
Abstract
Alzheimer’s disease (AD) is considered to be the most common neurodegenerative disease, with clinical symptoms encompassing progressive memory loss and cognitive impairment. Necroptosis is a form of programmed necrosis that promotes cell death and neuroinflammation, which further mediates the pathogenesis of several neurodegenerative diseases, especially AD. Current evidence has strongly suggested that necroptosis is activated in AD brains, resulting in neuronal death and cognitive impairment. We searched the PubMed database, screening all articles published before September 28, 2022 related to necroptosis in the context of AD pathology. The keywords in the search included: “necroptosis”, “Alzheimer’s disease”, “signaling pathways”, “Aβ”, Aβo”, “Tau”, “p-Tau”, “neuronal death”, “BBB damage”, “neuroinflammation”, “microglia”, “mitochondrial dysfunction”, “granulovacuolar degeneration”, “synaptic loss”, “axonal degeneration”, “Nec-1”, “Nec-1s”, “GSK872”, “NSA”, “OGA”, “RIPK1”, “RIPK3”, and “MLKL”. Results show that necroptosis has been involved in multiple pathological processes of AD, including amyloid-β aggregation, Tau accumulation, neuronal death, and blood-brain barrier damage, etc. More importantly, existing research on AD necroptosis interventions, including drug intervention and potential gene targets, as well as its current clinical development status, was discussed. Finally, the issues pertaining to necroptosis in AD were presented. Accordingly, this review may provide further insight into clinical perspectives and challenges for the future treatment of AD by targeting the necroptosis pathway.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), which is known to be the most common chronic neurodegenerative disease, clinically manifests as progressive memory loss, and cognitive impairment, which ultimately affects behavior, speech, visuospatial orientation, and the motor systems [1]. According to the World Alzheimer’s Disease Report, by 2050, 115 million people worldwide have been predicted to suffer from dementia, placing a heavy burden on societies and families [2]. To date, no effective treatment to cure or slow the progression of the disease exist. Neuropathologically, AD is characterized by the extracellular amyloid-β (Aβ) deposition, intracellular hyperphosphorylated Tau (p-Tau) formation of neurofibrillary tangles (NFTs), significant neuronal loss, and severe neuroinflammation [3–6]. However, the underlying mechanisms of these pathological processes have yet to be clarified. In light of recent research progress, the relationship between these pathological processes and necroptosis has garnered increased attention from scientists.
Necroptosis cell morphology resembles necrosis and is characterized by the loss of plasma membrane integrity, organelle swelling, chromatin fragmentation, and late cell lysis [7]. The execution of necroptosis involves three core components, namely receptor-interacting protein kinases (RIPK)1, RIPK3, and mixed lineage kinase domain-like protein (MLKL). Death receptors (DRs) activate RIPK1 kinase activity under the conditions of caspase inhibition, which alongside RIPK3 heterodimerization form amyloid-structured necrosomes and activate RIPK3 via phosphorylation. Activated RIPK3 subsequently recruits and phosphorylates MLKL, which leads to oligomerization and translocation of MLKL as well as the disruption of the plasma membrane, ultimately leading to necroptosis [8]. Currently, numerous studies have evidenced the involvement of necroptosis in AD pathology both in vitro and in vivo [9], which involved Aβ aggregation [10, 11], p-Tau and NFTs [12], neuronal death [8], blood-brain barrier (BBB) damage [13], granulovacuolar degeneration (GVD) [14], synaptic loss [15], restricted O-linked β-N-acetylglucosaminylation (O-GlcNAcylation) [16, 17], mitochondrial impairment [18], and neuroinflammation [19] leading to subsequent cognitive impairment [8]. In addition, the pharmacological and genetic inhibition of RIPK1/3 and MLKL have been shown to effectively ameliorate the pathological changes and cognitive deficits in associated AD models [10, 20].
The present review attempts to discuss the pathway that activates necroptosis and describes the mechanism of action of MLKL as an executor of necroptosis. Next, the mechanism by which necroptosis is involved in the corresponding pathological changes in AD are explained. More importantly, existing research pertaining to AD necroptosis intervention are summarized, including drug intervention and potential gene targets, as well as the current clinical development status. Finally, issues that warrant further discussion in necroptosis in AD are presented. The findings of this review may offer insight into the clinical perspectives and challenges involved in the future treatment of AD by targeting the necroptosis pathway.
METHODS
Literature retrieval and analysis were conducted in the PubMed database. The key words included a single search and combination search of “necroptosis”, “Alzheimer’s disease”, “signaling pathways”, “Aβ”, Aβo”, “Tau”, “p-Tau”, “neuronal death”, “BBB damage”, “neuroinflammation”, “microglia”, “mitochondrial dysfunction”, “granulovacuolar degeneration”, “synaptic loss”, “axonal degeneration”, “Nec-1”, “Nec-1s”, “GSK872”, “NSA”, “OGA”, “RIPK1”, “RIPK3”, and “MLKL”. Considering the novelty of the study as well as the latest research progress in the field, the retrieval time was set to be before September 28, 2022.
ACTIVATED PATHWAY OF NECROPTOSIS
In specific conditions, necroptosis is activated by DRs, including TNF receptor 1 (TNFR1), IFN receptor (IFNR), TRAIL receptors (TRAILR), and Toll like receptor (TLR)4/3, which upon stimulation by their cognate ligands, activate RIPK1. The subsequent section briefly introduces the activation of necroptosis according to two aspects of the different signaling pathways involved as well as the execution of downstream MLKL.
