
Abstract
Background:
While symptoms related to lower urinary tract dysfunction (LUTD) are common in individuals with Alzheimer’s disease (AD), pathophysiological links between AD and LUTD remain unclear.
Objective:
This study aimed to investigate whether AD neuropathology would cause autonomic dysfunction along the spinal cord-bladder axis, which could result in alterations in bladder muscle kinetics.
Methods:
We utilized APP NL-G-F/NL-G-F knock-in (APP KI) and APPwt/wt (wild-type) mice at two different ages, 4- and 10-month-old, to investigate how AD impacts bladder tissue function by immunohistochemistry, western blotting, and pharmacomyography.
Results:
We showed that the mucosal layer partially separated from the detrusor in 10-month-old APP KI mouse bladders. Although there was no detectable amyloid deposition in the APP KI bladder, we found amyloid plaques in APP KI lumbar spinal cord. Further immunoblot analysis revealed that tyrosine hydroxylase protein levels were significantly reduced in both 4- and 10-month-old bladder tissues, suggesting reduction of norepinephrine synthesis in APP KI mouse bladders. In contrast, the level of β2 adrenergic receptor was increased in 4-month-old but not 10-month-old APP KI bladders. In bladder strips, the adrenergic agonist isoproterenol induced increased relaxation in 4- but not 10-month-old APP KI bladders. With 10 Hz electrical field stimulation, 10-month-old APP KI bladder strips were more responsive than wild-type controls, with no differences observed in 4-month-old APP KI bladders.
Conclusions:
APP KI mice exhibit LUTD, which is likely arising from amyloid pathology in the spinal cord, and results in maturational declines in presynaptic activity combined with compensatory postsynaptic upregulation.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), the most common aging-dependent neurodegenerative disease, is characterized by the presence of amyloid deposition, neurofibrillary tangles, progressive loss of synapses, and severe cognitive dysfunction [1, 2]. While AD is best recognized for cognitive dysfunction such as impaired short term memory, lower urinary tract dysfunction (LUTD) is actually one of the most common symptoms seen in individuals with AD [3, 4], which often refers to urinary storage or voiding abnormalities. LUTD is more prevalent in nursing home patients with AD compared to those with normal cognition [5]. While it is commonly thought that LUTD is solely due to cognitive decline and awareness of social situations, recent studies have suggested that AD neurological lesions may impact micturition centers, specifically the frontal cortex, basal ganglia, and pontine micturition center in the brain [6]. Human AD urodynamics studies have been limited and do not provide sufficient information on the underlying mechanism of LUTD with AD since they mainly rely on self-reported symptoms, which may be erroneous due to memory and cognitive impairment in AD patients [4, 7]. Similarly, murine investigations concentrated primarily on the voiding behavior of AD mice [8, 9], have indicated that increased afferent activity in the lower urinary tract with anxiety leads to frequent urination, as previously reported in AD patients. However, these studies fail to detect the fundamental mechanism of impaired bladder function.
We have recently shown that APP/PS1 mice have altered urodynamics and increased variance in adrenergic pharmacomyography responses [10]. APP/PS1 mice are double transgenic AD model expressing a chimeric mouse/human amyloid-β protein precursor (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9) with mouse prion protein promoter to drive transgene expression predominantly by CNS neurons [11]. Since APP/PS1 mice have overexpression of both APP and PS1 transgenes, we here aim to use APP NL - G - F/NL - G - F knock-in (APP KI) mice to examine bladder pathophysiology to avoid potential effect associated with overexpression of transgenes and to discern etiology of LUTD with AD. APP KI mice express an APP construct with a humanized Aβ region includes the Swedish (KM670/671NL), Iberian (I716F), and Arctic (E693G) mutations [12]. The genetic approach used to insert those mutations was directly targeting the endogenous APP locus [13, 14]. Prior investigations [12, 15] in APP KI mice showed that neuropathology and memory deficits observed in this model are similar to human AD patients. Therefore, we postulate that APP KI mice are an ideal model to study AD associated bladder dysfunction without the caveat of overexpression of APP in transgenic mouse models.
In this study, we investigated whether AD neuropathology would cause autonomic dysfunction along the spinal cord-bladder axis, which could result in alterations in bladder muscle kinetics. To this end, we examined bladder histology, amyloid deposition, bladder neurotransmitters and receptors, and pharmacomyography in APP KI mice and age-matched wild-type littermates. We identified morphological changes in APP KI bladder and amyloid deposition in the lumbar spinal cord. Specifically, the sympathetic neurotransmitter norepinephrine was reduced in the APP KI bladder, which triggered compensatory elevation of β2 adrenergic receptors in the APP KI bladder. This change was confirmed by pharmacomyography, showing that the bladder strips increased relaxation responses to the adrenergic agonist isoproterenol only in 4-month-old APP KI mice. In addition, we showed that 10-month-old APP KI bladder strips were more responsive to 10 Hz electrical field stimulation. To our knowledge, this is the first evidence to uncover morphological alterations, reduced neurotransmitter norepinephrine, and pharmacomyographic changes in the bladder tissues of a knock-in AD mouse model.
MATERIALS AND METHODS
Animals
The APP NL-G-F/NL-G-F (APP KI) mice carrying Swedish, Arctic, and Iberian mutations were gifted by RIKEN Center for Brain Science, Japan. The colony of these mice was maintained in our animal houses at UConn Health. APP KI mice develop amyloid plaques in the brain beginning at two months and reaches saturation at seven months [12]. We utilized APP KI and APPwt/wt (wild-type, WT) mice at two different ages, 4- and 10-month-old, to investigate how AD impacts bladder tissue function. All mice were housed in a climate-controlled environment with 12 : 12 h light:dark cycle and fed standard rodent chow and water ad libitum. All animal use and procedures were performed according to the Institutional Animal Care and Use protocols at UConn Health, Farmington, and in compliance with the guidelines established by the Guide for the Care and Use of Laboratory Animals, as adopted by the National Institutes of Health.
