection (TBI+ n = 2; TBI–n = 6), only individuals with a duration of symptoms between 0–10 years were examined. Restricted cubic smoothing splines modelled the z-scored biomarkers non-linearly in the combined cognitively impaired groups as a function of time since cognitive symptom onset to compare the trajectory, timing, and order of biomarker increases between TBI+ and TBI–groups. All biomarker values presented are the log10 transformed values except for those displayed in figures (distribution plots depict raw measurements for biomarkers and trajectory maps depict z-score levels).
Characteristics of subjects with and without a history of TBI
Mean log concentration values (pg/mL) of serum biomarkers
aMCI>NC (p range <0.001–0.002). bDementia > NC and MCI (p range <0.001–0.008). cDementia > NC only (p = 0.001). dModerately Low > Normal to Mildly Low scores (p range <0.001–0.03). eSeverely Low > both other score categories (p range <0.001–0.02). fAPOE4 carrier > noncarrier (p range <0.001–0.04).
Mean log concentration values of serum biomarkers by TBI status
All comparisons based on history of TBI and its interaction with clinical classification (NC, MCI, Dementia), MMSE categories, and APOE4 status were non-significant (all ps > 0.05).
RESULTS
A total of 101 TARCC participants (NC, MCI, and dementia) had a reported history of TBI with LOC occurring more than 1 year prior to sample collection, and 303 participants were matched who had an absence of a history of TBI with LOC. For cognitively impaired participants, samples were collected an average of 3.55 years after symptom onset, and 100% of participants with dementia were diagnosed with an Alzheimer’s type dementia. Approximately half of the participants were female (48%) and a similar proportion identified as Hispanic (49%). The mean age of the cohort was 69.24 years (SD = 7.89), and 38% were APOE4 carriers. The cohort had a mean education level beyond high school (13.57 years; SD = 3.54), more than half had hypertension (57%), and 24% had diabetes. Demographic and clinical characteristics of the TBI+ and TBI–groups are displayed in Table 1. No significant group differences were seen based on age, education, sex, Hispanic ethnicity, APOE4 status, duration of cognitive symptoms, blood pressure, body mass index, presence of diabetes, or hypertension (all ps > 0.05), indicating that the two groups are well-matched on many factors that could influence biomarker concentrations.
Mean concentration values for biomarkers are listed in Tables 2 and 3. Serum GFAP, NFL, T-tau, and UCHL1 significantly differed across clinical diagnoses, with concentrations being greater for the dementia group relative to those with MCI (with the exception of UCHL1), and higher for both MCI and dementia groups relative to the NC group (Fig. 1). Similarly, GFAP, NFL, and T-tau were significantly elevated with poorer performances on the MMSE, as Severely Low scores were higher than Moderately Low scores and both were higher than Normal to Mildly Low scores. Whereas UCHL1 was significantly greater in those with Severely Low MMSE scores compared to higher scores, UCHL1 concentrations did not significantly differ between the other score categories. Also, serum GFAP, NFL, and T-tau were significantly higher among APOE4 carriers compared to non-carriers, but serum UCHL1 was not. The concentrations of GFAP, NFL, T-tau, and UCHL1 did not significantly differ between TBI+ and TBI–groups, and no significant associations were seen based on a history of TBI according to clinical diagnoses, MMSE scores, or APOE4 carrier status (Fig. 2). Also, when the cognitively impaired groups were combined, all serum biomarker comparisons remained non-significant between TBI+ and TBI–individuals.