Different signaling pathways involved
TNF-α-triggered necroptosis is the most widely studied mechanism of necroptosis, in which TNF-α binds primarily to two TNF receptor subtypes, TNFR1 and TNFR2 [21]. Unlike TNFR1, the latter does not contain the intracellular death domain (DD) necessary to mediate cell death responses [22]. Therefore, this review mainly discusses the TNFR1-mediated death signaling pathway (Fig. 1). Upon stimulation by TNF-α, TNFR1 interacts with TNFR1DD to recruit cellular inhibitor of apoptosis proteins (cIAP), RIPK1, TNF receptor associated factor (TRAF) and TNF receptor-associated death domain (TRADD) to the intracellular portion of TNFR1, thereby forming complex I [23]. Complex I then activates NF-κB and AP1 transcription factors in order to promote cell survival and pro-inflammatory gene expression [24]. When the ubiquitination of RIPK1 in complex I is inhibited, RIPK1 dissociates from the cell membrane and recruits TRADD and TRADD-FAS-associated DD protein (FADD) to form complex IIa, which then activates caspase-8 and leads to apoptosis [25]. When caspase activity is insufficient (via genetic ablation or pharmacological inhibition), RIPK1 and RIPK3 interact through the RIP homology-interacting domain (RHIM) to form RIPK1-RIPK3 necrosomes while phosphorylating RIPK3 (complex IIb) [26]. This is followed by p-RIPK3, which phosphorylates MLKL and facilitates its translocation to the plasma membrane, where MLKL triggers cell lysis, resulting in lytic cell death as well as the release of damage-associated molecular patterns (DAMPs) [24, 27].
IFN signaling is known as a signaling pathway that can activate necroptosis [28]. The IFN-I receptor consists of a heterodimer of two IFN-α receptor type I proteins (IFNAR1 and IFNAR2). Moreover, the binding of IFN-I to ubiquitous IFN receptor 1 (IFNAR1) activates Janus kinase (JAK), which further activates signal transducers and activators of transcription (STAT) 1/2 and interferon regulatory factor 9 (IRF9) in order to form IFN stimulation of the gene factor 3 (ISGF3) complexes (Fig. 1). The ISGF3 complex then promotes the transcriptional activation of interferon-inducible protein kinase R (PKR). Once activated, PKR recruits RIPK1 and RIPK3 to form a PKR necrosome, which phosphorylates MLKL to form RIPK1-RIPK3-MLKL to promote necroptosis [29]. Notably, PKR necrosomes are negatively regulated by FADD and caspase, and when FADD is silenced or when caspase is inhibited, RIPK kinase is overactivated, and ROS are overproduced and cannot be efficiently quenched in the mitochondria, leading to cell necroptosis [30].
Initiators of the TLR signaling pathway, including LPS-activated TLR4 signaling and TLR3 (which is activated by dsRNA in the endosome), are able to induce necroptosis following caspase inhibition via downstream TIR-domain-containing adapter-inducing interferon-β (TRIF). TRIF interacts with TLR3 or TLR4 through the RHIM domain in order to recruit RIPK1/3; when caspase is inhibited, necroptosis occurs [31].
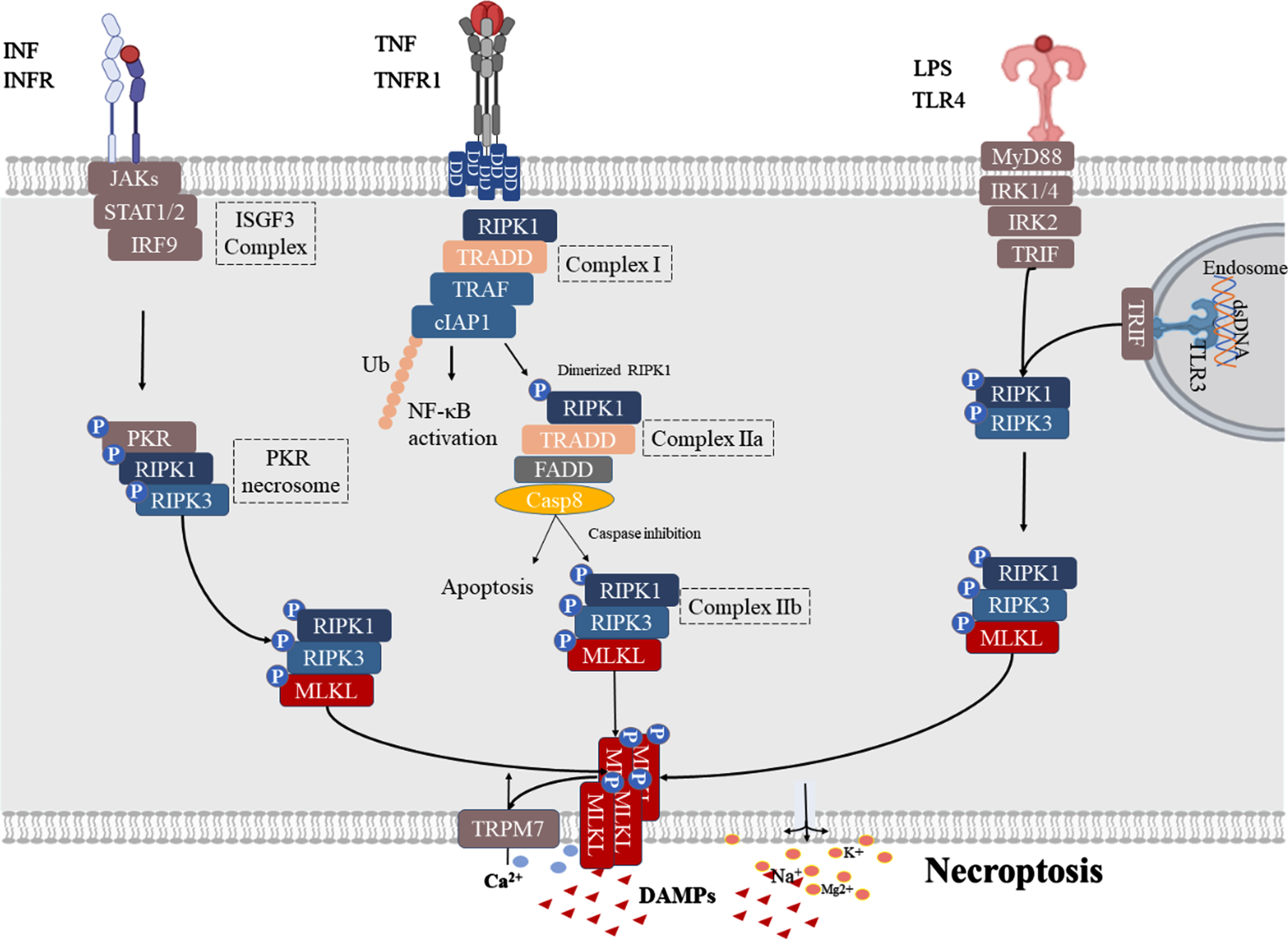