Immunohistochemistry
Mice at 4 and 10 months old (n = 6 per group) were euthanized and the tissues of bladder, spinal cord segment (T12-L3), and dorsal root ganglion were collected for histology and immunohistochemistry [16]. Tissues were fixed in 4% paraformaldehyde for 24 h, immersed in 20% sucrose overnight at 4°C, and sectioned at 16-μm thick on a cryostat microtome (Thermo HM525 NX). Sections on the slides were washed in PBS 3×for 5 min to remove OCT and then permeabilized with 0.3% Triton X-100 for 30 min, followed by washing with PBS (3×for 5 min). Antigen retrieval was performed by microwaving the sections in 0.05 M citrate-buffered saline (pH 6.0) for 3 min. The sections were blocked with 5% normal goat serum and incubated with the following primary antibodies at a 1 : 1000 dilution: human amyloid-β 1–16, 6E10 (BioLegend, cat# 803003), human amyloid-β1-40 (IBL, cat # 18580), APP-C (Sigma, cat# A8717), IBA1 (Wako, cat# 019-19741), GFAP (Covance, cat# SMI-22R), and ATG9A (Abcam, cat# ab108338). After washing with PBS (3×for 5 min), sections were incubated with Alexa Fluor-conjugated secondary antibody (1 : 400 in blocking buffer) at room temperature (RT) for 2 h. Slides were washed three times in PBS and mounted on a coverslip with Antifade mounting medium. Confocal images were acquired via Zeiss LSM800 confocal microscopy. AβPP fluorescence intensity in the mucosal layer was measured using ImageJ software (National Institutes of Health).
For the DAB staining, after the primary antibody, sections were incubated with universal biotinylated anti-mouse/rabbit IgG (1 : 200, Vector Laboratories) at RT for 2 h. After washing with PBS (3×for 5 min), sections were incubated with avidin-biotin peroxidase complex (1 : 200, Vector Laboratories) at RT for 1 h. Sections were then incubated with 0.05% DAB (Sigma) with 0.01% H2O2 in PBS for 5 min. Slides were washed three times in PBS and mounted on a coverslip with 60% glycerol. The spinal cord images were captured via Keyence BZ-X810 All-in-One microscope. Plaque numbers in the spinal cord were counted using ImageJ software (National Institutes of Health).
Western blotting
Protein extraction was performed according to previously described procedures [17]. Bladder samples were homogenized in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail [Roche]) and centrifuged at 13,200 rpm for 90 min. Protein concentrations were determined using a bicinchoninic acid (BCA) assay kit. Equal amounts of protein were loaded and resolved on a 4 to 12% SDS–polyacrylamide gel electrophoresis (NuPAGE system, Life Technologies) gels. Subsequently, blots were transferred to nitrocellulose membranes at 100 V for 2 h. The membranes were blocked with 5% BSA for 1 h at RT. The membranes were probed with the following primary antibodies and their dilutions: APP-C (1 : 1,000, Sigma, cat# A8717); BACE1 (1 : 1,000); tyrosine hydroxylase (TH) (1 : 500, Millipore, cat# AB152); ChAT (1 : 1,000, Cell Signaling, cat# 27269); nNOS (1 : 1,000, Cell Signaling, cat# 4231); M2mAChR (1 : 1,000, abcam, cat# ab109226); M3mAChR (1 : 1,000, Invitrogen, cat# PA5-85322); β2AR (1 : 500, Bioss, cat# bs-21455R); and β-actin (1 : 10,000, Sigma, cat# A5441). After 24 h, primary incubation at 4°C, blots were washed extensively and incubated with HRP-conjugated secondary antibodies and visualized using enhanced chemiluminescence (Thermo Scientific). The antibody-bound protein blots were detected by an iBright 1500 imaging system (Invitrogen). For quantification purposes, band intensities of immunoblots were analyzed using ImageJ software.
Quantification of Aβ42 using ELISA
Total Aβ1–42 was prepared from the frozen lumbar spinal cords by the guanidine hydrochloride method [16] and the levels of Aβ1–42 were quantified by the human Aβ42 ultrasensitive ELISA kit (ThermoFisher, cat # KHB3544) according to the kit described procedures. Results were obtained from eight female 10-month-old APP KI and eight age-sex-matched wild-type lumbar spinal cords.
Pharmacomyography
Bladder strip myography was utilized to investigate the differences in smooth muscle contraction and relaxation between the APP KI and WT mice. Mice (n = 8 per group) were euthanized with CO2, and bladders were excised immediately. Two longitudinal strips were prepared from each animal, and suture loops were used to tie each strip at the end, as previously described by our laboratory [18]. Then, strips were submerged in 37°C constantly aerated Krebs solution (NaCl 135 mM, KCl 5 mM, CaCl2 2 mM, MgCl2 1.2 mM, HEPES 10 mM, glucose 8.9 mM, pH 7.2) inside a horizontal tissue bath (Mayflower, Harvard Apparatus). One end of the strips was hooked to an isometric force transducer (HSE 372, Harvard Apparatus), and the second end was hooked to an anchor. Slowly and carefully, strips were stabilized at tension 10–12 mN for 30 min. We recorded the baseline in Krebs buffer for 5 min before adding drugs or initiating electrical field stimulation. Carbachol (0.1μM, and 1μM (108240050, Acros Organics)) and isoproterenol (0.1μM (15627, Sigma-Adrich)) were prepared in Krebs solution and utilized on one set of bladder strips to probe muscarinic and adrenergic responses, respectively. A separate bladder strip was used for the electrical field stimulation (EFS) to examine the evoked contractions of the bladder strips that would generally occur under nerve fibers excitation [19]. EFS triggers the nerve endings and caused a release of neurotransmitters in the detrusor smooth muscle tissues. The strips underwent gradating frequencies of EFS (10 Hz, 35 Hz, and 50 Hz) in aerated Krebs buffer for 5 s each at 5 min intervals with thorough flushes of aerated Krebs buffer after each stimulation. The change in tension, contraction, and relaxation responses were recorded on the LabChart software v8.0 (Powerlab, ADInstruments-Data Acquisition Systems for Life Sciences;RRID:SCR_001620). Potassium chloride (KCl 50μM) was flushed in at the end of the experiment to ensure the viability of all strips tested. If no contraction was observed, the strip was considered nonviable, and the data was not utilized.
Response ratios were calculated for each stimulus by dividing the tension (mN) after stimulation by the mean baseline (mN) prior to stimulation. More specifically, the response ratios for carbachol and EFS were calculated by dividing the maximum tension (mN) after stimulation by the mean baseline (mN) before stimulation. For adrenergic responses using isoproterenol, the mean tension (mN) during the last minute of isoproterenol stimulation was divided by the baseline tension (mN) prior to stimulation.
Statistical analysis
All data were analyzed via two-way ANOVA with Sidak posthoc multiple comparisons test with significance set at p < 0.05 or two-tailed Students t test. Statistical analysis was performed using GraphPad Prism V9.3.1. Data is presented as mean+/–standard error of measurement (SEM) unless otherwisenoted.
