
Abstract
High dietary intake of saturated fatty acids is a suspected risk factor for neurodegenerative diseases, including Alzheimer’s disease (AD). To decipher the causal link behind these associations, high-fat diets (HFD) have been repeatedly investigated in animal models. Preclinical studies allow full control over dietary composition, avoiding ethical concerns in clinical trials. The goal of the present article is to provide a narrative review of reports on HFD in animal models of AD. Eligibility criteria included mouse models of AD fed a HFD defined as > 35% of fat/weight and western diets containing > 1% cholesterol or > 15% sugar. MEDLINE and Embase databases were searched from 1946 to August 2022, and 32 preclinical studies were included in the review. HFD-induced obesity and metabolic disturbances such as insulin resistance and glucose intolerance have been replicated in most studies, but with methodological variability. Most studies have found an aggravating effect of HFD on brain Aβ pathology, whereas tau pathology has been much less studied, and results are more equivocal. While most reports show HFD-induced impairment on cognitive behavior, confounding factors may blur their interpretation. In summary, despite conflicting results, exposing rodents to diets highly enriched in saturated fat induces not only metabolic defects, but also cognitive impairment often accompanied by aggravated neuropathological markers, most notably Aβ burden. Although there are important variations between methods, particularly the lack of diet characterization, these studies collectively suggest that excessive intake of saturated fat should be avoided in order to lower the incidence of AD.
INTRODUCTION
Alzheimer’s disease (AD) remains the most prevalent cause of dementia, affecting over 40 million people worldwide [1–4]. From a neuropathological standpoint, two main hallmarks drive AD pathology: extracellular accumulation of amyloid peptides (Aβ) that form toxic senile plaques and the aggregation of neurofibrillary tangles (NFTs) made of hyperphosphorylated tau (ptau). Both Aβ and ptau are central for the diagnosis of AD [1, 4–7]. While these neuropathological markers are essential for a correct post-mortem diagnosis, their role on the etiopathophysiology of the disease is unclear. Decades of research has exposed the multifactorial nature of AD, which has led to a shift from targeting classical AD hallmarks separately to considering other risk factors that alter the progression of the pathology, including metabolic and cardiovascular deficits [6, 8–14]. Indeed, many clinical investigations now focus on improving brain metabolism by the implementation of insulin modulators or diets [15–18]. Though such investigations provide room for optimism, none has secured approval by health agencies.
In the last two decades, nutrition has risen to the forefront of research on neurodegenerative diseases (NDDs). Epidemiological and preclinical endeavors have provided solid evidence that an adequate nutrition may benefit cognition, at least in some population or experimental settings [19–21]. Dietary fats have received particular attention given the extent by which they are found in different human diets and because brain lipid concentrations, particularly fatty acids, can be altered by the composition of diet [20, 22–25]. In this sense, studies generally point to diets rich in poly- and monounsaturated fatty acids (PUFA, MUFA) to be associated with a lower incidence of NDDs. By contrast, diets rich in saturated fatty acids (SFA) are commonly deemed detrimental for cognition [26–28]. The link between brain function, lipid content, and diet composition has thus led to the hypothesis that dietary interventions may be used to prevent or modify disease progression in NDDs [2, 29–36].
Understanding the implications of nutritional factors on NDDs implies evaluating diets that can be preventive as well as those classically deemed detrimental for neurodegeneration. Whereas randomized controlled trials (RCTs) are essential to issue health recommendations and epidemiological studies are useful to detect associations, they remain silent on possible causal relationships. DHA for example, has been studied in RCTs of cognitive decline [29, 37] and high MUFA/DHA diets are now routinely incorporated into multidomain clinical trials [2, 31–33]. However, other types of fats cannot be investigated in clinical trials due to practical and ethical reasons. Dietary lipids such as SFA and trans-fatty acids cannot form part of RCTs given their possible detrimental effects on human health. In this sense, studies in animal models of NDDs offer a valuable opportunity to directly assess the impact of diets on mechanistic endpoints.
Data tell us that HFD have been extensively used to induce obesity and metabolic disease to model type 2 diabetes mellitus (T2DM) in preclinical models of AD (Table 1) [38]. However, the effect of HFD on AD related outcomes is still debatable, with a previous review already highlighting some inconsistency in the results published [39]. The present work seeks to provide a detailed overview of the HFD studies carried out in the last 30 years in animal models of AD, highlighting the consistent and less consistent results obtained. A focus will be set on cognitive endpoints and on the classical neuropathological markers of AD, but other AD-relevant outcomes will also briefly be discussed. We will emphasize the importance of detailing the nutritional content of experimental diets to generate reproducible data. Overall, this study responds to a need created by the multitude of studies published, with sometimes apparently contradictory results, carried out using a variety of methodological approaches.
ASSOCIATIONS BETWEEN DIETARY FATS AND THE INCIDENCE OF NEURODEGENERATIVE DISEASES
Saturated fatty acids
SFA are carbon chains containing a methyl group on one end of its structure and a carboxyl group at the other end. SFA, by definition, do not present double bonds in their structure and can be classified into two broad categories: long-chain saturated fatty acids (LCFA) and short-chain fatty acids (SCFA), although this is not a standardized definition [40, 41]. LCFA are typically found in dairy and red meat, but food sources contain a mixture of different fatty acids, which can influence their different physiological effects [40].
The health implications of high SFA intake have been studied mostly in respect to cardiovascular disease. Dietary saturated fats have been considered atherogenic, in part, due to their action on blood cholesterol [42, 43]. While recommendations to limit saturated fat intake have been widely accepted, recent studies conclude that saturated fats have no significant effect on cardiovascular or total mortality [40, 44]. This is particularly the case for high SFA foods such as dairy, unprocessed meat, and chocolate [40, 44]. Studies in animals also suggest that the actual processing of lipids is important for their health effects. For example, highly processed coconut oil raises serum cholesterol while virgin coconut oil does not [45, 46]. Thus, the physiological response to high SFA intake goes beyond a “black and white” scenario where different factors such as food source and processing must be considered.