Activated pathway of necroptosis. TNF signaling: Upon stimulation by TNFα, TNFR1 interacts with TNFR1 DD to recruit cellular inhibitor of apoptosis proteins (cIAP), RIPK1, TNF receptor associated factor (TRAF), and TNF receptor-associated death domain (TRADD) to the intracellular portion of TNFR1 to form complex I and activation NF-κB. When the ubiquitination of RIPK1 in complex I is inhibited, RIPK1 dissociates from the cell membrane and recruits TRADD, TRADD-FAS-associated DD protein (FADD) to form complex IIa that activates caspase-8, and leads to apoptosis. When caspase activity is insufficient, RIPK1 and RIPK3 interact through the RIP homology-interacting domain (RHIM) to form RIPK1-RIPK3 necrosomes and phosphorylate RIPK3 (complex IIb). This is followed by p-RIPK3 which can phosphorylate MLKL, facilitating its translocation to the plasma membrane where MLKL leads to cell lysis, resulting in Ca2 + influx, Na+, K+, and Mg2 + efflux, and DAMP release. IFN signaling: Binding of IFN-I to IFNAR1 further leads to PKR necrosome formation by activating the assembly of the ISGF3 complex, which phosphorylates MLKL to form RIPK1-RIPK3-MLKL for necroptosis. TLR signaling: TLR3/4 are activated by their respective ligands, and TLRs can activate necroptosis by recruiting RIPK1-RIPK3-MLKL necrosomes by linking TRIF.
The executor of necroptosis: MLKL
To date, MLKL has been identified as a downstream target of RIPK3, which further induces cell death by perforating the cell membrane leading to leakage of cellular contents and release of inflammatory factors [32]. Specifically, MLKL contains an N-terminal 4-helix bundle (4HB) domain, an intermediate brace region composed of two helices (Brace), and a C-terminal pseudo kinase domain (PsKD). Activation of MLKL requires RIPK3 to bind PsKD and phosphorylate MLKL, thereby inducing the release of the 4HB domain and formation of homo-oligomers through the Brace [33–35]. Upon activation, MLKL binds to the cytoplasmic membrane via an affinity site that binds phosphatidylinositol phosphate (PIP), and when MLKL undergoes translocation to the cytoplasmic membrane, it disrupts membrane integrity in a dose-dependent manner [36]. During this process, when MLKL binds to the cell membrane, its 4HB domain uses a “flip” mechanism to expose additional high-affinity PIP-binding sites, which adds another layer of different PIP-binding sites, making MLKL more plasma-membrane-bound [37]. Six helices (H1-H6) in the N-terminal domain following the translocation of MLKL form a cation channel that is permeable to Mg2 +, Na+, and K+ [38]. Moreover, oligomerized MLKL binds to transient receptor potential melastatin related 7 (TRPM7), allowing to Ca2 + influx, thereby leading to cell swelling and plasma membrane rupture in order to release DAMP [39].
NECROPTOSIS CONTRIBUTES TO AD PROGRESSION IN MULTIPLE PATHOLOGICAL PROCESSES
Necroptosis had been extensively explored in neurodegenerative diseases research, including multiple sclerosis (MS) [40, 41], AD [9, 11], Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [42, 43]. The activation of necroptosis has been detected in AD brains of both humans and mice, which plays a key role in AD progression in association with multiple pathological processes (Fig. 2).