RESULTS
Morphological change in 10-month-old APP KI bladder
The mouse bladder is a hollow organ, and the bladder wall consists of three layers from inner to outer, named mucosa, detrusor, and serosa. The mucosa, comprised of the urothelium and lamina propria, is arranged into numerous folds that disappear when the bladder is distended with urine. The detrusor, composed of smooth muscle, is the thickest part of the bladder. The serosa is the outermost part of the bladder covered by only one layer of mesothelium [20]. By examining the bladder histology in APP KI mice, we performed hematoxylin and eosin (H & E) staining of the cross section of bladder tissues. The bladder morphology was normal in 4-month-old APP KI mice compared to age-matched WT littermate controls. However, in 10-month-old APP KI bladders, we found that the mucosal layer was partially separated from the detrusor in five of six examined APP KI bladders (Fig. 1A).
This morphological change promoted us to further examine AβPP localization in the bladder tissues. We performed confocal examination of frozen bladder sections with an APP-C terminal antibody and found that AβPP was primarily localized in the urothelial cells (Fig. 1B). The number of AβPP positive cells were not visibly different between 4-month-old APP KI and littermate controls. However, in some mucosa regions, the AβPP positive urothelial cells were obviously reduced in 10-month-old APP KI bladder (Fig. 1B). The reduction of AβPP-positive cells was not due to the cell loss, as shown by H & E staining in Fig. 1A. We then used ImageJ to quantify AβPP florescence intensity in the urothelial cells. The average gray value was not different between 4-month-old APP KI and littermate controls, but significantly reduced in 10-month-old APP KI bladders compared to age-matched WT controls (Fig. 1C).
Increased AβPP processing in the APP KI bladder
APP KI mice express endogenous levels of AβPP with a humanized Aβ region including the Swedish, Iberian, and Arctic mutations. The AβPP processing is elevated in the brain of APP KI mice [12]. To examine AβPP processing in the bladder tissue by western blot, we noted that full-length AβPP (AβPP-fl) protein levels were visibly reduced in both 4- and 10-month-old APP KI bladders compared to age-matched controls. BACE1 protein levels were elevated in both age groups of APP KI bladders. We also measured the protein levels of the BACE1-cleaved AβPP cleavage product, CTF-99, which was elevated in both 4- and 10-month-old APP KI bladders (Fig. 2A, B). Together, we showed elevated processing of AβPP in the APP KI bladders compared to the controls, and this elevated processing may lead to reduction in AβPP-staining in cells.
Amyloid deposition in the lumbar spinal cord in APP KI mice
In APP KI mice, the amyloid deposition in the cerebral cortex begins by 2 months and is almost saturated by 7 months [12]. Since AβPP processing was increased in APP KI bladder tissues, we then examined amyloid deposition in fixed bladder sections using the antibody 6E10, which recognizes the first 16 residues of human Aβ. In spite of increased AβPP processing by elevated BACE1, we did not detect amyloid deposition in APP KI bladder sections (Fig. 3A).
We then used the antibody Aβ1-40, which recognizes C-terminal of human Aβ 1-40, to examine plaque load with DAB staining in the lumbar spinal cord (L1-2) and 6E10 immunofluorescence labelling in the dorsal root ganglion (DRG), which are upstream structures of bladder innervation. In the L1-2 spinal cord cross sections, we found a few plaques (2.67±0.21/section) in 4-month-old APP KI and more plaque loads in 10-month-old APP KI (9.00±0.45/section). These plaques were predominantly spread in the gray matter (Fig. 3B, C), and not seen in WT controls. Quantification of Aβ42 by ELISA showed a significant increase of total Aβ42 in 10-month-old APP KI lumbar spinal cords compared to age-matched WT controls (Fig. 3D; 207.25±2.01 ng/g tissue in APP KI versus 2.29±0.22 ng/g tissue in WT, n = 8, p < 0.001). Further double-staining of lumbar spinal cord sections to co-label 6E10-positive amyloid plaques and ionized calcium-binding adapter molecule 1 (Iba-1)-positive microglia, we found that the amyloid plaques were surrounded by activated microglia in APP KI mice (Fig. 3E). To co-label AβPP-positive neurons and glial fibrillary acidic protein (GFAP)-positive astrocytes, we found that reactive astrocytes were in close contact with neuronal dystrophic neurites in APP KI mice (Fig. 3E). Previous study we found that ATG9A, an early autophagy protein, was accumulated in dystrophic neurites that were surrounding amyloid plaques in mice brain [21]. Here we found massive ATG9A-positve dystrophic neurites surrounding 6E10-positive amyloid plaques in 10-month-old APP KI lumbar spinal cords (Fig. 3F). In the dorsal root ganglion, neither amyloid deposition nor microglia and astrocyte were found in APP KI mice (Fig. 3G).
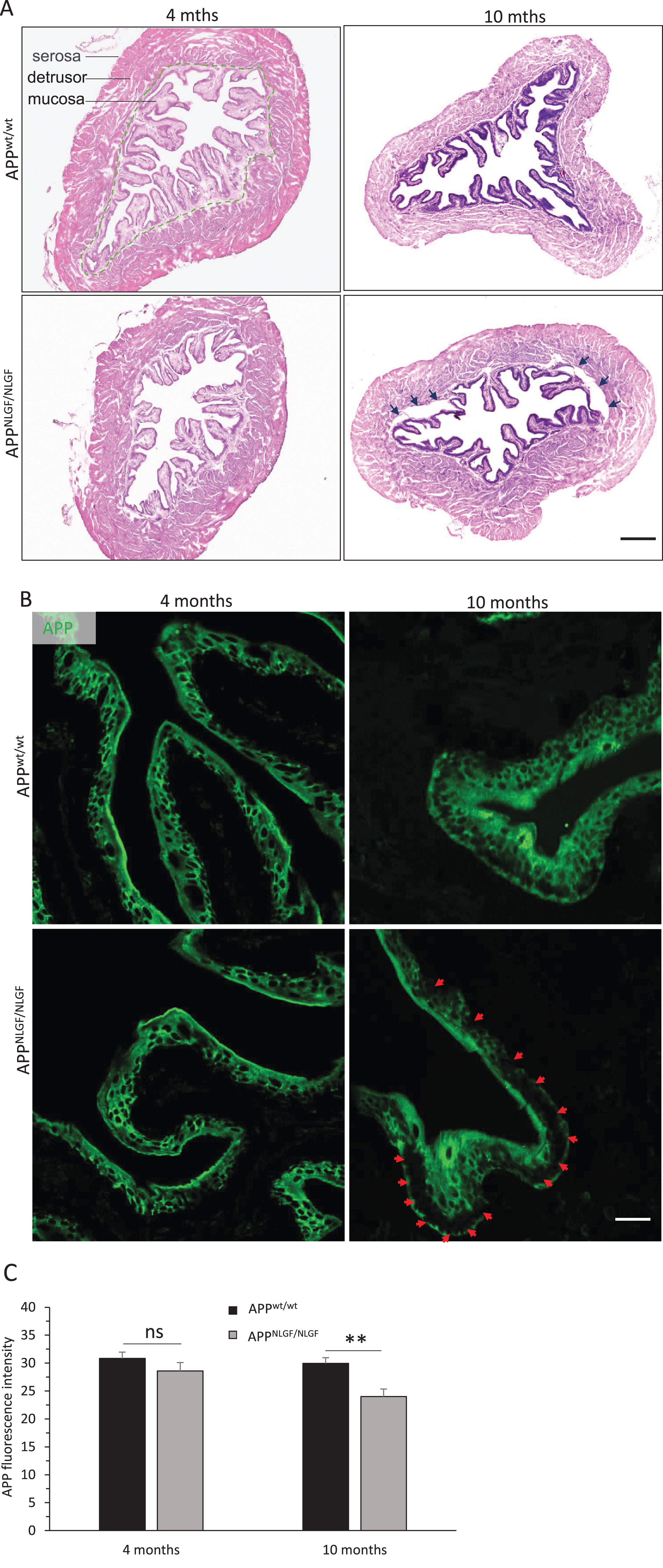
Morphology alteration in APP KI bladder. A) Representative images of hematoxylin and eosin staining and B) AβPP immunostaining in 4- and 10-month-old APP KI bladders and age-matched WT controls (n = 6 per group). The dashed curve separates the mucosal layer from the detrusor in A, and black arrows indicate that the mucosal layer was partially separated from the detrusor in 10-month-old APP KI bladders. Red arrows indicate reduced AβPP expression in the urothelial cells in B. Scale bar, 250μm in A; 40μm in B. C) Bar graph shows quantification of AβPP fluorescence intensity in the mucosal layer in 4- and 10-month-old APP KI bladders and age-matched WT controls (n = 6 mice per group, **p < 0.01). Values are expressed as mean±SEM.