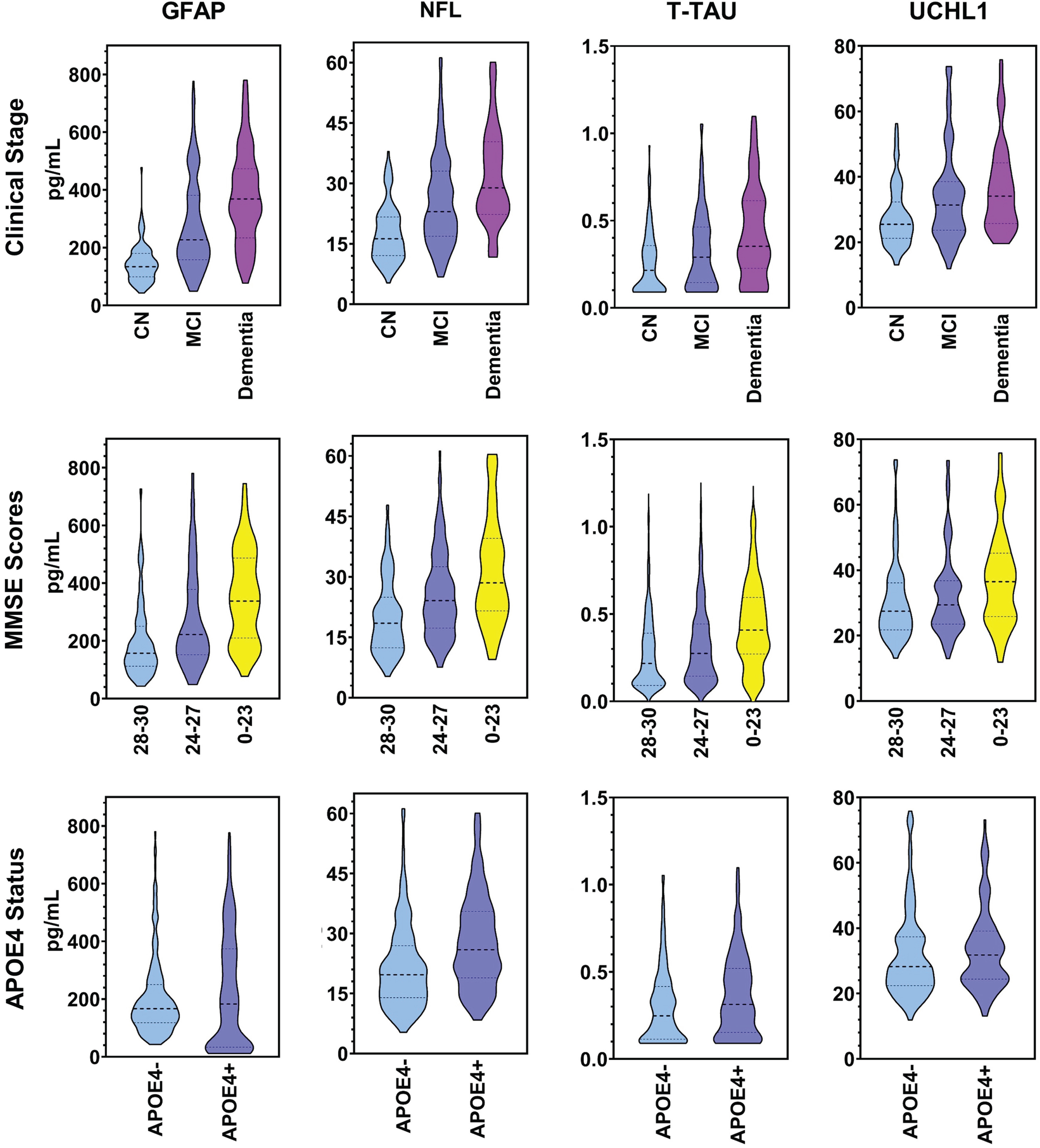
Serum biomarkers by clinical stages, MMSE, and APOE4 status. Dashed lines represent the median value. Dotted lines represent the first and third quartile values. NC, cognitively normal; MCI, mild cognitive impairment; APOE4, apolipoprotein ɛ4.