Variation in the dietary uptake of fatty acids have also been implicated in risk of dementia [47–50]. Clinical cohorts, including the Chicago Health and Aging Project (CHAP), the Cardiovascular Risk Factors, Aging and Dementia (CAIDE), and the Three-City study, associate multi-nutrient diets low in PUFA and high in SFA with increased risk of dementia and cognitive deficits [47, 51–53]. Further associations between multi-nutrient diets low in SFA and high in fruit, vegetables, olive oil, and fish strengthen the link between low SFA intake and better cognitive health [25, 54–64]). Albeit some exceptions [65, 66], circulating lipids including LCFA and PUFA also appear to be modulated in dementia and have been regarded as possible biomarkers of cognitive status [67–72]. Thus, lipid metabolism, including that of SFA, remains a critical aspect of dementia that should be taken into account in new interventions for neurodegenerative diseases.
Mono- and poly-unsaturated fatty acids
Contrary to SFA, monounsaturated fatty acids (MUFA) and polyunsaturated fatty acids (PUFA) have one or more carbon-carbon double bonds in their structures. Both MUFA, such as oleic acid (18 : 1), and n-3 PUFA, such as DHA and EPA, have received increasing attention in dementia research as they play fundamental roles in brain structure and function [73–75]. Associative studies consistently report a healthier lifestyle and high PUFA intake with lower risks of dementia [20, 76]. These observations have been studied in registered RCTs where results have been less conclusive [21, 77–80]. Still, multi-domain interventions aiming at preventing dementia via modification of lifestyle factors include high ratios of MUFA and PUFA as part of their dietary patterns and have shown some benefits on cognitive scores [2, 81]. Altogether, while preventive measures based on dietary patterns including PUFA and MUFA, combined with changes in other lifestyle habits, attract a growing interest as a mode of intervention in dementia [2, 81–86], there are many gaps in our understanding of their causal association with brain function that deserve further study.
ANIMAL MODELS USED IN AD RESEARCH
Animal models constitute a critical part of AD research that serve as the basis to decipher pathological mechanisms and test therapeutic targets. Thus, preclinical models that accurately modulate AD pathology are needed [87–90]. Of these, mice are the most commonly used given their relative ease of breeding and genetic manipulation [90, 91]. They also allow the control of study variables to an extent that is not possible in humans. For example, inducing obesity through HFD to determine its impact on AD pathology in humans would be unethical. Mouse models thus, provide an essential tool to bridge the gap in translating preclinical to clinical outcomes in AD research [88, 92]. That being said, one must keep in mind that, because of their short lifespans, mouse models cannot emulate the aging process as it occurs in elderly humans and thus cannot fully incorporate the main risk factor of AD, namely old age. As AD-like pathology does not spontaneously develop in mice, these transgenic mice are rather models of living brains exposed to the progressive build up of molecular hallmarks of AD. The objective behind the use of these models is not to discover what is the initiating culprit that triggers the development of AD (i.e., etiology), but rather to study how an experimental intervention impacts the classical features of AD, in a fully controlled setting. In this perspective, risk factors associated with AD are anticipated to exacerbate AD-related pathologies. Conversely, potential treatments are expected to either mitigate this pathology or delay its onset.
WHAT CONSTITUTES A GOOD MODEL FOR AD?
AD models should display neuropathological hallmarks that are central for diagnosis; that is Aβ pathology and ptau, with ideally some degree of synaptic and neuronal damage [91]. So far, no model fully achieves every feature, most recapitulating only one or a limited set of pathological hallmarks [93, 94]. Nonetheless, a milestone in the development of rodent models of AD came with the implementation of transgenic technology in the early 1990 s. Since murine Aβ and tau are not as prone to form insoluble deposits as human Aβ and tau, the introduction of human transgenes becomes necessary to generate mouse models of AD neuropathology. This made it possible to study in vivo genetic mutations known to cause AD in humans, giving rise to transgenic models bearing one or several phenotypes of the disease. These models have been valuable tools in supporting the launching of clinical trials. The database Alzforum reports 162 mouse models of AD, summarizing the numerous options available and reflecting the progression of new technologies in this area. Defining a good model for AD depends much on the hypothesis investigated. Some models are characterized by a fast induction of AD neuropathology (e.g., intracerebral infusion of Aβ) [95] while in other models this happens more progressively (e.g., chronic transgene expression) [96–98]. The following section briefly covers some of the most prominent mouse models that have been used to investigate HFD, but more extensive reviews can be found elsewhere [90–92, 99–101].
TRANSGENIC MODELS FOR AD
Transgenic models expressing human APP mutations
One of the earliest models of transgenic mice harboring a human APP mutation is the PDAPP mouse model [102, 103]. PDAPP mice exhibit cognitive deficits in spatial and recognition memory from a young age (at 4 and 6 months) [104, 105] and a 10-fold increase in APP, extracellular plaque formation, gliosis, and synaptic deficits [104]. The generation of the Tg2576 model followed PDAPP and is considered one of the most well characterized mouse models of AD [96]. This model overexpresses human APP containing the Swedish mutations (K670 N, M671 L) and develops age-associated cognitive deficits, Aβ plaques and vascular amyloid by 11-13 months [106]. However, a limitation associated with these transgenic models is the lack of extensive neuronal loss and brain atrophy that is seen in AD patients [106].
Efforts to induce a more severe neuropathological phenotype resulted in mice expressing multiple mutations of the familial form of AD (FAD). The J20 model carries two FAD mutations in the APP gene (Swedish and Indiana) and expresses high levels of Aβ [107] while 5xFAD mice harbors five mutations in the APP and PSEN1, showing an aggressive form of the pathology as early as 2 months of age [93]. Mice expressing FAD mutations can partially induce AD-pathology and are useful to study Aβ plaque formation and cognitive deficits. However, a complete recapitulation of AD still lacks in these models given the limited neurodegeneration observed and the absence of tau pathology [108, 109].