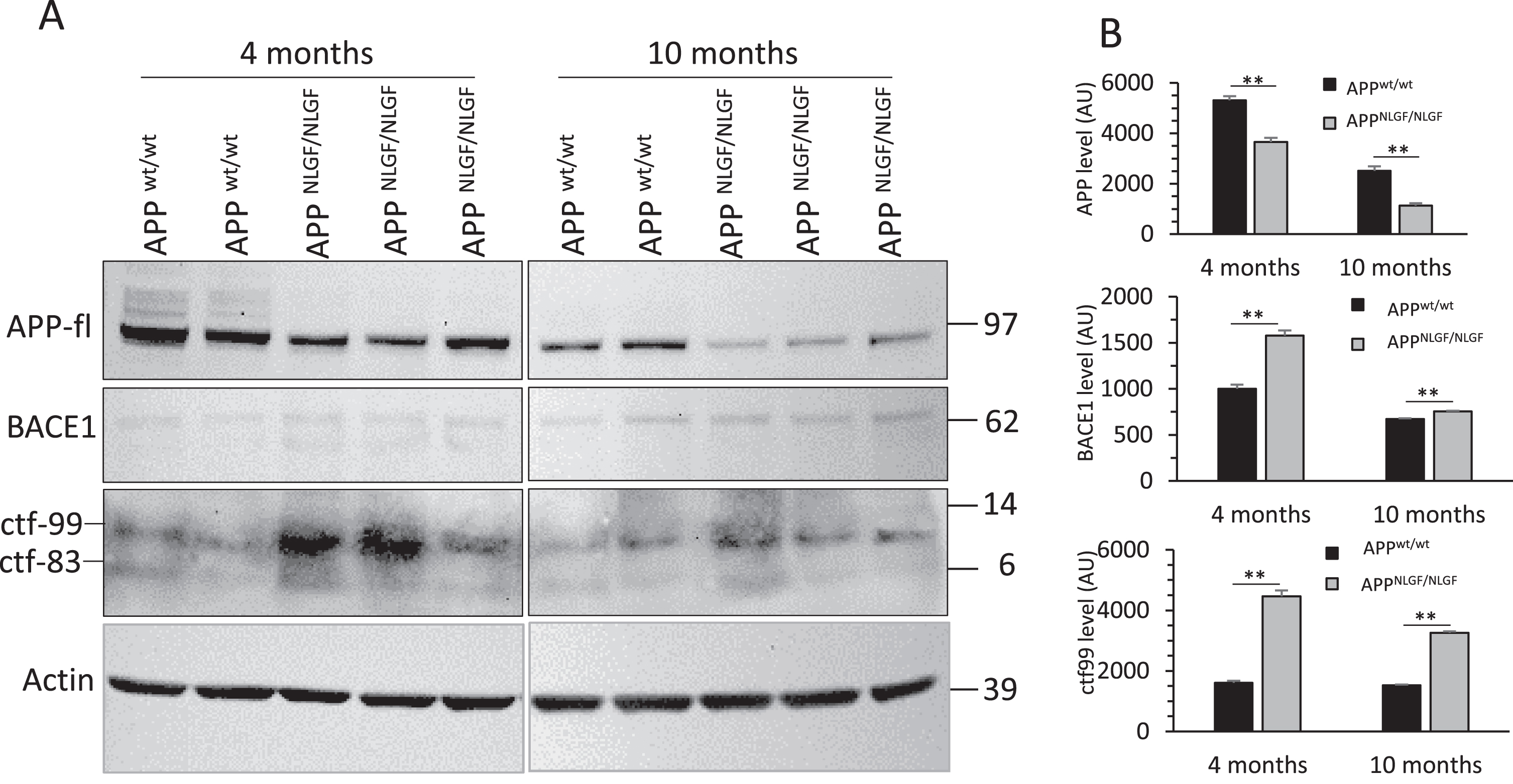
Increased AβPP processing in APP KI bladder. A) BACE1 protein levels and AβPP processing products in 4- and 10-month-old APP KI bladders and age-matched WT controls were examined by western blot. CTF-99 is a BACE1-cleaved APP C-terminal fragment, and CTF-83 is a product resulting from α-secretase cleavage of AβPP. β-actin was used to verify equal loading. Blot measurements are in kilodaltons (kDa). B) Bar graphs show quantification of relative protein levels based on western blots shown in A. Bar graphs are from at least 6 animals in each group. n = 3 independent experiments; two or three animals in each group were compared side by side; **p < 0.01; two-way ANOVA with Sidak posthoc multiple comparisons test. Values are expressed as mean±SEM.
Reduced tyrosine hydroxylase in APP KI bladder
The bladder is innervated by both autonomic and somatic nerves. Sympathetic postganglionic nerves release norepinephrine (NE), which excites adrenoceptors in bladder smooth muscles to elicit contraction of bladder base and urethral smooth muscle and relaxation of the bladder body. Parasympathetic postganglionic nerves release acetylcholine (ACh), which excites muscarinic receptors to contract bladder muscles. Parasympathetic nerves also release nitric oxide (NO), which has been implicated as an important neurotransmitter in urethral relaxation [22]. The somatic nerve releases ACh to excite the external urethral sphincter (Fig. 4A).