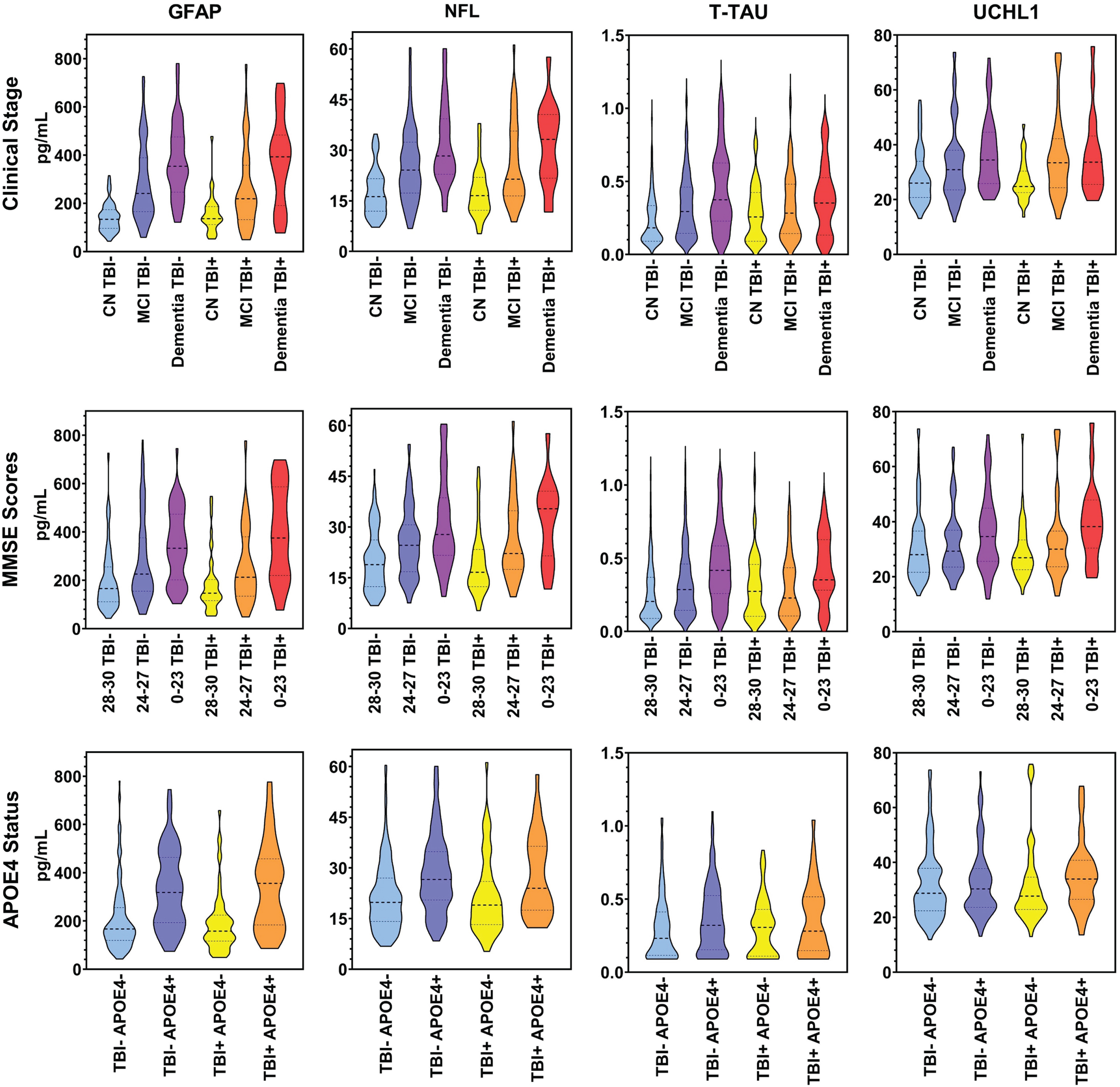
Serum biomarkers by history of TBI status. Dashed lines represent the median value. Dotted lines represent the first and third quartile values. NC, cognitively normal; MCI, mild cognitive impairment; APOE4, apolipoprotein ɛ4.
Duration of cognitive symptoms for MCI/dementia was linearly associated with serum T-Tau only in TBI–cognitively impaired individuals (beta = 0.32; p = 0.04), with every increase of almost a half a standard deviation (z-score) being related to a one-year increase in symptom duration. Unexpectedly, no biomarkers were linearly associated with the duration of cognitive symptoms in the TBI+ group. Figure 3 displays the trajectory of each serum biomarker as a function of time since cognitive symptom onset for the groups. For the TBI–group, serum GFAP showed an early modest elevation followed by NFL and then both plateaued around the point T-Tau increased, despite T-Tau being flat in the early phase of symptom duration. UCHL1 was flat for the TBI–group across the duration of cognitive symptoms. For the TBI+ group, in contrast, GFAP and UCHL1 had early modest elevations followed by a drop, and then marked increases at later durations more than twice the levels seen in the TBI–group. NFL showed the reverse trajectory of GFAP and UCHL1, while T-Tau exhibited a flat curve across for the TBI+ group.