Transgenic models with tau pathology
Contrary to FAD mutations in the APP gene, genetic mutations for tau protein have not been described in AD thus far. Instead, models expressing mutations associated with frontotemporal lobar degeneration (FTLD) in the MAPT gene are used to mimic tau pathology observed in AD [110]. The JNPL3 mouse is the first model reported in this category and harbors the P301 L mutation, developing motor and behavioral deficits with age-dependent accumulation of NFTs [111]. In general, tau transgenic mice exhibit NFT-like pathology, neuronal and synaptic loss and cognitive impairment in an age-dependent manner [110]. However, the phenotype induced in these models is not entirely representative of that in human AD since NFT development in these mice requires FTLD mutations, which are not found in AD [112]. An exception is the hTau mouse, which expresses only non-mutated human tau isoforms in the absence of the murine orthologs but nonetheless develops tangles and cognitive deficits [113].
Transgenic models with both Aβ and tau pathologies
A more representative way to study AD pathology is through preclinical models that express both Aβ and NFT pathology. Achieving such a task relies on the simultaneous expression of mutated APP, microtubule-associated protein tau (MAPT) and sometimes PSEN1 or PSEN2. The 3xTg-AD model combines familial mutations for AD (APP Swedish, and PSEN1 M146 V) and FTLD (MAPT P301 L) and is one of the most used transgenic models of AD [90, 115]. These mice develop progressive accumulation of Aβ and NFTs in the cortex and hippocampus, as well as deficits in learning and memory by 6 months [98, 116]. In addition, several sexual dimorphisms have been noted in the 3xTg-AD model as female mice tend to exhibit more pronounced Aβ pathology and age-dependent deterioration in glucose metabolism [115, 117–121]. Altogether, this transgenic model offers the opportunity to study AD in a context of both Aβ and tau pathologies while also considering a degree of metabolic dysfunction.
Knock-in models
Most of the above cited models depend not only on APP mutations to generate Aβ pathology but also in overexpression of APP driven by an exogenous promoter. Since the transgene can be integrated at random locations and in multiple copies, an artificial phenotype of AD can be induced [89, 122]. To overcome setbacks of APP overexpression, knock-in models were developed to produce pathogenic Aβ42 [88, 122–125]. In such models, Aβ is humanized by changing the three amino acids that differ between mice and humans (G676 R, F681Y, H684 R) and two FAD mutations are introduced into the App murine gene [108]. The pathology is generally less aggressive than in previous transgenic models and the timing of pathology depends on the mutation expressed (https://www.model-ad.org/resources/). These mice may represent a step towards a more physiologically accurate model of AD in mice, but defining appropriate controls in knock-in models is complicated [88]. In other words, models with overexpressed genes can be compared with their wildtype controls while it is likely that the human KI gene does not behave like its endogenous murine counterpart [126].
Other rodent models
Mouse models that introduce genetic risk factors for sporadic AD, such as the ɛ4 allele of APOE have also been developed, but many of them have not been exposed to HFD protocols yet [127, 128]. Transgenic mice expressing either human APOE ɛ3, or ɛ4 under the regulation of different promoters, including the neuro-specific enolase (NSE) and glial fibrillary acidic protein (GFAP) have been used for more than a decade [129, 130]. APOE ɛ4 mice have shown age and sex-dependent deficits in learning and high phosphorylation of tau in neurons [99, 132]. The development of mice with different mutations related to AD (APP, PS1, MAPT, and APOE) has also allowed for the creation of double mutant mouse lines. Models like the 5xFAD mice and TgCRND8x, for example, harbor mutations on APP+PS1, and APP+APOE, respectively [93, 133]. Other models have made it possible to study neurovascular dynamics. Transgenic mouse strains with tamoxifen-inducible deletion of Lrp 1 (Slco1c1-CreERT2 Lrp1fl/fl mice) in brain endothelial cells have been used to study the role of LRP1 in AD pathology [134, 135]. In addition to transgenic and knock-in models, cognitive deficits can be induced in mice using Aβ peptides, streptozotocin (STZ), okadaic acid, ibotenic acid, and quinolinic acid. Of these, intracerebroventricular (ICV) injection of Aβ peptides remains the most representative, showing neuronal loss and cognitive deficits similar to AD [136]. Though these mouse models continue to be used, the development of transgenic models have progressively replaced their use in AD research as they mimic more closely the slow progression of AD pathology [100]. Of note, rat and rabbit models have also been developed, at greater cost but with the advantage of allowing for more in-depth behavioral evaluations [137–139]. Finally, many research teams have tried to identify spontaneous models of AD with relatively limited success, particularly because of their short lifespan [90, 141]. When the research in progress will bear fruit, we will develop a better understanding of the impact of HFD on these promising new models.
WHICH FAT MAKES A HFD?
The typical American or European diet contains 36–40% fat by calories [142]. A human HFD contains somewhere between 50% and 60% of fats relative to total caloric intake [143] and may include other nutrients such as cholesterol, sugars, and sodium [144]. HFD in preclinical research aim to replicate as close as possible a human diet enriched in fat and a variety of HFD have been used with differences in fat percentage, fatty acid composition and fat sources [145]. In the scope of this review, mouse diets include a range between 35–60% kcal from fat (Table 1). Fatty acid composition in such diets can also vary in terms of saturation percentage and fat source. Altogether, the common consensus of the definition of a HFD is limited to an elevated percentage of kcal in fat, leaving a gap for variability in terms of fat sources and other macro- and micronutrients.
