
Abstract
Using APP/PS1 mice that overproduce amyloid-β (Aβ) peptides, we investigated whether intranasal infection with a neurovirulent clinical strain of herpes simplex virus 1 (HSV-1) before Aβ deposition could accelerate or increase Alzheimer’s disease-like pathology. After HSV-1 infection, APP/PS1 mice presented a similar disease as wild type animals based on body weight changes, clinical symptoms, and survival rates. The number and volume of Aβ plaques, the number of microglia, and the percentages of circulating monocyte subsets were similar in APP/PS1 mice infected or not with HSV-1. Thus, intranasal infection with HSV-1 does not alter Aβ pathology in this mouse model.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder. Early-onset AD is associated with genetic mutations in amyloid protein precursor (APP) and the γ-secretase subunits presenilin-1 and -2 (PSEN1 and PSEN2) [1]. This results in aberrant amyloid-β protein precursor processing and overproduction of amyloid-β (Aβ) peptides in the brain. Late-onset AD may be triggered by the aggregation and deposition of Aβ peptides into senile plaques, which represent the foundation of the amyloid cascade hypothesis [2]. Viral or microbial infection of the central nervous system could be involved in the etiology of AD or contribute to its progression [3]. Herpes simplex virus 1 (HSV-1) is one of the most plausible candidates based on postmortem brain studies, genome-wide association studies, epidemiological evidences, and pre-clinical studies [4]. A retrospective study also showed that treatment of HSV-infected patients with antiviral drugs reduced the risk for the development of dementia including AD [5] strengthening this hypothesis. It is proposed that periodic subacute reactivations of latent HSV-1 in the brain over the lifetime could lead to chronic neuroinflammation that could trigger the onset and/or accelerate the progression of AD [6]. Aβ peptides, produced to bind to the virus and neutralize it as a defense mechanism against the infection, could trigger the amyloid cascade [7]. Carrying the well-established risk factor for AD, apolipoprotein E (APOE) ɛ4 genotype [8], is also a major risk factor for recurrent herpes labialis [9]. An association between HSV-1 and AD is thus suggested in APOE ɛ4 carriers. It is not excluded that other host risk factors may be involved in such an association [10]. For instance, it is not known whether HSV-1 contributes to the onset and/or progression of AD in carriers of genetic mutations in APP and PSEN1, which are implicated in Aβ peptide overproduction. In this paper, we compared the effects of an intranasal infection with a neurovirulent clinical strain of HSV-1 on Aβ pathology in double transgenic APP/PS1 and wild type (WT) mice. Our results show that intranasal infection with HSV-1 does not increase Aβ pathology in APP/PS1 mice.
MATERIALS AND METHODS
Infection of mice with HSV-1
APP/PS1 mice harboring a chimeric human/mouse APP gene (APPSwe) and a mutant human PSEN1 gene (PS1-dE9) were originally purchased from The Jackson Laboratory [B6C3-Tg(APP695)3Dbo Tg(PSEN1)5Dbo/J], backcrossed and maintained in a C57BL/6J background. Three month-old male and female APP/PS1 (n = 12) and C57BL/6J (WT; n = 8) mice were infected intranasally with 750,000 plaque forming units (PFUs) of a clinical HSV-1 strain H25 [11] in 20μL of minimum essential medium (MEM) equally distributed into both nostrils. Uninfected male and female APP/PS1 (n = 10) and WT (n = 8) mice received 20μL of MEM intranasally.
Mice were weighed and monitored for clinical signs of infection including swollen forehead, modified posture, neurological symptoms (i.e., shaking movements, modified gait and convulsions) and mortality daily for 14 days [12]. Animals were sacrificed when a body weight loss ≥20% or obvious neurological signs were observed.
The Canadian Council on Animal Care guidelines were followed for mouse experiments. The protocol was approved by the Animal Care Ethics Committee of Laval University (#20-515).