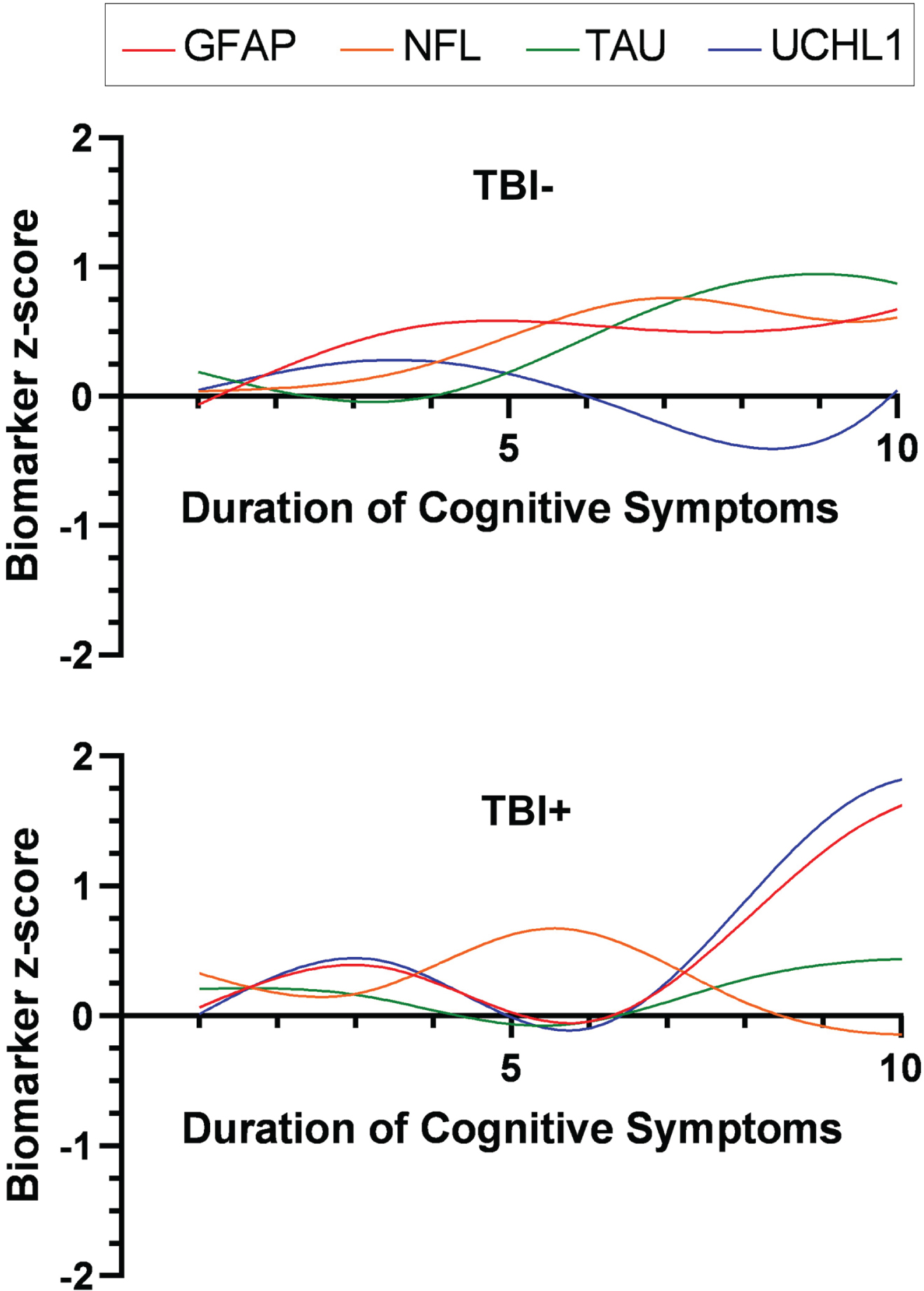
Model of serum biomarker trajectories across cognitive stages of ADRD based on history of TBI status. Duration of cognitive symptoms since ADRD onset was a proxy to represent progression as a function of time. TBI–, no history of traumatic brain injury with loss of consciousness; TBI+, a history of traumatic brain injury with loss of consciousness.