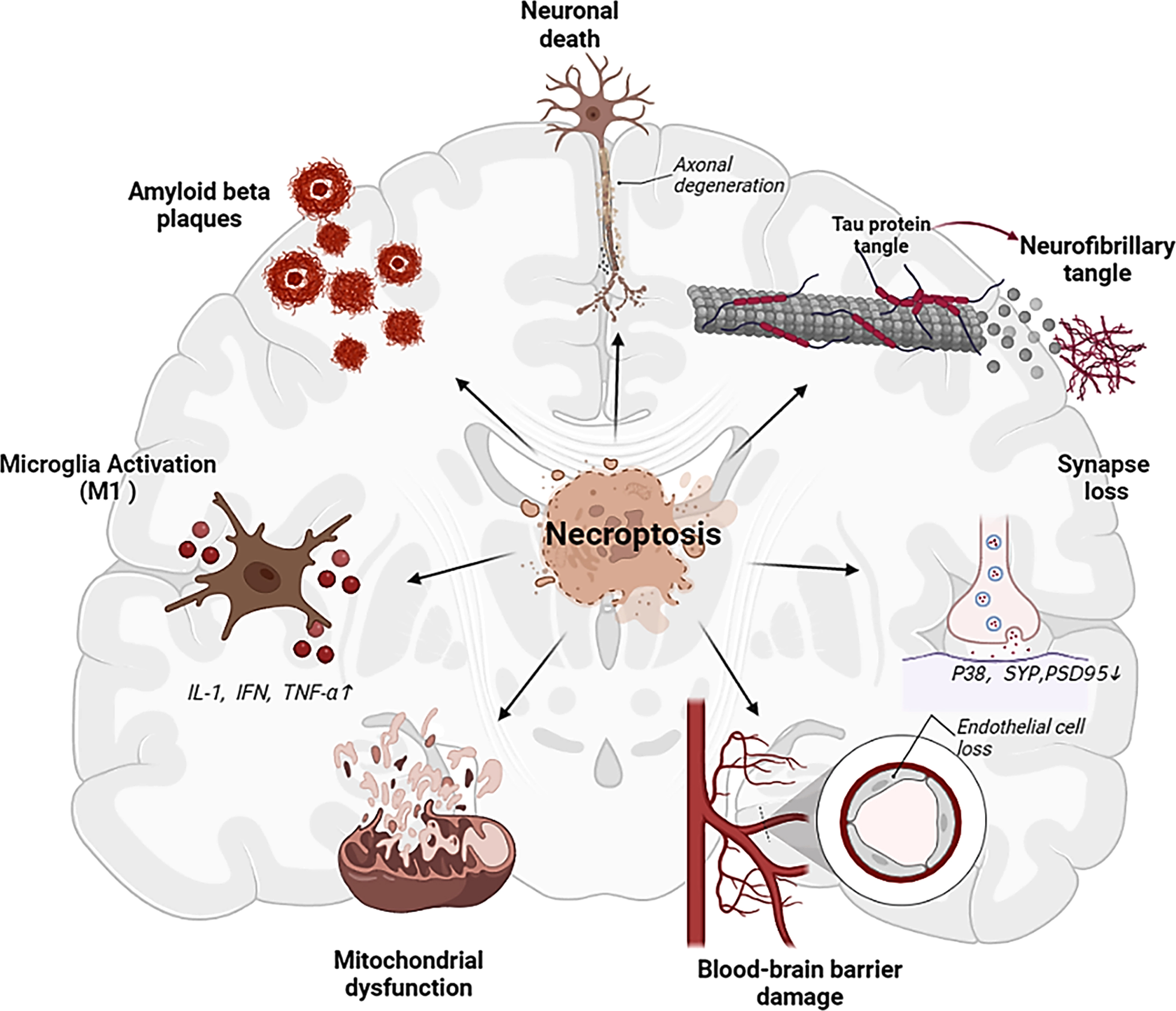
Multiple influences of necroptosis activation on AD related factors, which may serve as important triggers of cognitive impairment.
Necroptosis and Aβ aggregation
Aβ is deposited in the brain decades before the manifestation of clinical symptoms and induces senile plaque formation at a later stage [44, 45], thereby facilitating neuronal cell death and dementia [46]. Aβ is the product of hydrolysis of amyloid-β protein precursor (AβPP), which is first sorted in the endoplasmic reticulum (ER) and then transported from the Golgi apparatus to the trans-Golgi-network (TGN) [47–49]. Next, a portion of AβPP is transported as secretory vesicles to the cell surface or endosomal compartment where it is hydrolyzed by α-secretase in order to generate neuroprotective sAβPPα through a non-amyloid pathway [50–52]. The other part is cleaved by β-site amyloid cleavage enzyme 1 (BACE1) into β-N-terminal fragment (sAβPPβ) and membrane-bound C99. The latter acts on the 40/42 amino acid position of the Aβ sequence by γ secretase to produce a 39–42 amino acid peptide-Aβ and another intracellular fragment AβPP intracellular domain (AICD) [53, 54]. Aβ peptides exist in the form of monomers, oligomers (Aβo), and fibers/aggregates. Aβ monomers are relatively non-pathogenic, though they aggregate into toxic insoluble fibers that eventually assemble into amyloid plaques, of which Aβo is the most toxic type of Aβ [55–58]. Aβ insoluble fibers and oligomers often synergistically produce multiple cytotoxic effects, leading to neuronal death, immune cell activation, and inflammatory cascade [53, 60].