To examine whether amyloid deposition in APP KI lumbar cord affects expression of neurotransmitters and their receptors in the bladder, we performed immunoblot assays of TH, choline acetyltransferase (ChAT), neuronal nitric oxide synthase (nNOS), M2 and M3 muscarinic receptors, and β2 and β3 adrenoceptors in the bladder tissue (Fig. 4B). TH levels were significantly reduced in both 4- and 10-month-old APP KI bladders compared to age-matched WT controls (Fig. 4B, C). TH is the rate limiting enzyme involved in catecholamine synthesis which is responsible for catalyzing the conversion of L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) [23], and L-DOPA is a precursor for dopamine, which in turn is a precursor for NE. Thus, reduction of TH suggests that less NE is produced in APP KIbladder.
ChAT is responsible for the synthesis of ACh, and nNOS drives NO synthesis from L-arginine. We found that the levels of ChAT and nNOS were not visibly different between APP KI and littermate controls in both 4- and 10-month-old, suggesting that APP KI mice do not alter ACh and NO signaling in the bladder tissues. In addition, levels of ACh receptors, muscarinic acetylcholine receptor M2 and M3, were not altered in APP KI bladders (Fig. 4B, C).
In contrast, levels of β2 adrenergic receptors were increased in 4-month-old APP KI bladders and restored to normal levels in 10-month-old bladders when compared to age-matched littermate WT controls (Fig. 4B, C), suggesting that reduction of NE likely triggers compensatory elevation of β2 adrenergic receptor early in the APP KI bladder. We also observed that while the β3 adrenergic receptor was expressed in the mouse brain, it was almost undetectable in bladder tissues (data not shown).
Altered postsynaptic adrenergic signaling and EFS responses in APP KI bladder
We next sought to determine if the altered morphology and protein levels would influence relaxation or contractile properties of bladder tissues in APP KI mice. Thus, we performed bladder pharmacomyography, which are often used to evaluate changes in myogenic activity and neuronal factors that modulate bladder smooth muscle under pathophysiological conditions to determine any receptors and/or intracellular pathways involved in alterations of smooth muscle contraction or relaxation [19].
To examine the bladder tissue relaxation responses, we subjected bladder tissue strips to 0.1μM of isoproterenol, an adrenergic agonist drug that works by binding to the adrenergic receptors (β2 and β3) in the smooth muscle to cause hyperpolarization and relaxation in a condition established in our previous work [10]. We observed increased relaxation in 4-month-old APP KI bladder strips compared to age-matched littermate WT controls (Fig. 5A). Interestingly, no differences were observed at 10 months of age. This response pattern appears consistent with the altered levels of β2 adrenergic receptor in APP KI bladder (Fig. 4B, C). Likely, decreased TH in APP KI bladder tissues caused a compensatory increase in β2 adrenergic receptors in APP KI bladders, and correspondingly, an increase in adrenergic-induced relaxation responses in 4-month-old APP KI bladder strips. Interestingly, the compensatory effect was not observed in β2 adrenergic receptor levels or adrenergic-induced relaxation responses in 10-month-old APP KI bladder strips.
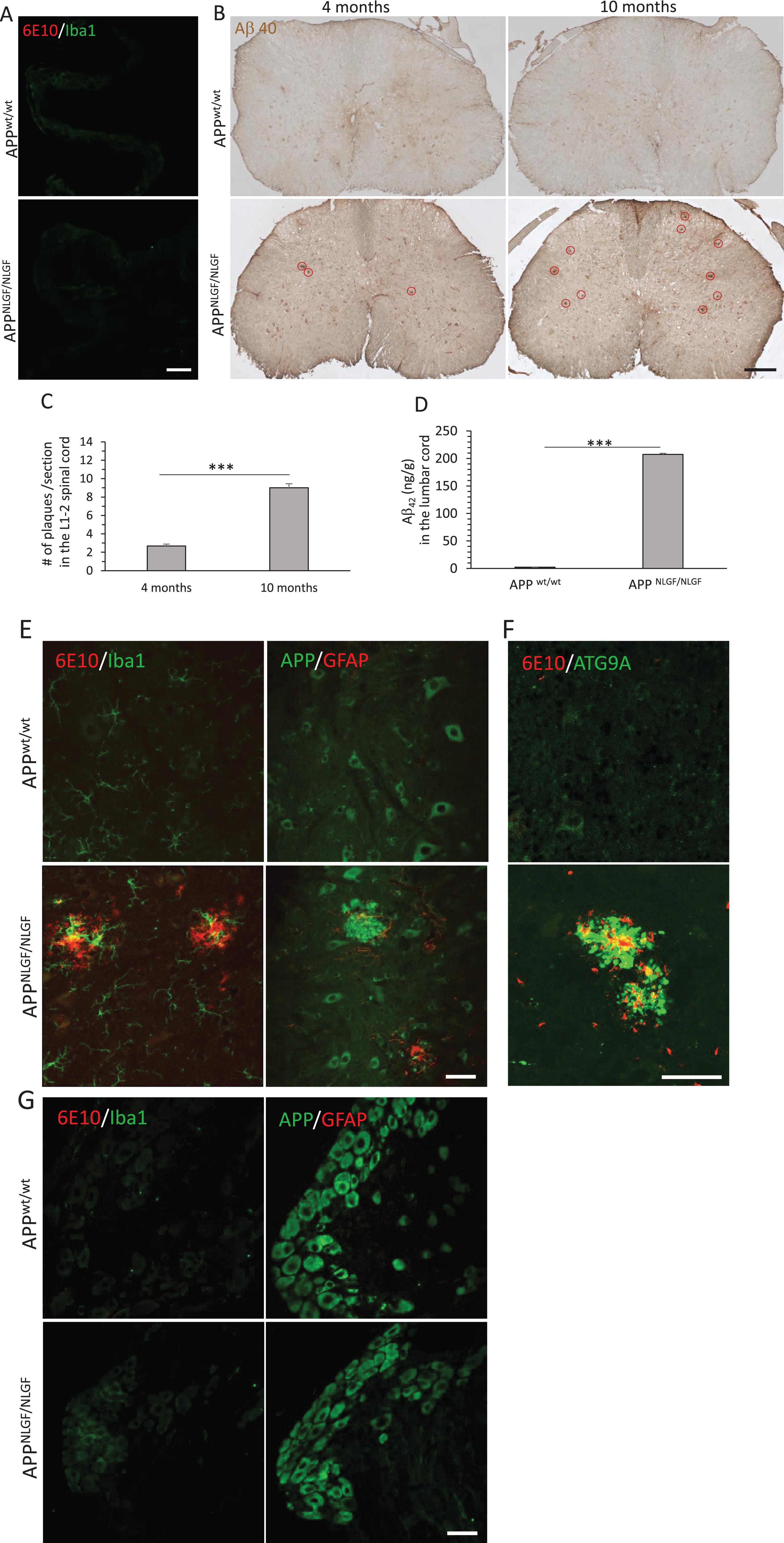
Plaque load in the APP KI lumbar spinal cord. A) Double staining of 6E10 and Iba-1 in 10-month-old APP KI bladders and age-matched WT controls. B) Representative images of DAB staining of Aβ1 - 40-positive amyloid plaques indicated with red circles in 4- and 10-month-old APP KI lumbar spinal cords (L1-2) and age-matched WT controls. C) Quantification of plaque load in DAB-stained sections from the lumbar cords in APP KI mice (n = 6 mice per group, ten sections were selected in every 10th per mouse, two-tailed Students t test, ***p < 0.001). D) Total Aβ42 from mice lumbar cords was extracted and measured by the human Aβ42 ultrasensitive ELISA kit (n = 8 mice per group, two-tailed Students t test, ***p < 0.001). Values are expressed as mean±SEM. E, F) Double staining of the lumbar spinal cord sections to co-label 6E10-positive amyloid plaques and Iba-1-positive microglia, AβPP-positive neurons and GFAP-positive astrocytes, and ATG9A-positive dystrophic neurites surrounding 6E10-positive amyloid plaques in 10-month-old APP KI mice and age-matched WT controls. G) Double staining of dorsal root ganglion sections to co-label 6E10/Iba1 and AβPP/GFAP in 10-month-old APP KI mice and age-matched WT controls. Scale bar, 250μm in B; 40μm in A and E–G.