DISCUSSION
GFAP, NFL, T-tau, and UCHL1 are considered to reflect different yet related processes in the pathophysiological cascade of ADRD. Tau is mainly expressed in neuronal axons and is a multifaceted protein that not only promotes the polymerization of microtubules during axonal growth but also has implications in regulating cell signaling, molecular trafficking, neuronal excitability, and synaptic functions [45–48]. Whereas the accumulation of tau may differ between TBI and distinct neurodegenerative conditions, it is thought that a potential pathway that might overlap both may relate to neuroinflammatory/infectious agents and/or degeneration of acetylcholinergic neuronal pathways [49]. Tau pathology in ADRD, nonetheless, results in prominent cytotoxicity downstream including mitochondrial dysfunction, synaptic impairment, impaired cellular transport mechanisms, defective protein degradation, and neuronal death. Similarly, the NFL protein chain is a major component of neuronal axons, but with the highest concentrations in long myelinated axons of the cerebral deep white matter. NFL serves to maintain axonal stability via interactions with organelles such as microtubules and mitochondria and are critical for radial growth and effective nerve conduction [50–53]. NFL levels in biofluids increase 2.5-fold between age 20 and 50 years in cerebrospinal fluid and by 2.2% per year between ages 18 and 70 years [54, 55], likely from age-related structural damage and metabolic changes of protein turnover. However, pathological increases in biofluids such as serum and cerebrospinal fluid in TBI and neurodegenerative syndromes are well documented, indicating loss of axonal integrity [56, 57]. GFAP is an intermediate filament that stabilizes the astrocytic cytoskeleton, and as a result, has implications in metabolic support to neurons, control of tissue homeostasis, recovery of neurotransmitters, and maintenance of synapses [58–60]. In pathological states such as TBI and ADRD, both astroglial atrophy and astrogliosis (i.e., proliferation of astrocytes) are observed, in which astrocytes become reactive and release cytokines, reactive oxygen species, and other neuroinflammatory mediators [59, 61]. A number of studies have revealed that reactive astrocytes can precede hallmarks of AD pathology (i.e., amyloid plaques and NFTs), and may thus represent an early sign of brain injury [61]. In addition, pathophysiological lesions have been shown to activate astrocytes and stimulate abnormal astrocytic signaling that may have toxic effects, thus creating a vicious circle. Ubiquitin C-terminal hydrolase-1 (UCHL1) is a protein with abundant expression in neurons, and as a deubiquitinating or hydrolase enzyme its function is to remove misfolded, excessive, and oxidized proteins via the ubiquitin-proteasome pathway [62]. The structure and function of UCHL1 can be altered by several factors such as lipid species and post-translational modifications that occur in the setting of oxidative stress and pro-inflammatory states such as brain injury and ADRD [63]. The sequence of mechanistic events leading to the release of these soluble markers into the blood circulatory system is unclear, but a popular concept is that T-tau, UCHL1, NFL, and GFAP in biofluids are considered markers of neuronal, axonal, and astrocyte injury. However, some view GFAP as a marker of reactive astrocytosis instead given its role [64], and the connection UCHL1 has as a marker is less clear as there have been mixed reports on the direction of expression in ADRD (over- versus under-expressed) [65–67].