RELEVANCE OF HFD IN ANIMAL MODELS OF AD
Obesity and T2DM are major health problems that result from high consumption of fat and calories along with a sedentary lifestyle [146]. Besides affecting peripheral organs, both obesity and T2DM have been associated with higher risks of AD [147–153]. Indeed, mid-life obesity has been associated with nearly a three-fold higher risk of developing dementia and with a decreased hippocampal volume [147, 154]. Metabolic dysfunctions in glucose metabolism, insulin signaling, and inflammation resulting from T2DM have also been proposed to contribute to AD pathology while metabolic determinants are considered potential therapeutic targets in AD [8, 155–159]. Causative relations and mechanistic processes, however, are more elusive. Therefore, testing HFD in animal models of AD adds the opportunity to decipher overlapping mechanisms linking obesity, T2DM, and AD. Moreover, long-term HFD, which can be proinflammatory, may also enhance “inflammaging”, a pathway correlating aging and age-related disease with chronic inflammation that is hypothesized to be an initiator of AD [160–165]. Of note, differences regarding the way HFD modulate metabolic indices and AD-like pathology have been linked to biological sex. Male rodents fed a HFD have shown worse glucose metabolism, inflammation, anxiogenic and cognitive deficits [120, 167]. On the other hand, higher inflammation and learning deficits have also been reported in female mice on a HFD [168, 169]. All in all, it is important to consider sex-specific mechanisms when implementing dietary protocols as they may be affected differently by HFD. The following section summarizes evidence from HFD studies in mouse models of AD with outcomes focused on glucose metabolism, insulin sensitivity, Aβ and tau pathology, cognition, and behavior.
EFFECTS OF HFD ON THE METABOLIC PROFILE IN THE MOUSE: RELEVANCE TO AD
Glucose metabolism
Defects of brain glucose uptake is one of the most replicated observations in AD patients and one of the most key information brought by positron emission tomography (PET) over the last decades [170–173]. Relative to its size, the energy required by the brain is high. It is estimated that 20% of energy uptake, mostly in the form of glucose, is destined for this organ [18]. Thus, any disruption of glucose homeostasis may consequently impact brain function. Deficits in normal glucose metabolism may result from the normal aging process but are more pronounced in people with dementia [18, 174–176]. Glucose intolerance and hyperglycemia are prevalent conditions among AD patients and correlate with AD pathology, even without the development of diabetes [177–180].
In mice, metabolic deficits are typically assessed through the glucose tolerance test (GTT), which involves measuring blood glucose concentration at 15-min intervals for 2 h after the administration of a bolus of glucose (1 g/kg, i.p.) to fasted mice [117, 181–184]. The AUC (area under the curve) is then determined and compared between groups. Overall, these tests give an indication of the metabolized glucose during the 2 h period and serve as a tool to evaluate changes in glucose metabolism in AD mice fed a HFD. Imaging techniques such as the 18FDG-PET, and magnetic resonance imaging (MRI), including chemical exchange saturation transfer (CEST)-MRI, have been used in mouse models to assess 2D-glucose uptake, and should be further explored to assess the impact of dietary lipids in glucose metabolism [185–188].
HFD typically induces deficits in peripheral glucose metabolism such as hyperglycemia and aggravated glucose tolerance across distinct AD models (Table 1) [189–194]. In addition, abnormal Aβ peptide metabolism in the brain and periphery of AD mice may also contribute to metabolic disturbances. For example, the genetic induction of AD-like pathology in the brain has been shown to lead to age-dependent deterioration of peripheral glucose tolerance in female 3xTgAD mice at 14 months in comparison to NonTg mice [117, 195], reflecting an overlap between glucose dysmetabolism and AD pathology.
Insulin
While the main role of insulin in peripheral tissues is that of facilitating glucose uptake, its physiological role in the CNS goes beyond maintaining metabolic homeostasis. Insulin receptors in the hippocampus may modulate central pathways involved in neuronal plasticity, learning and memory [196]. Both central and systemic deficits in insulin signaling have been suggested to contribute to AD pathology with therapeutic efforts focusing on improving insulin metabolism in the brain [197–202]. In addition, preclinical and clinical trials show that insulin may improve cognitive functions [159, 203–206], making it an important target linking metabolism and cognition.
Different techniques may be utilized to evaluate insulin sensitivity in animal models. The “gold-standard” to carefully evaluate insulin sensitivity is the hyperinsulinemic-euglycemic (HIE) clamp [207]. When combined with radioactive hexose tracers, the HIE clamp allows to assess whole-body hepatic and peripheral insulin sensitivity in awake mice [208]. This is the only technique that allows the simultaneous determination of both hepatic and peripheral insulin sensitivity in vivo. However, such methodology is invasive and requires specialized technical skills, limiting its implementation in animal models of AD, which require aging [210].
The insulin tolerance test (ITT) and the homeostatic model assessment-IR (HOMA-IR) are simpler alternatives to evaluate insulin action in mice. ITT measures the rate of fall of glucose every 15 min during 1 h after an i.p. injection of an insulin bolus (1 U/kg) [210]. The drop in glucose levels reflects the action of insulin in peripheral tissues, indicating an index of insulin sensitivity [211]. Furthermore, the HOMA-IR assesses β-cell function and insulin resistance from basal glucose and insulin or C-peptide concentrations [212]. Both insulin and C-peptide are produced in equimolar concentrations by pancreatic β-cells and can thus inform on insulin production [213]. However, reproducible results rely on careful manipulation of mice to avoid causing stress and anxiety as well as having exact experimental conditions. Age and sexual dimorphisms have also been reported and it is thus recommended to use age- and sex-matched mice in these tests [211, 214].
Besides deficits in glucose metabolism, high fat intake typically induces deficits in insulin metabolism (hyperinsulinemia and insulin resistance) across mouse models of AD (Table 1) [178, 216]. However, this is less frequently seen in mice expressing tau pathology [217, 218]. Of note, it has been reported that tau can modulate both brain and peripheral insulin metabolism [219, 220], suggesting common pathophysiological pathways between T2DM and tau pathology.