Flow cytometry analyses
Seven months after HSV-1 infection, blood sample (60μL) was incubated with 2 mL of 1x red blood cell lysis buffer (BioLegend) for 15 min at room temperature (RT). Nonspecific binding was blocked with purified rat anti-mouse CD16/CD32 antibody (1/100; BD Biosciences) for 10 min on ice. Cells were labeled at 4°C for 40 min with anti-CD45, anti-CD11b, anti-Ly6G and anti-Ly6C antibodies described in Supplementary Table 1. Cells were then fixed in 250μL of fixation solution (Invitrogen) for 20 min at RT. Precision count beads (50μL; BioLegend) were added to each sample to obtain absolute cell counts. Flow cytometry, data acquisition and analyses were respectively performed using BD SORP LSR II, BD FACS Diva software and FlowJo software v10 (all from BD Biosciences).
Immunofluorescence analyses
On month 7 post-infection (p.i.), deeply anesthetized mice were perfused transcardially with 0.9% saline followed by paraformaldehyde 4% (pH 7.6). Whole brains were collected, post-fixed in paraformaldehyde 4% containing 20% sucrose for 24 h at 4°C. Brains were cut in 25μm coronal sections using a freezing microtome (Leica Biosystems) and kept in a cryoprotectant with glycerol at – 20°C until used.
Free floating sections were washed and incubated in blocking/permeabilization solution containing 1% bovine serum albumin, 1% Triton X-100, and 4% normal donkey serum (Abcam) in potassium phosphate-buffered saline for 20 min at RT. Sections were incubated with anti-Aβ 6E10 and anti-Iba1 primary antibodies (Supplementary Table 1) in the same blocking/permeabilization solution under agitation overnight at 4°C. Incubation with fluorochrome-conjugated secondary antibodies (Supplementary Table 1) were then done in the dark for 2 h at RT. Nuclear staining with DAPI (1/10,000; Molecular Probes) was done for 20 min at RT. Brain sections were mounted onto MicroSlides Superfrost® (Fisherbrand) and coverslipped with Fluoromount-G® (Electron Microscopy Sciences).
Image acquisition and stereological count
Immunofluorescence images were captured using a Zeiss LSM800 confocal microscope equipped with ZEN imaging software (2.6 systems). Images were processed using Fiji (ImageJ Version 2.0.0-rc-43/1.51n). The number and volume of 6E10+ Aβ plaques and the number of Iba1+ microglia in the hippocampus region of APP/PS1 mice were blindly quantified using a Stereologer TM2000 MFC V.11.0 (SRC Biosciences) as previously described [13].
Statistical analyses
All statistical analyses were performed using GraphPad Prism software v9 (GraphPad Software). A p value of <0.05 was considered statistically significant.
RESULTS
We evaluated whether infection with a neurovirulent clinical strain of HSV-1 could contribute to the progression of AD pathology in mice carrying mutations in APP and PSEN1 genes. Male and female WT and APP/PS1 mice were infected intranasally with 750,000 PFUs of HSV-1 strain H25 at 3 months of age (before the onset of Aβ deposition in APP/PS1 mice at 4 months) [14]). The survival rates were similar in WT (Fig. 1A) and APP/PS1 (Fig. 1B) mice infected with HSV-1 (60% and 70%, respectively) and no statistically significant differences were observed compared to the non-infected groups. Both infected mouse strains started to lose weight at day 5 p.i. Compared to their non-infected counterparts, the differences in body weight losses were statistically significant on days 6 (p < 0.01), 7 (p < 0.01), and 13 (p < 0.05) p.i. in WT group (Fig. 1C) and from day 5 (p < 0.01) to day 14 (all others p < 0.001) p.i. in APP/PS1 mice (Fig. 1D). At the peak of infection (days 5 to 7 p.i.), WT and APP/PS1 mice exhibited similar clinical signs with most animals showing a swollen forehead starting at day 5 p.i. whereas a modified posture and neurological symptoms started to occur at day 6 p.i. (Fig. 1E, F). Thus, APP/PS1 and WT mice demonstrate a similar susceptibility to infection with a neurovirulent clinical HSV-1 strain H25.