Four serum markers that overlap the neurobiological cascades for both TBI and ADRD were investigated in a well-matched, clinically-characterized, diverse cohort to inform if the presence of these markers across the cognitive spectrum may relate to a history of TBI. While GFAP, NFL, T-tau, and UCHL1 were higher across the stages of cognitive impairment, characterized by clinical diagnosis and MMSE performance, a history of TBI was not associated with differences in concentrations of the markers. There have been limited studies on the influence of a history of TBI on the presence of ADRD biomarkers later-in-life within blood [68, 69], with slightly different results due to markers measured. For instance, GFAP, NFL, and T-tau (along with amyloid-beta markers 40 and 42) in older adults with a remote mild TBI were not significantly different from similarly aged adults with no history of mild TBI [69]. Whereas GFAP and NFL also did not differ between cognitively impaired older adults with a history of any TBI and those having cognitive impairment without a TBI history in another study [68], phosphorylated tau levels were significantly elevated for the TBI group. However, it is important to note that cognitive impairment resulting from the TBI was not isolated from deficits associated with a neurodegenerative disorder in that study. As such, to our knowledge, the current study is the first to examine blood markers within a TBI cohort without persistent injury-induced deficits along the cognitive continuum of ADRD.
The results from our study corroborate growing research on the viability of blood-based biomarkers being associated with cognitive impairment [35, 70–72]. In our sample, all four biomarkers examined, GFAP, NFL, T-tau, and UCHL1, were higher among individuals with cognitive impairment and associated with poorer cognitive function. As the impetus grows to incorporate blood-based markers in primary and tertiary clinics as well as for screening for inclusion in clinical trials, there is an urgent need to validate their performance in diverse cohorts, reflective of the broader population [73, 74]. Our community-dwelling cohort had approximately 50% Hispanic representation— a demographic that will demonstrate tremendous growth in the oldest age groups in the coming years [28]. Since Hispanic adults tend to be diagnosed later or more frequently misdiagnosed than non-Hispanic Whites [75], validation of biological disease markers may in the long run translate to better identification and treatment in those with higher risk for ADRD. Several studies have shown that demographic and medical factors may affect blood-based biomarker levels [76–78], and while TBI is prevalent, it is understudied in ADRD research [79]. Because there were no significant differences in the biomarker values based on a history of TBI with advancing ADRD clinical staging, the results might suggest that these biomarkers have similar discriminability for cognitive impairment regardless of remote TBI history, which has important implications for the feasibility of blood-based biomarker implementation in real-world settings.
The APOE4 allele is linked to defective cholesterol transport and lipid metabolism, and many studies have shown an association with accumulation of amyloid beta and tau pathology in AD, along with multiple other pathophysiological processes [80]. Nearly all of the markers examined (GFAP, NFL, T-tau) were higher among APOE4 carriers relative to non-carriers, which is not surprising and in alignment with previous research indicating an early vulnerability of those with the allele for ADRD pathophysiological cascades [81]. However, there were no additive or synergistic effects between a history of TBI and APOE4 for any of the markers in serum. Moreover, the lack of association between APOE4 and UCHL1 elevation was unexpected, and could suggest that APOE4 does not affect protein clearance mechanisms through UCHL1. Interestingly, to our knowledge, this is the first study to report on the association between APOE4 status and UCHL-1. Our data indicate that long-term neurodegeneration in APOE4 carriers could be independent of UCHL1 mediated deubiquitination, albeit detailed mechanistic studies will be required to test this hypothesis.
Although no serum marker was elevated based on a history of TBI in group comparisons, an association might be more apparent when examining how biomarkers relate to the timing and course of cognitive decline. A potential reason for such might relate to there being narrow differences in the broad literature between groups having normal cognition and dementia on blood-based biomarkers [44], which are the two extremes on the cognitive spectrum. Thus, it is reasonable to assume that any potential effect associated with a history of TBI would be constricted within such a small window, which would make it more challenging than cerebrospinal fluid to identify when comparing well-matched clinical groups (e.g., TBI+ MCI versus TBI–MCI). This could explain why no relationship was identified in this study, when previously, an association was detected with some ADRD markers derived from cerebrospinal fluid [36]. Nonetheless, those findings still await replication to confirm a link between remote mild TBI and levels of tau in those having cognitive impairment.