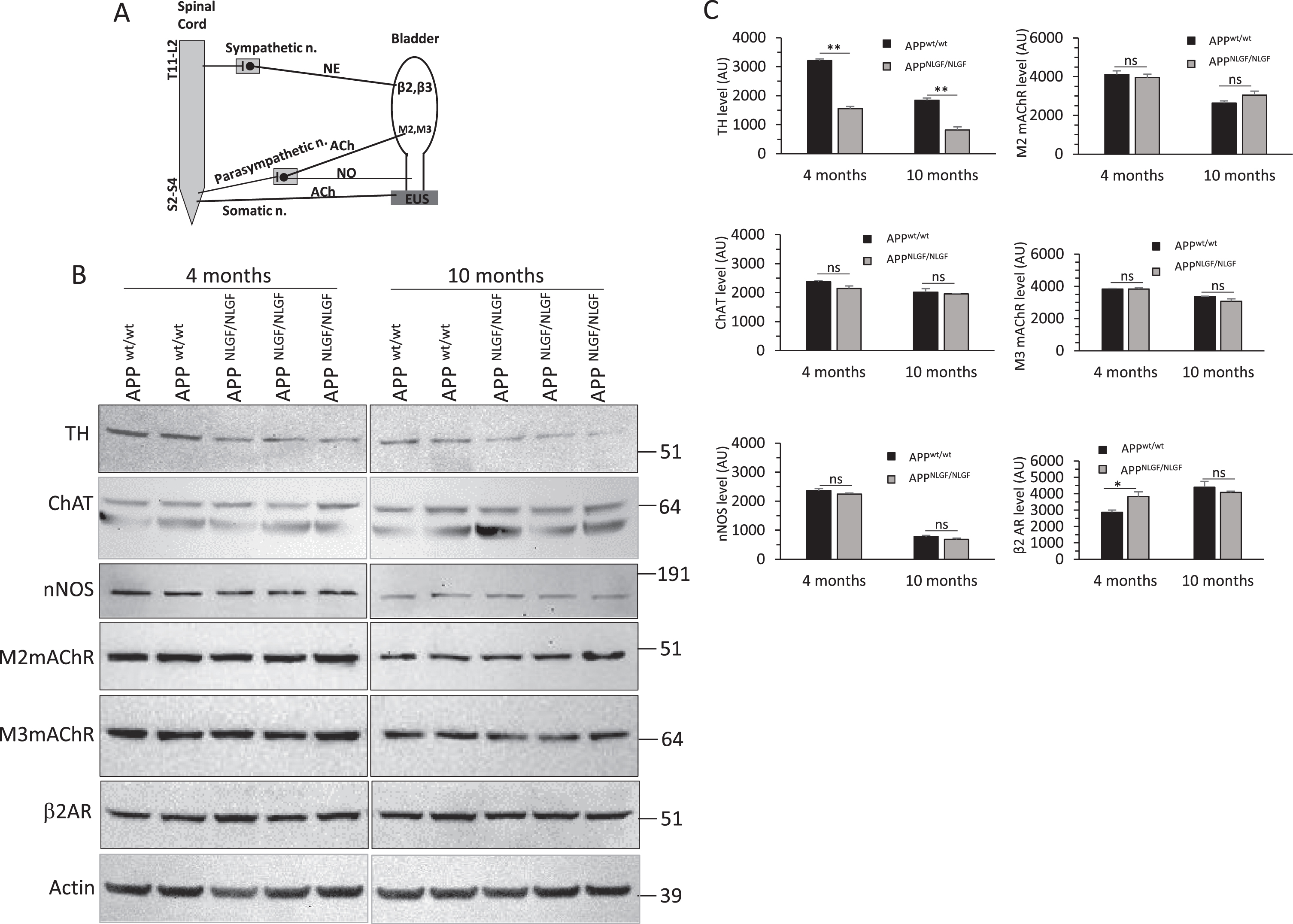
Immunoblot analysis of bladder neurotransmitters and receptors. A) schematic illustration of bladder innervation, neurotransmitters, and receptors. B) Protein levels of TH, ChAT, nNOS, M2mAChR, M3mAChR, and β2AR in 4- and 10-month-old APP KI bladders and age-matched WT controls were examined by western blot. β-actin was used to verify equal loading. Blot measurements are in kilodaltons (kDa). C) Bar graphs show quantification of relative protein levels based on western blots shown in B. Bar graphs are from at least 6 animals in each group. n = 3 independent experiments; two or three animals in each group were compared side by side; *p < 0.05, **p < 0.01; two-way ANOVA with Sidak posthoc multiple comparisons test. Values are expressed as mean±SEM.