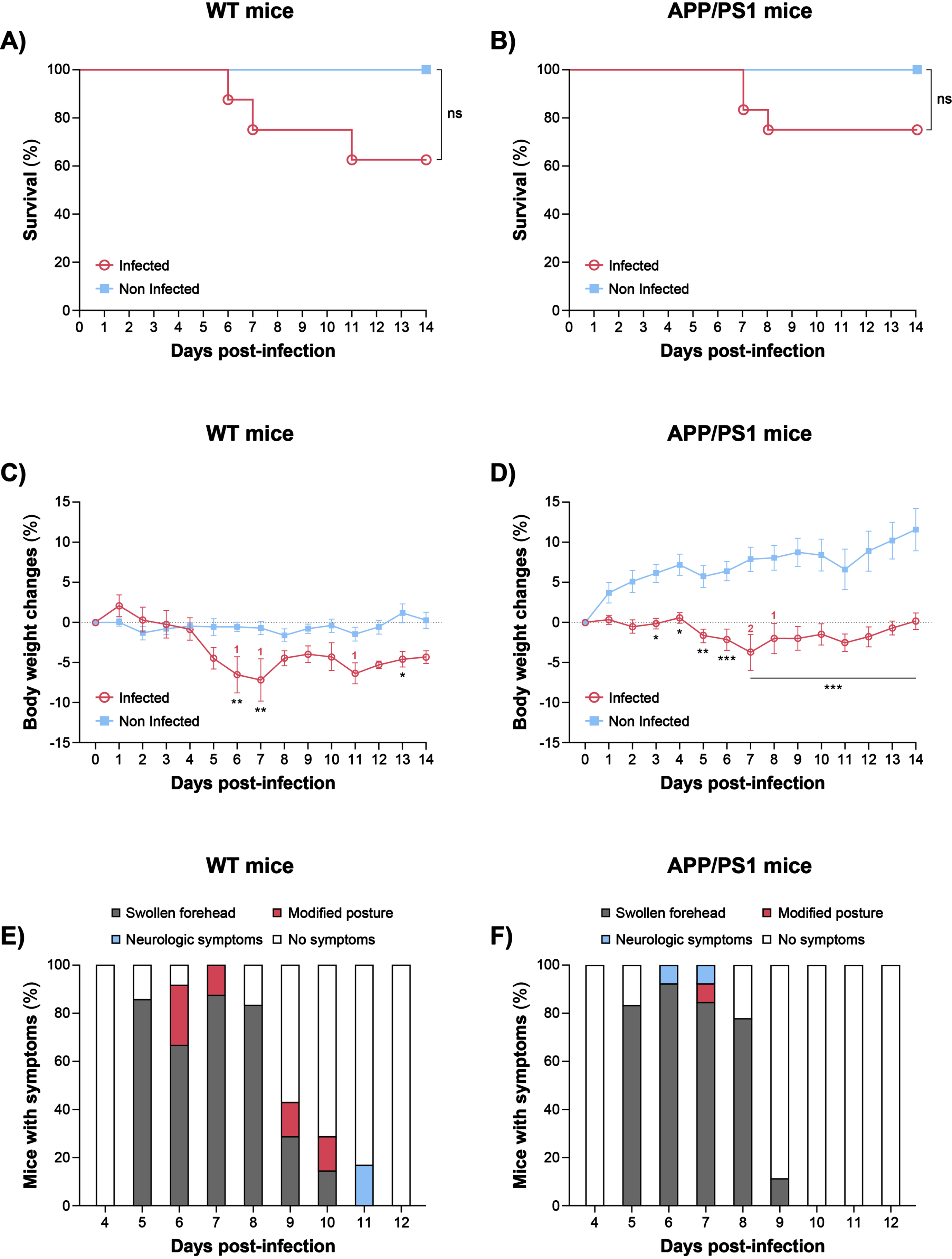
Clinical outcome of 3-month-old WT and APP/PS1 mice infected or not with HSV-1. Survival curves of WT (A) and APP/PS1 (B) mice that received 20μL of 750,000 plaque forming units of HSV-1 strain H25 (n = 8 for WT and n = 10 for APP/PS1) in minimum essential medium (MEM) or MEM only (non-infected controls; n = 8 for WT and n = 12 for APP/PS1) equally distributed in both nostrils. Survival rates were analyzed using a log-rank (Mantel– Cox) test. ns, not significant. Percentage of body weight changes of WT (C) and APP/PS1 (D) mice infected or not with HSV-1. Digits correspond to the numbers of dead mice at the indicated days. Results represent the mean±SEM. Outliers were first excluded by Grubb’s test and statistical analyses were performed using a two-way ANOVA with a Sidak’s multiple comparison test. *p < 0.05; **p < 0.01; ***p < 0.001. Cumulative percentage of HSV-1-infected WT (E) and APP/PS1 (F) mice showing no symptoms or clinical signs of infection such as a swollen forehead, a modified posture and/or neurological symptoms.