Through the activation of necroptosis, the RIPK1/RIPK3 complex forms amyloid fibril structures [26]. These cross-β-structured amyloids result in a rise level of full-length AβPP and sAβPPβ at the cell surface [61, 62] (Fig. 3). Consistent with this, in Aβ-treated SH-SY5Y cells, RIPK1 on Aβ-induced cell death and endogenous AβPP protein stability was demonstrated [63]. Nec-1, the first RIPK1 kinase inhibitor discovered, has been shown to inhibit the downstream activation of necroptosis by binding to the hydrophobic pocket between the N- and C-termini of the RIPK1 kinase domain [64, 65]. Studies have also shown that Nec-1 small molecules can dock in several hydrophobic pockets in the Aβ fibril structure and may bind to hydrophobic sites to dissociate Aβ aggregates [11]. This finding indicated that necroptosis promotes the aggregation of Aβ through the formation of insoluble amyloid complexes [61]. Inhibiting RIPK1 kinase activity via pharmacology and genetic techniques has been found to reduce this phenomenon in APP/PS1 mice [11, 19].
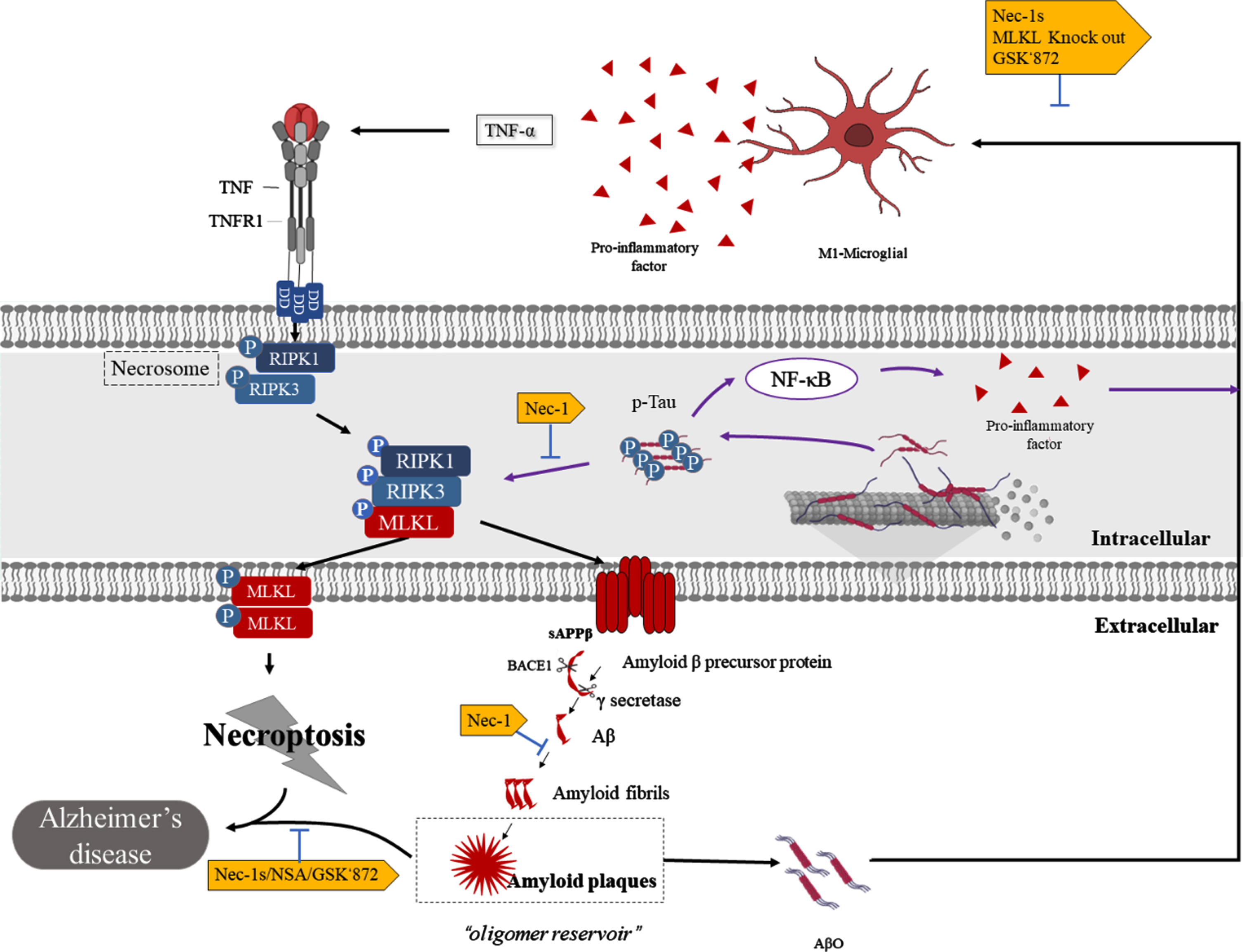
The interaction of necroptosis and Aβ formation and Tau. Binding of the death receptor TNF and the ligand TNFR1 stimulates the formation of necrosomes, which consist of RIPK1 and RIPK3 and allow autophosphorylation. Then, p-RIPK3 recruits and phosphorylates MLKL, promoting its translocation to the plasma membrane, leading to necroptosis. In this pathway, the RIPK1/RIPK3 complex forms amyloid fibril structures and increases cell surface levels of full-length AβPP and sAβPPβ, promoting Aβ plaque aggregation. Furthermore, Aβ plaques act as a reservoir for Aβo, and when Aβo is released, it stimulates microglia to secrete inflammatory factors, including TNF-α, and to undergo necroptosis. On the other hand, hyperphosphorylated Tau promotes necroptosis by activating the RIPK1/RIPK3/MLKL axis and simultaneously activates the NF-κB pathway, leading to M1microglial activation and neuroinflammation.