Second, to assess how AD pathology affects bladder tissue contraction, we utilized carbachol, a muscarinic agonist drug that works by binding to the muscarinic receptors (M2 and M3) in the smooth muscle to cause depolarization and contraction, at two different concentrations as previously described [19] (0.1 and 1μM). No significant differences were identified between the APP KI and age-matched WT controls at either age group (Fig. 5B, C). This appears consistent with no changes in M2 and M3 levels in APP KI bladders as aforementioned (Fig. 4B).
Finally, we utilized increasing frequencies (10, 35, and 50 Hz) EFS to assess contraction responses. EFS stimulates both purinergic and muscarinic receptors involved in the contractile response of the bladder tissue to mimic the descending neural system (parasympathetic) that controls the bladder contraction during voiding. At 10 Hz, the most physiologically relevant frequency [19], 10-month-old APP KI mice had increased contractile response compared to the littermate WT controls, while the younger 4-month-old mice did not demonstrate any significant alterations in the contractile response (Fig. 5D). Additionally, no alterations in responses to the higher EFS frequencies (35 and 50 Hz) were observed (Fig. 5E, F).
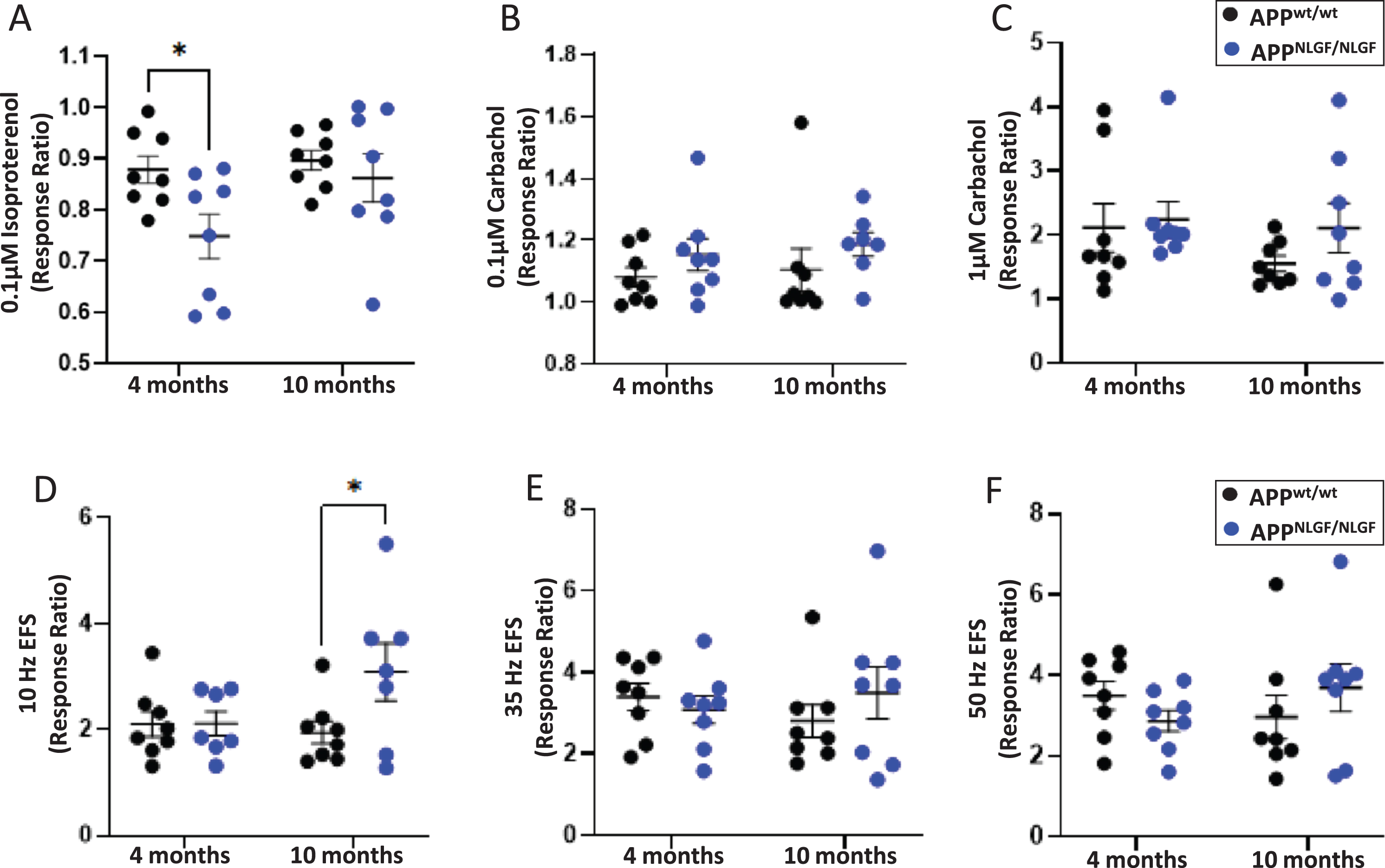
Bladder strips from 4- and 10-month-old APP KI mice and age-matched WT controls were subjected to pharmacomyography to assess adrenergic, muscarinic, and electrical field stimulation (EFS) responses. A) Bladder strips were stimulated with 0.1μM isoproterenol and responses ratios were calculated. B, C) Bladder strips were stimulated with 0.1 and 1μM carbachol and responses ratios were calculated. D–F) Bladder strips were subjected to EFS at 10, 35, and 50 Hz and response ratios were calculated. Results were analyzed using two-way ANOVA with Sidak posthoc multiple comparisons test. n = 8 mice per group. *p < 0.05, Values are expressed as mean±SEM.
DISCUSSION
The manner in which AD is associated with and contributes to LUTD represents a question of vital importance, which nonetheless remains to be fully understood. AD-induced cognitive impairments have often been viewed as directly leading to dysfunctional voiding behaviors in AD patients. Amyloid plaques and neurofibrillary tangles damage to the micturition centers of the brain could cause a loss of signal to the bladder, resulting in LUTD [6]. In late AD stages, patients may lose the ability to have proper void.
In this study, we provided evidence that the pathophysiological mechanism is multifactorial and many factors outside and inside the lower urinary tract contribute to this LUTD. Thus, LUTD with AD is not solely due to AD-induced cognitive impairment. Our morphological study in the APP KI mouse model revealed for the first time the existence of partial detachment of the mucosal layer from the detrusor in fixed bladder tissues (Fig. 1), as well as evidence of amyloid deposition in the lumbar spinal cord (L1-2) (Fig. 3) contributing to LUTD in APP KI mice, which unlike other AD transgenic mice express transgenes.