The aggregation and deposition of Aβ peptides in the brain is one of the hallmarks of AD pathology. Microglia were shown to play an important role in Aβ clearance and AD progression [15]. To assess the impact of HSV-1 infection on Aβ peptide accumulation and the recruitment of microglia around Aβ plaques, brain sections of APP/PS1 mice infected or not with HSV-1 and obtained on month 7 p.i. (i.e., 10 months of age) were stained with 6E10 and Iba1 antibodies that specifically recognize Aβ and microglia. Immunofluorescence images showed a high number of 6E10+ amyloid plaques in the hippocampus of both infected and non-infected APP/PS1 mice whereas no plaque was seen in WT group (Fig. 2A). Aβ plaques were surrounded by Iba1+ microglial cells in both infected and uninfected APP/PS1 mice (Fig. 2B). Similar findings were observed in brain cortex (data not shown). The number and volume of amyloid plaques and the number of microglia were quantified in the hippocampus of infected and uninfected APP/PS1 mice but no statistical differences were observed between the two groups (Fig. 2C). There was no evidence of a more pronounced cognitive decline in HSV-1-infected APP/PS1 mice compared to non-infected animals on month 6 p.i. as assessed by the novel object recognition test [16] (data not shown). Similar findings were made with APP/PS1 mice infected with HSV-1 at 5 months of age (after Aβ plaque deposition) (data not shown). These results suggest that HSV-1 infection does not exacerbate Aβ burden and cognitive decline in the APP/PS1 mouse model.
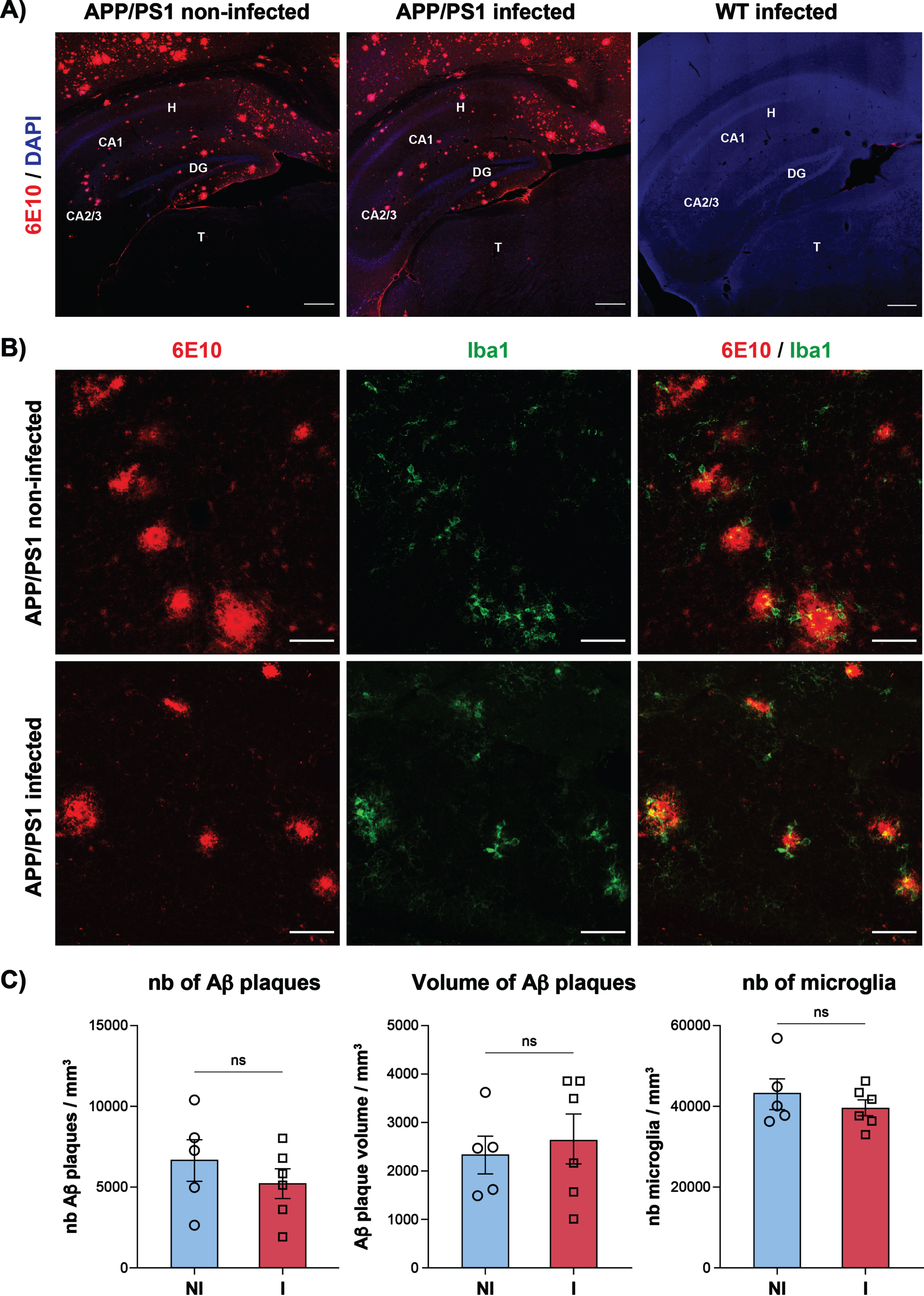
Amyloid plaques and microglia in the hippocampus region of APP/PS1 mice infected or not with HSV-1. Immunofluorescence images showing amyloid plaques labeled with anti-6E10 antibody (in red) in the hippocampus region (selected Bregma – 2.91; [23]) of APP/PS1 mice infected or not with HSV-1 (at a 10 X magnification; left and middle) and