Aβ aggregates and Aβo, in turn, promote neuronal necroptosis. Studies have shown that Aβ aggregates induce neuronal necrosis through the RIPK1-MLKL axis and are inhibited by Nec-1 [11, 63]. It has been hypothesized that Aβ plaques act as a reservoir for toxic oligomers, where over time the plaques saturate and Aβo diffuses, exerting toxicity on peripheral neurons [56, 66–68]. This has been found to be consistent with the findings in which Aβ plaques frequently localize around necroptotic complexes [8]. In fact, Aβo has been shown to activate necroptosis and neurodegeneration in microglia through the promotion of TNF-α release from microglia, with Infliximab protecting it. Interestingly, no neurodegeneration has been found in neurons treated with TNF-α alone [10]. Meanwhile, the knockout of MLKL, or pharmacological inhibition of RIPK3 (GSK’872), has been shown to reverse this phenomenon and improve the cognitive impairment of Aβo-induced mice [10]. Therefore, necroptosis and Aβ may form a cascading effect in AD; that is, necroptosis complexes promote Aβ aggregation to form plaques, and over time, AβO diffusion further exacerbates neurotoxicity and disease severity.
Necroptosis and p-Tau
Tau protein is known to serve as a major microtubule-associated protein (MAPT) in the mammalian nervous system. In AD, abnormal post-translational modifications have been shown to give rise to hyperphosphorylation of Tau (p-Tau) [69, 70]. p-Tau accumulates in neurons in order to form NFTs at several residues, including Ser393/404, Ser202, and Thr 205 [71]. This process destroys the microtubule structure of neurons, resulting in a loss of communication between the neurons. Braak et al. demonstrated in their clinicopathological correlation study that AD brain pathological changes can be divided into six stages, which are referred to as “Braak stages”, according to the amount of NFT accumulation and regions involved [72]. In addition, p-Tau has been detected in the cerebrospinal fluid and blood of AD patients, with the amount detected correlating with the degree of cognitive impairment [73].
In a study on GVDs, the co-expression of pMLKL and p-Tau in GVD-bearing neurons has been observed in AD and pre-AD human brains, suggesting that Tau accumulation may serve as a key necroptosis-activated trigger [14], which have been confirmed by recent studies [12]. Specifically, in three cell lines (HT22 nerve cells, HEK293T cells, and SH-SY5Y cell line) transfected with p-Tau, Tau was found to mediate necroptosis and inflammation via activation of the RIPK1/RIPK3/MLKL and NF-κB pathways in promoting cell death [12] (Fig. 3). The in vitro findings were then further validated in separate in vivo studies in TauP301S mice. Remarkably, Nec-1stable (Nec-1s, a Nec-1 analog) was found to inhibit p-Tau-induced neuronal death and microglial hyperactivation in TauP301S mice, significantly downregulate cytokine expression, and ameliorate cognitive deficits in an AD model [12]. Similarly, in APP/PS1 mice, Nec-1 has been shown to reduce p-Tau by blocking p-Tau S199 and inhibiting aggregation through direct interaction [9, 11]. Furthermore, in an AlCl3-induced AD rat model, treatment with necrosulfonamide (NSA), a pharmacological inhibitor of MLKL, has been found to reduce p-Tau levels and improve associated spatial learning and memory deficits [20]. These findings suggest a close relationship between p-Tau and necroptosis. Furthermore, pharmacological intervention of RIPK1 and MLKL can reduce p-Tau-induced cell death and amend cognitive deficits in AD models.
Necroptosis and neuronal death
Neuronal death in the hippocampus serves as the main pathological hallmark of AD and is closely related to memory loss. Neuronal loss in certain regions, such as layer II of the entorhinal cortex, Meynert basal ganglia, and locus coeruleus has been shown to occur even before the onset of AD symptoms [74].
Conventional wisdom posits that neuronal death in AD is usually based on two categories: caspase-mediated apoptosis and DRs-mediated necroptosis [75, 76]. Apoptosis is characterized by cell shrinkage, membrane blebbing, as well as the formation of apoptotic bodies. In various studies, the caspase-mediated apoptosis signaling pathway has been found to be inactive in AD [8, 78]. Instead, these cells exhibited a necroptotic swollen morphology and were found to be positive for DNA fragmentation, demonstrating that apoptosis does not sufficiently explain neuronal loss in AD. In addition, according to disease development, apoptosis is acute, and cells are lost within hours or days after apoptosis, while AD is a progressive process lasting decade [79, 80]. In terms of cellular immune response, apoptosis is immune silent, and its activation does not cause an inflammatory response [81–83]. Necroptosis typically results in the rupture of the plasma membrane as well as the subsequent release of cellular content into the surrounding environment, leading to a severe inflammatory response that may contribute to neuroinflammation in AD [84]. In view of these findings, apoptosis may not serve as the only factor that can lead to neuronal death as necroptosis is also involved.