Summary of rodent’s studies and one human study investigating the effect of HFD on AD-related outcomes
Most studies in rodents have investigated the effect of HFD on metabolic endpoints, behavior, and brain amyloid-β load. Some have studied the effect on tau pathology, synaptic and/or neuroinflammatory markers. Diets were various but most included≥40% kcal from fat. Diet composition enabling replication was available in about 33% of studies. Metabolic impairments were confirmed in a majority of studies, with assessment of weight and deficits in peripheral glucose/insulin metabolism. AA, Homozygous carriers of the rs9472159 polymorphism in SLC2A1 gene; Aβ, amyloid-beta; AVLT, Auditory verbal learning task; Chol., Cholesterol; Comp., Composition; GFAP, Glial fibrillary acidic protein; HDL, High density lipoprotein cholesterol; HF/HC, High Fat+High Cholesterol; HFD, High Fat Diet; IBA-1, Ionized calcium-binding adapter molecule 1; LDL, Low-density lipoprotein cholesterol; LFD, Low Fat Diet; ND, Non-determined; pTau, phosphorylated tau; Neuroinflamm., Neuroinflammation; Syp, synaptophysin; TC, total cholesterol; VLDL, Very low density lipoprotein cholesterol. Mouse models cited: Tg2576 and TgAPPSwe: mice overexpressing a mutant form of APP (APPSwedish); TgCRND8: mice overexpressing 3 human mutations in the APP gene (K670M/N671 L and V171F); APP/PS1: double transgenic mice expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9); THY-Tau 22: model of tauopathy with Tau 2 mutations (G272 V and P301 S); PS19: model of tauopathy with hMAPT mutation on P301 S; hTau: mice lack endogenous murine microtubule-associated protein tau (Mapt) gene expression, and express all six isoforms (including both 3 R and 4 R forms) of hMAPT; APPSwe-Tau: Tg2576/Tau(P301 L) generated by crossing Tg2576 mice, which have the transgene for human APP (isoform 695) carrying the Swedish mutation with JNPL3 mice expressing human MAPT (4 repeat) with the P301 L mutation.; 3xTg-AD: Triple transgenic mice expressing 3 human mutations APPswe, PSIMI46 V on APP and tauP301 L hMAPT to generate Aβ and tau pathologies; MxD: Carotid artery occlusion model.
EFFECTS ON BEHAVIOR AND COGNITION
The effect on cognitive behavior is more challenging to detect in animal models that are already impaired due to a threshold effect and is further complicated by the weight gained after HFD. Obese mice may perform differently in tests requiring movement and exploration such as the Morris Water Maze or the open field test, imposing an “obesity” bias [221]. Thus, the aggravating effects of HFD on cognition and behavior can be hard to demonstrate. For instance, transgenic APP/PS1 mice show impairment in short-term memory, irrespective of diet, while HFD seems to drive impaired memory in wild-type animals [222]. Memory deficits caused by HFD are more pronounced in 3xTg-AD mice, as shown by higher escape latency in comparison to other groups [223]. In addition, the strongly decreased scores in various behavioral tests, such as the Y-maze, Novel Object Recognition, Morris Water Maze, and Barnes maze, observed in 3xTg-AD mice with aging [195], were worsened by a HFD [178, 224]. In some occasions, these changes happened without aggravating Aβ or tau pathology [195, 223]. Overall, albeit some exceptions [222, 226], most studies listed in Table 1 report some sort of additional cognitive and behavioral impairment, but results should be interpreted carefully when working with obese mice.
EFFECTS OF A HFD ON AD NEUROPATHOLOGY
Amyloid-β
Genetic data provide a strong support for the hypothesis that accumulation of Aβ peptides is one of the initiators of AD pathology [227–232]. Increased production and reduced clearance of Aβ result in toxic aggregates of Aβ42 and Aβ40 and may promote the onset of the disease [233–236]. Other data point to Aβ accumulation as the consequence from a complex interplay between neurons, astrocytes, microglia, and vasculature that drives neurodegeneration [6, 237]. From a neuropathological perspective, the extent of neuritic plaques and Aβ aggregates in the brain remain the cornerstone of AD diagnosis, which make Aβ levels an inescapable parameter when assessing AD pathology [1, 238].
Perhaps the most consistently reported impact of a HFD is an aggravation of Aβ pathology in the brain (Table 1). Soluble and insoluble forms of Aβ42, Aβ40, the Aβ42/40 ratio, and plaques increased in the hippocampus or cortex after high-fat consumption [239–242]. Still, as shown in Table 1, HFD may not always induce such changes. Impairments in learning and memory were reported in AD-HFD mice without significant changes on Aβ proteins [191, 223]. Data in animal models support an aggravating effect of HFD on several forms of Aβ but its deleterious impact on cognitive function may also stem from additional mechanisms that are not limited to Aβ pathology.
Hyperphosphorylated tau
Tau is a microtubule associated protein with high susceptibility to post-translational modifications, one of which is phosphorylation [243–245]. Accumulation of tau aggregates that include ptau and NFTs is a classical feature observed in AD brains and strongly correlated with cognitive function [232, 247]. Results on the effect of HFD on tau pathology in mice are more equivocal than with Aβ. As seen in Table 1, some studies report increased forms of ptau in HFD-fed mice, notably in mice expressing transgenes-induced tau pathologies [217, 248–250]. HFD also leads to slight increases limited to specific forms of tau, like cortical ptau, [242], certain tau epitopes [217, 251], and insoluble [249] or total human tau [250]. Additional studies in models carrying both APP and tau mutations show that tau phosphorylation is not markedly affected by HFD consumption or is even decreased (Table 1) [120, 252–254]. Overall, given the dynamic nature of tau, data on tau pathology is highly variable with differences in genotypes, protein forms and anatomical regions. Further studies, with more controlled settings, are thus necessary.