infected WT mice (at a 20 X magnification; right) at month 7 post-infection (A). CA, cornu ammonis 1 and 2; DG, dentate gyrus; H, hippocampus; T, thalamus. Scale bar, 250μM. Staining of amyloid plaques and microglia with anti-6E10 (in red) and anti-Iba1 (in green) antibodies in the hippocampus of APP/PS1 mice infected or not with HSV-1 at a 20×magnification (B). Scale bar, 50μM. Unbiased stereological analysis of the number (nb; left) and volume (middle) of amyloid plaques and the number of microglia (right) per mm3 in the hippocampus region of APP/PS1 mice infected (I) or not (NI) with HSV-1 (C). Results represent the means±SEM for 5-6 animals per group with both males and females. Outliers were first excluded by Grubb’s test and statistical analyses were performed using a nonparametric Mann– Whitney comparison test. ns, non-significant.
Circulating monocyte subsets are also involved in the regulation of AD pathology [17]. Indeed, LyChigh inflammatory monocytes infiltrate the brain where they differentiate into microglia-like cells and contribute to Aβ clearance whereas Ly6Clow patrolling monocytes are implicated in the clearance of Aβ that deposits on cerebral blood vessels [18]. On month 7 p.i., the blood monocyte populations were analyzed in WT and APP/PS1 mice infected or not with HSV-1 by flow cytometry based on the gating strategy described in Fig. 3A. The percentages of neutrophils (Fig. 3B1) and total monocytes (Fig. 3B2) were not affected by HSV-1 infection in both mouse strains. The percentages of the monocyte subsets, i.e., Ly6Chigh inflammatory monocytes, Ly6Cint intermediate monocytes, and Ly6Clow patrolling monocytes were similar in infected and uninfected APP/PS1 and WT mice (Fig. 3C1-C3), except for a slight increase of intermediate monocytes in infected WT group (p < 0.05). These results demonstrate that HSV-1 infection does not affect circulating monocyte populations in APP/PS1 mice.