Previous studies have demonstrated that necroptosis is activated in AD and that two key necroptosis proteins, RIPK1 and MLKL, are significantly increased in the AD postmortem brain, which leads to decreased neuronal survival and brain weight [8, 9]. Furthermore, TNFR1/RIPK1 signaling and altered expression of endosomal sorting complexes required for transport group III (ESCRT III) have been found to be activated in the AD postmortem brain, which may serve as a possible mechanism underlying neuronal necroptosis [8]. The latter has been identified to be an antagonist of necroptosis as well as a possible mechanism by which cells can survive after necroptosis has been triggered [85]. However, the small molecule inhibitors of RIPK1, RIPK3, and MLKL have been observed to significantly reverse this effect [5, 86]. These studies demonstrated that neuronal death may play a role in degeneration as an independent factor rather than a downstream product of pathogenic proteins.
In fact, we do not deny the existence of other types of programmed cell death in AD including pyroptosis, ferroptosis, oxytosis, autophagy, etc. Moreover, there may be extensive crosstalk between these mechanisms in chronic degenerative diseases, where they may interact with each other intracellularly, with the dominant mechanism ultimately leading to cell death and further giving rise to inflammation and contributing to further neurotoxicity.
Necroptosis and BBB damage
The BBB is mainly composed of continuous capillary endothelial cells (capECs), continuous basement membrane, pericytes, and astrocytes, of which endothelial cells (ECs) are the main components of the BBB. This structure has selective permeability and transport functions for peripheral substances, protecting neurons from various factors in the systemic circulation, thereby maintaining a highly regulated internal homeostasis of the central nervous system. A dynamic contrast-enhanced (DCE) MRI study of individuals with mild cognitive impairment described BBB injury in the hippocampus, suggesting that BBB damage occurs early in AD [87]. Studies have shown that BBB damage and Aβ act independently and/or synergistically in order to promote AD progression [88–91]. BBB injury is also affected by certain factors, such as genetics (i.e., carrier of apolipoprotein E (APOE ɛ4) E4 allele), cardiovascular and cerebrovascular disease (i.e., hypertension, diabetes, and dyslipidemia) [89].
ECs play a key role in the BBB, and damage to ECs results in BBB injury with concomitant infiltration. A recent study has demonstrated that electron microscopy and immunohistochemistry can reveal the selective loss of venous and capECs in APP/PS1 mice [13]. Necroptosis in venule ECs and capECs from AD mice and postmortem human AD brain samples was found to be a major contributor to EC loss and BBB leakage. Research has shown that the inhibition of RIPK1 kinase activity ameliorates TNFα-induced increases in vascular permeability in TNFα/Z-ValAla-Asp (OMe)-fluoromethyl ketone (ZVAD, a caspase inhibitor) treated mice [92]. In other acute brain injuries, Nec1 has been found to prevent brain swelling and disruption of tight junctions in subarachnoid hemorrhage as well as avert BBB injury triggered by necroptosis in mice models of cerebral ischemia/reperfusion injury [93, 94]. Accordingly, the corresponding studies confirmed that necroptosis plays an important role in BBB injury. Furthermore, previous studies have shown that necroptosis promotes the aggregation of Aβ via the formation of amyloid complexes [11], whereas Aβ promotes AD progression synergistically with BBB injury [95]. Therefore, it is necessary to elucidate how necroptosis-induced Aβ deposition leads to BBB damage in other animal models.
Necroptosis and neuroinflammation
Neuroinflammation is a common AD feature. Microglia, which constitute the main immune cells of the brain, work with oligodendrocytes and astrocytes in order to maintain central nervous system homeostasis and have been shown to coordinate neuroinflammation and play a key role [96, 97]. Through the influence of different environments, M1-type microglia usually exhibit a pro-inflammatory phenotype, whereas M2-type microglia have an anti-inflammatory phenotype. In AD, M1-type microglia release inflammatory factors, such as IL-1, IFN, and TNF-α, which have been shown to be involved in neuroinflammation and further lead to peripheral neuronal death [98, 99]. M2-type microglia is induced by IL-4 and/or IL-10 and are associated with clearance of Aβ [100, 101]. Microglial responses have been initially thought to be serendipitously triggered by Aβ amyloid plaques and dystrophic neurites. Genome-wide association studies have also shown that many AD risk genomes are highly expressed at or near genes unique to microglia [97]. Recently, due to progress in necroptosis research, numerous studies have illustrated that necroptosis activates microglial activation.
Specifically, necroptosis has been described to be active in the microglia and can induce the M1 polarization of microglia/macrophages [102–104]. In rd1 mice with retinal degeneration, as well as in mice with acute retinal nerve injury, microglia have been shown to undergo RIPK1- and RIPK3-dependent necroptosis and release multiple proinflammatory cytokines and chemokines via TLR4 signaling pathway [105]. Likewise, in the macrophages/microglia of mice, MLKL and the key necroptosis regulator Z-DNA/RNA-binding protein 1 were found to be mainly induced in M1 but not M2 macrophages/microglia [106]. Ablating RIPK3 or MLKL could switch the activation of microglia/macrophages from M1 to the M2 type in the ischemic cortex [107]. Studies have shown that RIPK1 is highly expressed in the microglia of AD patients [19]. By inhibiting RIPK1 kinase expression, a significant reduction in M1 microglia and levels of proinflammatory cytokines have been demonstrated. Specifically, the kinase activity of RIPK1 has been described to mediate the transcriptional upregulation of Cystatin 7 and Cholesterol-25-Hydroxylase, leading to the inhibition of cathepsin activity and impairment in the microglial lysosomal pathway in AD mice microglia [19]. These studies further demonstrate that the activation of necroptosis mediated the inflammatory response and phagocytic capacity damage of microglia. Therefore, targeting necroptosis can promote the transformation of M1 to M2 phenotype and restore the phagocytic function of microglia, which may potentially serve as a therapeutic strategy for AD.