Synaptic markers
Synaptic dysfunction is a common pathological feature in AD that correlates with cognitive decline [232, 255–257]. Synaptophysin, drebrin, PSD-95, SNAP25, and syntaxin are often reported to be downregulated in the AD brain, with synaptophysin distinguishing mild cognitive impairment and AD cases from controls [243, 259]. Prominent decreases in postsynaptic drebrin and PSD-95 have also been reported in the cortex of AD patients [258, 260] while biomarkers of synapse dysfunction have surged as promising measures of cognitive status [255, 261–263]. There is also some evidence linking pathological Aβ and tau to synaptic loss [264–266], further making synaptic dysfunction a key aspect of AD physiopathology.
When reported, high fat intake does not consistently induce significant changes on synaptic proteins (Table 1) [178, 250]. Still, reduced levels of drebrin, synaptophysin, or syntaxin have been reported after high fat consumption [249, 267]. Of note, drebrin is also markedly reduced after n-3 PUFA deprivation [258] and upregulated by dietary DHA in the hippocampus [268] and the cortex [258], perhaps revealing a susceptibility of this protein to specific dietary fats. Though synaptic dysfunction is not routinely included as an endpoint in the studies reviewed, data suggest a tendency for individual synaptic markers to be more strongly affected by the type of fat in diets. It is important to consider that HFD have different ratios of n-3 PUFA that, although found in low levels, can modulate synaptic markers. Thus, it is the proportion of each fatty acid in a diet, rather than their total amount, that appears to exert an influence on synaptic proteins.
Neuroinflammatory markers and oxidative damage
Neuroinflammation and oxidative damage are important features in NDD [269–271]. In the AD brain, microglia bind to misfolded proteins that trigger neuroinflammatory responses, resulting in elevated biomarkers of lipid peroxidation in postmortem brain tissues of AD patients [272, 273]. Obesity and metabolic syndrome are associated with higher markers of inflammation in the periphery while the opposite seems to be true for diets low in fat or carbohydrate [274, 275]. This is consistent with mice fed HFD where inflammatory responses are activated, both in the brain and in peripheral organs (Table 1). APP/PS1 mice fed a HFD have a greater increase in hippocampal microglial activation than transgenic mice fed a normal diet or non-transgenics fed a HFD [191, 192]. Other studies have also reported elevated inflammatory markers in the cortex or hippocampus of mice as a response to HFD and irrespective of genotype [195, 276–278]. Oxidative damage is also aggravated in different organs following a HFD, including the microvasculature [278], cortex [223], and liver [192, 279]. In essence, inflammatory, and oxidative responses in HFD-fed mice remain underlying mechanisms linking nutrition, metabolic dysfunction and AD pathology, but that can be hard to pinpoint methodologically.
HFD WITH SUGAR OR SODIUM
A distinction should be made between diets with a high content of saturated fat only and those that include other nutrients such as salt or sugar. Indeed, besides being high in saturated fat, westernized diets (WD) are associated with overprocessed foods and may incorporate a high content of certain types of proteins, trans fats, refined grains, sugar, alcohol, salt, and high-fructose syrup with a reduction in fruits and vegetables [280]. To recapitulate a more accurate version of a WD in humans, HFD used in animals sometimes include elevated quantities of sugar or sodium [184, 282]. However, the incorporation of these two nutrients may inevitably impose an additional variable for the outcomes seen on AD mice. For example, in 5xFAD mice, a WD led to higher Aβ deposits in hippocampal microvessels with no difference in parenchymal Aβ pathology [283]. Meanwhile, a WD containing 57% kcal in sugars increased hippocampal Aβ42 in APP/PS1 mice [284]. Genotype and sex differences may also be present as WD-fed E3FAD female mice showed higher hippocampal Aβ load whereas no alterations in Aβ pathology were seen in E4FAD mice [285]. The opposite seems to be true for males where a worsening effect of Aβ burden by WD is seen in E4FAD versus the E3FAD genotype [286]. Altogether, when comparing the effect of various HFD on AD endpoints, it is important to consider the inclusion of other nutrients frequently found in WD.
HOW CAN SFA CONTRIBUTE TO AD PATHOLOGY?
Direct effect on the brain
Fifty-five percent (dry weight) of the human brain is composed of lipids. In mammals, PUFA accounts for 30% of brain fatty acids, at least half from DHA, while SFA and MUFA make up the rest (70%) [287–291]. Although the composition in PUFA in the brain was repeatedly shown to be altered by dietary shifts in fatty acids, brain SFA composition is more stable and less dependent on the relative dietary intake [19, 292–295]. Still, specific dietary manipulations high in SFA have shown to increase SFA levels in the cortex and hippocampus [250, 297], showing a degree of susceptibility of brain SFA to dietary manipulations. However, it is not clear yet whether such small changes in membrane SFA can influence physiological properties of neurons, such as those observed with PUFA and MUFA [73, 298–303]. Evidence gathered over the years suggest that HFD may impact the endothelium of brain microvessels, cerebral blood flow, and neuroinflammation while disrupting the equilibrium between the uptake of nutrients versus clearance of toxic proteins. As the brain’s energy requirement is relatively high compared to other organs, any defects in energy uptake would also compromise brain functionality [18]. It has been shown that HFD can downregulate cerebrovascular levels of the glucose transporter-1 (GLUT-1) and decrease insulin degrading enzyme (IDE), resulting in blunted brain glucose uptake and accumulation of toxic Aβ aggregates [155, 304–306]. HFD may also promote central insulin resistance, which is detected in the brain itself in AD, particularly at the vascular level [157, 202] and negatively affect neurovascular coupling and cerebrovascular function even in the absence of dyslipidemia [307]. These metabolic abnormalities would then induce alterations in neuronal morphology and physiology that translate in decreased long-term potentiation and reduced markers of synaptic plasticity [41]. Several lines of studies also suggest that HFD generate a proinflammatory environment, which may further favor Aβ deposition and tau phosphorylation and negatively affect synaptic plasticity [41, 308–312]. Increased lipid peroxidation and reduced autophagy have also been reported in the brain of obese mice while mitochondrial regulators including SIRT3 and PGC1α are reduced by saturated fats and cholesterol consumption, which may promote the accumulation of neurotoxic proteins and lead to neuronal death [313–316]. Furthermore, serotonergic and dopaminergic pathways are susceptible to the excess of fat and sugar intake which can induce in depressive-like behaviors [317–319]. Alterations in insulin metabolism [318] and neuroinflammatory responses in the hypothalamus and hippocampus have been described as likely mechanisms [320–324]. Altogether, changes in the brain lipid profile and downstream metabolites, alterations in central glucose metabolism, and neuroinflammatory responses, all caused by high fat intake, can form part of a pathological loop linking fatty acid consumption and neurodegeneration in AD (Fig. 1).