Turning to how the biomarkers related to timing of cognitive decline, only serum T-tau was linearly related to duration of ADRD symptoms among TBI–individuals with cognitive impairment, which follows multiple reports of tau being tied to the timing as well as rate of cognitive decline in ADRD [82, 83]. Failing to see any biomarker have a linear association with duration of ADRD symptoms in the TBI+ group was unexpected. One possible hypothesis is that a history of TBI may be related to differences in the trajectory, timing, and/or order of one or more pathophysiologic mechanisms underlying ADRD. While longitudinal data is ideal for exploring trajectories, modelling the trajectories of each biomarker across the cognitive stages of ADRD using duration of symptoms as a proxy for progression over time provides a lens for examining this idea. GFAP, UCHL1, and T-tau displayed different trajectories between the TBI+ and TBI–groups. GFAP and UCHL1 showed earlier elevations with onset of symptoms for the TBI+ group relative to those that were TBI–, which peaked and decreased prior to elevating to twice the levels of the TBI-group at longer durations. Furthermore, T-tau remained flat for TBI+ individuals, unlike for TBI–persons. The modelled trajectory of GFAP and UCHL1 could be posited to reflect a significant activation of protective inflammatory and cell recycling processes early on to defend against ADRD pathophysiologic processes, but with progression, the protective role may diminish and eventually become dysregulated, increasing to substantial levels that induce toxic effects. Similar trajectories and effects have been previously described in ADRD [84], but a rationale for why this may be altered or influenced by a history of TBI is lacking. One theory is that given GFAP and UCHL1 increase in the acute period after TBI and then normalize over time, the processes tied to both markers may prime such individuals to have a slightly magnified response later-in-life to ADRD pathogenesis. However, the current results are preliminary and need to be explored through longitudinal investigation.
This study has several strengths, including being the first of its kind to examine the influence of a history of TBI on blood-based biomarkers across the cognitive continuum of ADRD as well as within a diverse cohort that is often underrepresented in biomarker research. However, the study is not without limitations. As with many aging/dementia databases, TBI information recorded in the TARCC dataset was limited. As a result, biomarker levels could not be examined in relation to TBI severity, multiple injuries, or timing. Thus, potential links specifically with moderate-to-severe TBI and mild TBI could not be examined. Moreover, TBI details and the estimation of duration of cognitive decline were collected from retrospective self or informant-report methods, which has been relied on for most aging and neurodegeneration studies, allowing potential for recall bias that may limit accuracy in reporting. TARCC participants, while clinically well-characterized, lack biomarker information regarding amyloid and phosphorylated tau to biotype individuals using the amyloid, tau, and neurodegeneration (ATN) research framework [85]. As such, it is uncertain how many in the sample would have evidence of underlying AD pathology, thereby potentially limiting generalization of the findings to the neurobiology of this condition specifically. A final limitation is that the biomarker trajectories were modelled from cross-sectional data rather than longitudinal changes over time.
Serum GFAP, NFL, T-tau, and UCHL1 were higher across each stage and level of cognitive impairment in this diverse cohort comprised of nearly 50% of individuals identifying as Hispanic. Furthermore, each biomarker except UCHL1 was elevated in carriers of APOE4, which has not been reported on to our knowledge and indicates that the functional role of UCHL1 in ADRD is separate from APOE4 and merits further research to elucidate how and why these are distinct. The concentrations of GFAP, NFL, T-tau, and UCHL1 in serum did not significantly differ based on having a history of TBI, suggesting their association with cognitive impairment may not vary based on this prevalent risk factor. However, future studies with larger samples, longitudinal measurements, and more detailed inventories of TBI history will be necessary to investigate this further.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
This work was supported by a Texas Alzheimer’s Research and Care Consortium grant [2018-28-81-JI] and also developed under grants from the Alzheimer’s Association [2019-AARG643558], US Army Medical Research and Development Program [W81XWH-20-1-0493], and the National Institute of Aging [1K23AG075261-01A1, R01AG077472].
CONFLICT OF INTEREST
Dr. Cullum serves as the Scientific Director of the TARCC. Dr. LoBue is an Editorial Board member of this journal but was not involved in the peer review process nor had access to any information regarding its peer review.