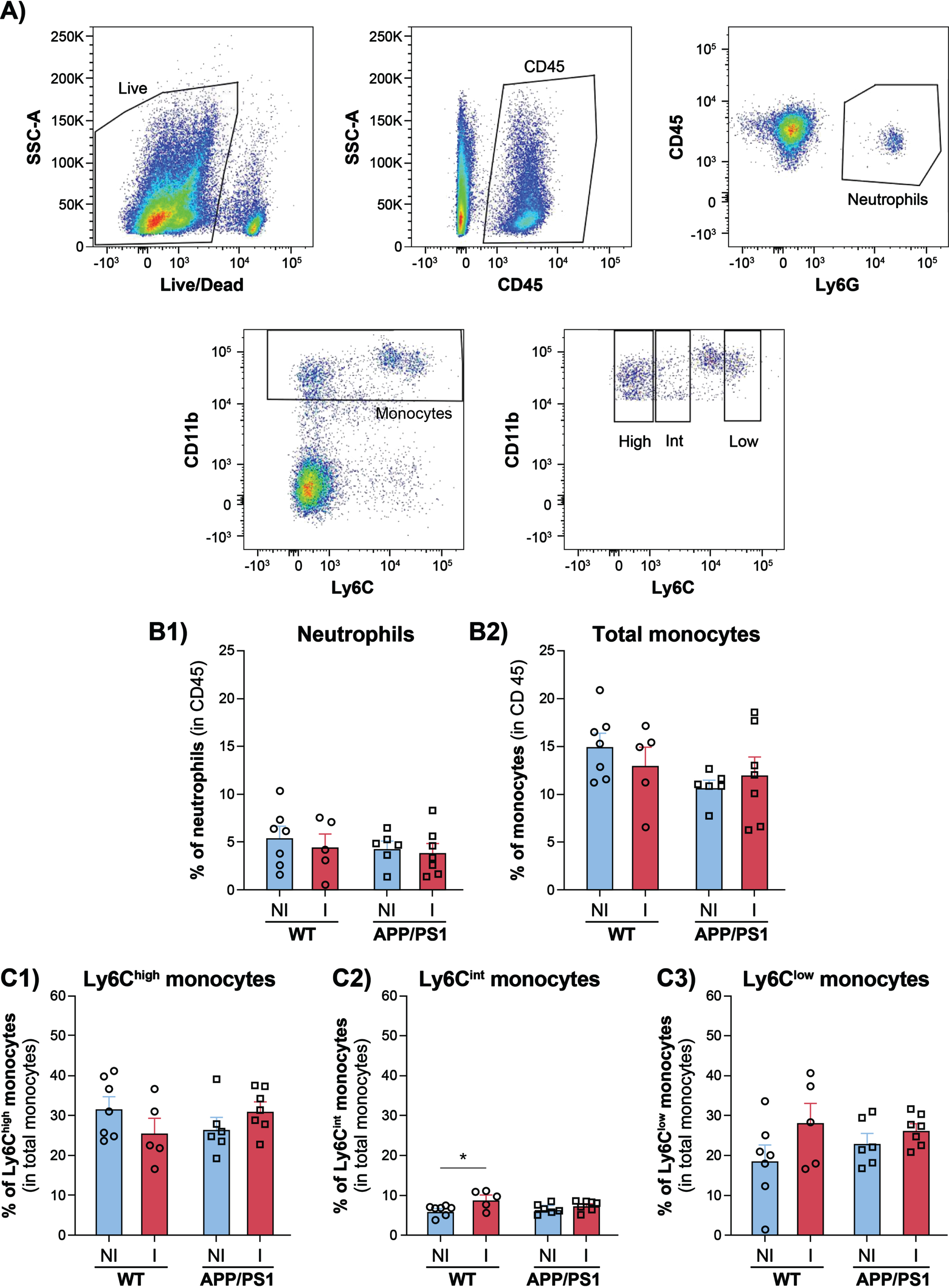
Neutrophils, total monocytes and monocyte subsets in the blood of WT and APP/PS1 mice infected or not with HSV-1. Gating strategy used to discriminate live leukocytes (CD45+), neutrophils (CD11b+, Ly6G+), total monocytes (CD11b+, Ly6C+), and monocyte subsets, i.e., inflammatory monocytes (CD11b+, Ly6Chigh), intermediate monocytes (CD11b+, Ly6Cint), and patrolling monocytes (CD11b+, Ly6Clow) by flow cytometry analyses (A). Percentages of neutrophils (B1) and total monocytes (B2) in CD45+ leukocytes of WT and APP/PS1 mice infected (I) or not (NI) with HSV-1 at month 7 post-infection. Percentages of Ly6Chigh inflammatory monocytes (C1), Ly6Cint intermediate monocytes (C2), and Ly6Clow patrolling monocytes (C3) in total monocytes of WT and APP/PS1 mice infected (I) or not (NI) with HSV-1. Results represent the mean±SEM for 5 to 7 mice per group with both males and females. Outliers were first excluded by Grubb’s test and statistical analyses were performed using a one way ANOVA with a Tukey’s multiple comparison test. *p < 0.05.