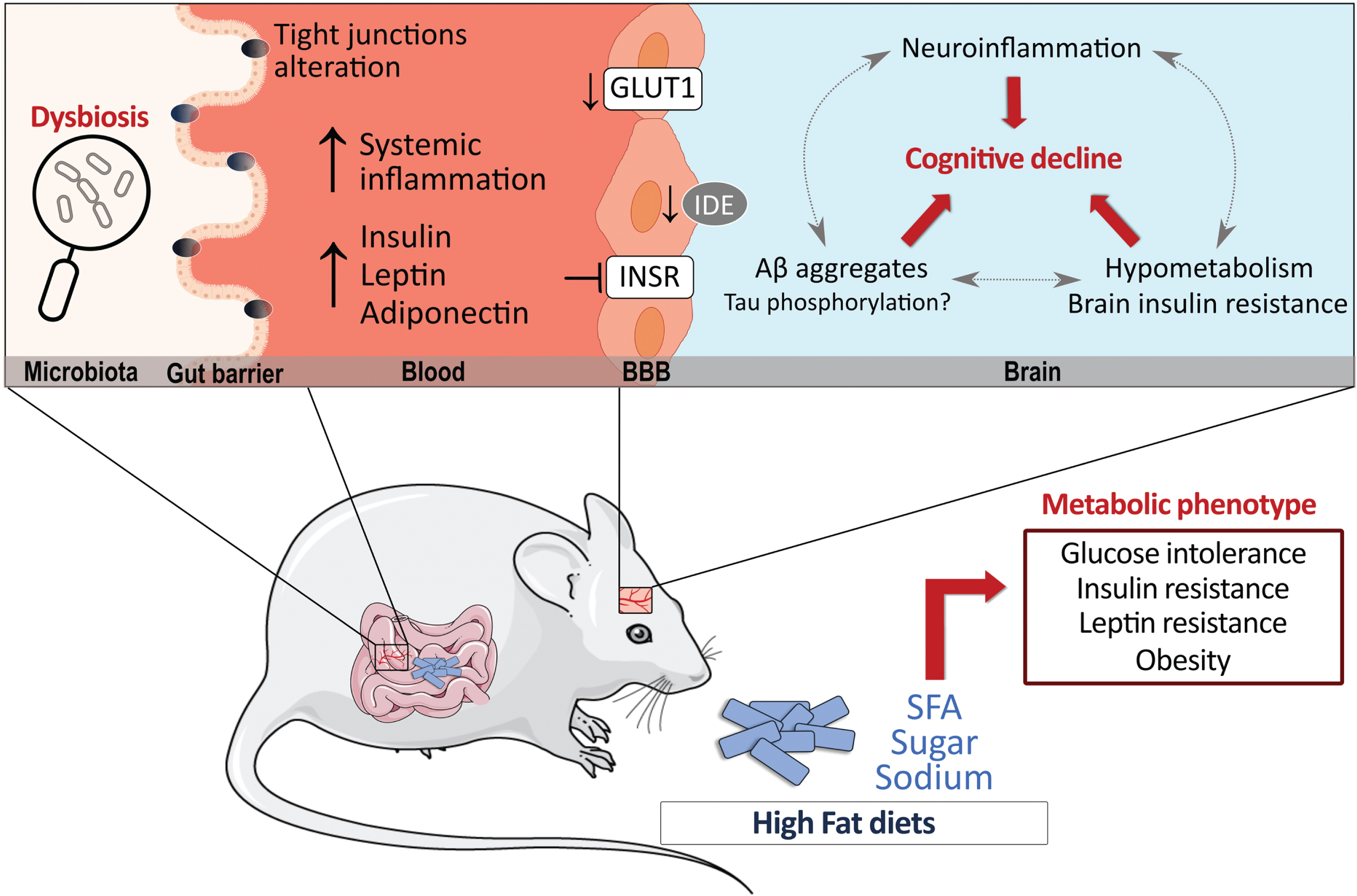
High-fat diets (HFD) have significant metabolic effects and can contribute to Alzheimer’s disease (AD) pathology. Possible mechanisms by which HFD can modulate AD pathology are shown, whether directly in the brain or indirectly in the periphery. Data suggest that HFD may promote insulin resistance and decrease GLUT-1 and IDE at the cerebrovascular level while also triggering an inflammatory response in the brain. In the periphery, the alteration of insulin metabolism, adipokines and gut microbiota can indirectly affect brain processes. Finally, the majority of reports indicate that a prolonged exposure to a HFD increases brain Aβ levels, whether its effects in tau pathology is less clear. Aβ, amyloid peptides; BBB, blood-brain barrier; GLUT1, glucose transporter 1; IDE, insulin degrading enzyme; INSR, insulin receptor; SFA, saturated fatty acids.