In APP KI mice, human mutations in APP were engineered in the mouse genome using a knock-in approach [12]. This mouse model has increased levels of Aβ peptides due to the Swedish mutation at the N-terminal end of Aβ region. In APP KI bladders, we noted increased expression of BACE1, a known β-secretase required for Aβ generation [24, 25]. Consistently, levels of BACE1-cleaved CTF99 were correlatively increased while AβPP staining was correspondingly reduced in the mucosa layer due to enhanced processing of AβPP (Fig. 2). One intriguing speculation is that reduced levels of AβPP in the mucosa likely contributed to partial separation of the mucosa layer from the detrusor smooth muscles (Fig. 1) because AβPP is known to also function as a cell adhesion molecule by binding to collagen type I [26]. Reduction of AβPP in the peripheral organs may affect collagens function in connecting other tissues.
We also explored whether morphological alterations in APP KI mice or other changes could affect bladder function. The bladder urothelium has for a long time been thought to act solely as a barrier protecting the underlying detrusor smooth muscle. There is, however, a growing body of evidence to indicate that this tissue plays a far more active role in bladder function [27, 28]. The bladder urothelium may be able to modulate bladder contractile function through local secretion of bioactive substances into the detrusor adjacent to the urothelium. Removal of the mucosal layer has been shown to significantly increase muscle contractile responses to EFS or many other contractile agents [29–31]. Indeed, we found that 10-month-old APP KI mice had increased responses to 10 Hz EFS, but no differences were observed at higher frequencies. Importantly, higher frequencies of 35 and 50 Hz are supraphysiological, and changes in 10 Hz responses are more physiologically relevant [19]. Previous research in humans found that the purinergic component of bladder signaling was less than 3% in normal settings, but increased to 40% in pathological conditions such as the neuropathic bladder [32]. Since the EFS contractile responses involve both muscarinic and purinergic receptors, and we observed no differences in muscarinic responses to carbachol, it is likely that increased responses to 10 Hz EFS are due to increased purinergic responses in 10-month-old APP KI mice. Previous study discovered that with aging purinergic signaling in the bladder is increased [33]. This altered signaling could enhance the excitation at the afferent terminals or increase the production of neurotransmitters such as ATP at the bladder tissue level. It is possible that 10-month-old APP KI mice are more akin to an aged mouse as AD accelerates other age-related dysfunctions, and similarly increased pathological purinergic signaling may lead to increased bladder EFS responses at 10 Hz. Future studies should more closely examine how the purinergic responses are altered in AD bladders.
Elevated AβPP processing in the APP KI bladder by BACE1 prompted us to examine amyloid deposition in this organ. Contrary to our expectation, we did not observe amyloid plaques in the APP KI bladder. Instead, we detected amyloid plaques in the lumbar spinal cord, the upstream structure of bladder innervation, in 4-month-old APP KI mice, and the plaque number was even more in 10-month-old mice. Amyloid plaques were found in the spinal cords of human AD patients [34]. It is likely that this neuropathology in the lumbar cord may affect neurotransmission to the bladder. By western blot analysis, we indeed observed that the TH levels were reduced by about 52% in 4-month-old and 56% in 10-month-old APP KI bladder. We speculate that the reason of reduced TH protein levels in APP KI bladder is probably coming from either loss of sympathetic postganglionic nerves innervating the bladder or reducing TH synthesis in the sympathetic axons. To clarify this, we need to examine morphology and biochemistry of sympathetic postganglionic neurons in the pelvic ganglion, which is located in soft floating protective tissue deep within the pelvic cavity. However, this experiment is difficult to perform, mainly because of technical challenges to surgically dissect out the pelvic ganglion. We believe that this can be addressed in our future experiments when we can acquire the related skills. As a compensatory reaction, β2 adrenergic receptors were elevated in the 4-month-old APP KI bladder. Interestingly, this compensation was not persistent over time as we noted that the β2 adrenergic receptor level was back to normal in the 10-month-old bladder. To our knowledge, this is the first evidence of reduced indicators of neurotransmitter NE synthesis in the AD bladder, which could reduce relaxation of bladder for urine storage.
Importantly, we identified changes in bladder smooth muscle tissue relaxation responses in APP KI mice, suggesting that AD urinary dysfunction involves bladder tissue-specific alterations and is not simply due to cognitive impairments. In contrast to decreased TH in the bladder, bladder strips pharmacomyography demonstrated that 4-month-old APP KI mice increased adrenergic responses; however, this was not evident at 10-month-old mice. These responses were consistent with our finding of increased levels of β2 adrenergic receptor in APP KI bladders, suggesting that early plaque deposition in the lumbar spinal cord and/or neuronal synaptic loss triggers compensatory mechanisms in the bladder tissue. However, this compensation is lost at late diseases stages with severe neuropathology in the AD mice. Decreased TH observed in APP KI bladder likely leads to decreased NE signaling, yet compensatory elevated β2 adrenergic receptor in 4-month-old bladder tissue result in increased adrenergic responses to lower stimuli. However, this compensatory mechanism is only transient and diminishes with maturation, as evidenced by the lack of difference in adrenergic responses between 10-month-old APP KI and WT controls. These results are consistent with previous data from our lab in APP/PS1 murine model [10].
The loss of cholinergic neurons has been linked to the amyloid deposition in the CNS with AD progression in both humans and AD murine models [35]. While the impact of AD pathology on cholinergic innervation and function in peripheral tissues is not well understood, our data showed no significant alterations in the contractile response of the bladder tissue to carbachol between APP KI and WT mice. These data aligned well with previous observation in the APP/PS1 mice that showed no structural difference in the cholinergic innervation of the bladder smooth muscle in APP/PS1 mice [36]. Taken together, this data suggests that sympathetic adrenergic nerve may be more susceptible to AD pathology in the spinal cord and resulting in bladder dysfunction than the parasympathetic cholinergic nerve in APP KI mice. Further investigation of in vivo contraction and relaxation responses is necessary to better tease apart differences in AD effects on adrenergic or cholinergic dependent micturition responses.
In summary, our study provides novel insights into impairments of bladder morphology, reduced neurotransmitter norepinephrine, and physiological changes in APP KI bladder, suggesting that AD could disrupt the neuronal regulation of the bladder resulting in abnormal bladder tissue function.
Footnotes
ACKNOWLEDGMENTS
We would like to acknowledge the late Dr. Phillip P. Smith for his tireless efforts to elucidate mechanisms of bladder dysfunction over his career and for obtaining the funding for this project.
We also thank Dr. Riqiang Yan for unwavering support to this study and many fruitful discussions.
FUNDING
This study is supported by the NIH grant R21AG061609 to P.P. Smith and X. Hu. Y. Ge was fully supported by NIH grant RF1AG025493 and X. Hu was also partially supported by the grant RFA NS074256 to R. Yan.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
DATA AVAILABILITY
All original data presented in the paper will be made available for review when needed. Research materials will be also made available when required.