DISCUSSION
Male and female APP/PS1 mice infected intranasally with a neurovirulent clinical strain of HSV-1 at 3 months of age, before the onset of Aβ deposition, had similar disease outcome (body weight loss, clinical symptoms, and survival rates) compared to age- and sex-matched WT animals. It was also reported that the survival rates of young (5-6-week-old) 5XFAD mice (that overexpress the familial AD forms of human APP and PSEN1 transgenes with 5 AD-linked mutations) infected intracerebrally with HSV-1 strain 17syn+ or McKrae were not affected compared to WT animals [19]. This suggests that overproduction of Aβ peptides does not affect the course of HSV-1 infection in these transgenic mice. Furthermore, we observed that the survival rate of APP/PS1 mice infected at 5 months of age (after the deposition of Aβ peptides) was lower than that of WT animals (70% versus 100%; data not shown) but the difference was not statistically significant. The survival rates of aged (7-14-month-old) 5XFAD mice infected intracerebrally or by eye scarification with HSV-1 strain McKrae were also similar compared to WT. This suggests that the presence of Aβ aggregates in the brain does not confer protection against HSV-1 infection in these mouse models [19, 20].
Our results also showed that HSV-1 infection did not exacerbate Aβ accumulation in the hippocampus and cortex regions of APP/PS1 mice compared to non-infected animals. Intraperitoneal infection of neonatal or 9-week-old 5XFAD mice (before and after the formation of extracellular Aβ plaques) with murine roseolovirus did not increase Aβ deposition and/or progression of Aβ pathology in the cortex on month 6 p.i. despite an acute inflammatory response in the brain [21]. We found that the number and volume of Aβ plaques were similar in the hippocampus of infected and non-infected APP/PS1 mice. These Aβ plaques were surrounded by microglial cells. Reactive microglia and infiltrating Ly6Chigh inflammatory monocytes are involved in the phagocytosis and removal of Aβ plaques in the brain [17]. However, the number of Iba1+ microglia in the hippocampus and the percentages of monocyte subsets in the blood were not altered in infected APP/PS1 mice suggesting that the absence of effects of HSV-1 infection on Aβ accumulation is not due to a higher rate of elimination by these phagocyticcells.
A limitation of our study is that the effects of HSV-1 on Aβ pathology were tested in transgenic APP/PS1 mice overexpressing human APP and PSEN1 after a single acute infection. We did not evaluate whether multiple reactivations of latent HSV-1 could trigger or accelerate Aβ accumulation. Indeed, repetitive reactivations of latent HSV-1 in BALB/c mice subjected to thermal stresses resulted in cognitive decline associated with Aβ accumulation, tau phosphorylation and neuroinflammation [22].
In conclusion, our results suggest that intranasal HSV-1 infection does not increase the progression of Aβ burden and cognitive decline in the context of predisposition factors associated to APP and PSEN1 mutations.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research [CIHR foundation scheme program no 148361 to G.B. and no 143279 to S.R.] and the Fonds de recherche du Québec, Santé (FRQS) via the research center funding grant to S.R. S.R. is supported by a Canada Research Chair in Neuroimmunology. We would like to thank Pierre-Alexandre Piec and Nataly Laflamme in the neuroscience laboratory for training on the Stereologer and managing mouse colonies, respectively.
CONFLICT OF INTEREST
The authors have no conflict of interests to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article, its supplementary material or on request from the corresponding author.