Indirect effect via the periphery
HFD can also impact the brain indirectly through its effect on the periphery [319]. As evidenced by Table 1, when correctly assessed, insulin resistance is often detected in HFD-fed animal models of AD (Table 1). Notably, insulin administration reversed HFD-induced metabolic and cognitive deficits in 3xTg-AD mice while age-dependent deficits in insulin and glucose metabolism have been reported in the 3xTgAD mice, suggesting a vicious cycle between insulin metabolism and AD pathology [117, 178]. The altered metabolism of other adipokines including adiponectin and leptin also stand as key pathways linking T2DM and AD pathology (reviewed elsewhere: [325–327]). Furthermore, specific nutrients have the potential to change gut microbiota and influence CNS diseases upward through the gut-brain axis [328–334]. For example, SFA metabolism has been reported to increase intestinal permeability by affecting tight junction proteins and inducing dysbiosis of gut microbiota, processes that have been implicated in the pathology of AD [335–342]. Therefore, it is likely that exaggerated intake of HFD disrupts energy-regulating processes in the periphery that potentiate the detrimental effects on metabolic, cognitive, and neuropathological dysfunction in AD [8, 343–345].
VARIATIONS IN HFD COMPOSITION AS COFOUNDING FACTORS IN PRECLINICAL STUDIES
The present review highlights some inconsistency in results regarding the effect of HFD on AD-relevant endpoints in animal models (Table 1). An important factor to consider when comparing preclinical studies is the difference between HFD in terms of fat percentage and fat composition. Fat content in HFD can vary from 45% fat kcal to 60% fat kcal [120, 218]. As the definition of HFD is limited to a fat content between 40–60% kcal in fat, using different percentages of fat in diets may inevitably induce different degrees of metabolic deficits in mice, which could affect cognitive, behavioral, and neuropathological markers differently. We have found that complete characterization of the HFD used is reported in a little less than half of published papers, making comparisons even more difficult.
Fat source and composition are also important to consider when comparing HFD studies. HFD are typically made up of a combination of SFA, MUFA, and PUFA, which often varies between diets. For instance, Bracko et al. report a SFA:MUFA:PUFA proportion of 62% : 27% : 4.7%, respectively [222], whereas another HFD rather comprises 22% : 66% : 10.4% of each FA species [191]. Fat source in HFD further contributes to this variability [40]. Such variability in the proportion of fatty acids is critical when using HFD in AD models as PUFA intake, such as DHA, is typically associated with beneficial effects on cognition [20, 76] and MUFA could play a similar role [300, 346]. Thus, a HFD composed mostly of high SFA will yield different results than a HFD based on elevated MUFA or PUFA. Lastly, the impact of dietary treatment on actual fatty acid content in the brain is rarely disclosed in studies. HFD can change the lipid profile in the brain [250, 347], which remains an essential information to draw mechanistic conclusions at the molecular level. Overall, to fully benefit from the advantages of preclinical studies, a complete characterization of the fatty acid profiles both in the diet and in target tissues is necessary and should always be recommended.
LIMITATIONS
The present study provides an overview of the impact of HFD in animal models of AD, but is limited first by the information available in published papers. Although studies that included only female or only male mice were detailed in Table 1, the effects reported did not account for sex-specific effects in many instances. When a study does not report sex-specific interactions, it is difficult to know if they were non-existent or because male and females were not separated in the analysis. However, considerations were made in the text regarding sex-specific effects. Secondly, while we report the duration of diet in Table 1, the age of mice when starting diet consumption was not included, making comparisons between age groups more challenging. Lastly, our focus was on studies in the mouse, but we are aware that there might be HFD studies in other mammals, such as rats, or smaller organisms that were outside the scope of this review. Future studies that aim to fill these gaps would be valuable.
CONCLUSIONS, IMPLICATIONS FOR THE HUMAN CONDITION AND FUTURE STUDIES
Nutrition has become an important factor to consider when developing therapeutic strategies in AD. Diets have the potential to play a protective role, alter positively or negatively disease progression or interact with treatment. Consumption of high amounts of SFA and other energy-dense nutrients is highly prevalent in our society, and this has contributed to higher incidence of not only obesity and T2DM, but also neurodegenerative diseases. Controlled, RCT would be essential to issue recommendations regarding mono- and poly-unsaturated fatty acids. However, testing the effect of HFD in humans would not only be unethical, but would also be plagued by many confounding variables. On the other hand, these diets can be investigated in animal models in fully controlled settings in a shorter timeframe. There are over 30 reports of HFD in animal models of AD that can be found on PubMed since 2002. In rodent models, the intake of a HFD generally aggravates cognitive performance, but most study paradigms can be biased by the weight gain inevitably associated with it. The majority of studies have found an aggravating effect of a HFD intake on brain Aβ pathology. Tau has been much less studied, and results are more equivocal due to the complexity of post-transcriptional changes, while other key AD markers have been even less consistently reported. Discrepancies between studies can be explained by differences in the diet composition, the age and duration of exposure, the exact model used and endpoints assessed, among other variables discussed above. We have found that more than half of reports do not even fully disclose the diets used. Future studies should consider that distinct fatty acids, despite small chemical differences, may modulate pathophysiological pathways very differently. More specifically, when considering HFD protocols, forthcoming studies need to document percentages, sources, and types of fatty acids; aspects that are not consistently reported across laboratories. These are important steps towards harmonizing data regarding the effect of HFD on the different animals in AD, which is pivotal to issue sound conclusions, and to generate evidence that can be compared and replicated, serving as a basis for the launching of clinical trials and discovery of new preventive strategies and therapies in AD.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to Vincent Émond for his critical comments on the manuscript.
FUNDING
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) to F.C. [grant numbers PJT 168927, PJT 156054]. F.C. is a Fonds de recherche du Québec-Santé (FRQ-S) senior research scholar. J. V-E. was supported by scholarships from Fonds d’Enseignement et de la Recherche (FER) from the Faculty of Pharmacy, Laval University, from the Fondation du CHU de Québec, from the Fondation Lemaire and the Consortium pour l’Identification Précoce de la Maladie d’Alzheimer (CIMA-Q). M.L. was supported by scholarships from the Fondation du CHU de Québec and from FRQ-S.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.