Necroptosis and mitochondrial dysfunction
The brain is characterized by high energy metabolic demands and is extremely dependent on mitochondria for its energy supply; therefore, it is extremely sensitive to mitochondrial dysfunction. Extensive research has demonstrated that mitochondrial dysfunction in AD is closely related, which can manifest in mitochondrial fission-fusion, abnormal mitochondrial dynamics, and oxidative damage [108–110]. Studies have shown that mitochondria in AD have decreased activity in all respiratory chain complexes, having the most significant decrease in complex IV, which results in increased ROS production, decreased ATP production, and DNA damage. These factors contribute to the processing and aggregation of Aβ, thus triggering synaptic degeneration and cognitive decline [111–113]. p-Tau also leads to mitochondrial dysfunction, affecting the axonal transport of organelles in AD neurons and ultimately leading to neuronal dysfunction [114].
Past studies have found that necroptosis is involved in mitochondrial dysfunction. Specifically, the mitochondrial protein phosphatase PGAM5 is a component of the RIPK1/RIPK3 complex and manifests as two splice variants, PGAM5L (long form) and PGAM5S (short form). Following the induction of necroptosis, PGAM5S further induces mitochondrial fragmentation through the recruitment of dynamin-related protein 1 (Drp1) and activation of its GTPase activity by dephosphorylation, which are key steps in the subsequent execution of necroptosis [18]. On the contrary, RIPK1/MLKL/PGAM5L interaction is blocked by another necroptosis inhibitor, NSA. A recent study has reported that active necrosomes are translocated to the mitochondria in an MLKL-dependent manner, in which RIPK3 directly phosphorylates the E3 subunit of the pyruvate dehydrogenase complex (PDC) at T135, leading to PDC activation, and subsequent rise in aerobic respiration and mitochondrial reactive oxygen species production [115]. Interestingly, mitochondrial ROS in turn induces autophosphorylation of RIPK1 at Ser161, which is critical for the formation of RIPK1-RIPK3 necrosomes [116, 117]. These results may suggest that mitochondrial dysfunction is greatly involved or tends to form a feedback loop in order to induce necroptosis. Essentially, the inhibition of necroptosis was shown to aid in the recovery of mitochondrial dysfunction in neurons, ultimately alleviating cognitive impairment.
Necroptosis and granulovacuolar degeneration
GVD was first proposed in 1911. When GVD forms up to 3–5μm in diameter, a membrane-bound structure of edged vacuoles with a dense argyrophilic core is present. In AD pathology, it is often characterized by the presence of abundant GVDs in neurons with ultrastructural resemblance to lysosomes [118]. These GVDs bodies have been initially and predominantly found in hippocampal pyramidal neurons in CA1 and CA2. As AD progresses, the systemic spread of GVD lesions in the brain, such as the temporal lobe, hypothalamus, and amygdala, has been observed [119, 120]. Accordingly, in vitro induction of intracellular aggregation of Tau has been found to lead to the formation of GVDs in mouse primary neurons [121]. In the AD hippocampus, the number of neurons with GVDs increases according to the Braak stage of NFTs, which may participate in the formation of NFTs [122–125].
Koper’s team was the first to describe the relationship between necroptosis and GVDs in the context of AD-associated neuronal loss, proposing the hypothesis of GVDs as a morphological correlate of necrosome activation in AD [14]. In addition, their study examined areas with significant neuronal loss in pre-AD and AD patient, in which the presence of large numbers of necrosome-positive GVDs were found. In other words, GVD-mediated necroptosis leads to massive neuronal loss in pre-AD and AD cases. Recently, Koper et al. have found that hippocampal transactive response DNA binding protein 43 (TDP-43) inclusion bodies are closely related to GVD-mediated necroptosis [126]. TDP-43 inclusion bodies have been frequently detected in up to 57% of AD cases and have been associated with worsening brain atrophy and greater memory loss in AD patients [127, 128]. Notably, a large number of necrosome-positive GVDs were found in the hippocampus of AD patients with TDP-43 compared with AD patients without TDP-43 [126, 128]. Consistent with this, in a study of C9ORF72-mutated ALS/frontotemporal dementia (FTLD) cases, TDP-43-related necrosome-positive GVD was also observed in the post-mortem hippocampus [129]. Taken together, TDP-43 aggravates GVD-mediated necroptosis in AD and ALS/FTLD cases and leads to more severe dementia. However, further elucidation of the mechanism by which TDP-43 activates GVD-mediated necroptosis in AD is needed.
Necroptosis and other AD pathological events
Synaptic loss
Studies have suggested that between 27% and 42% of synapses in the frontal cortex of the brain are lost in AD patients [130–135], in which the absence of key elements in interneuron communication has been thought to constitute the main morphological counterpart of cognitive deficits in AD. Cornel Iridoid Glycoside (CIG), the main component of cornus officinalis, which is widely used in China to treat age-related diseases and dementia, has been shown to increase the expression of synaptophysin, postsynaptic density-95 (PSD95), and Glutamate receptor 1 (GluR1) in the hippocampus of SAMP8 mice by inhibiting the RIPK1/MLKL pathway [15]. However, whether necroptosis inhibitors rescue AD synaptic damage has yet to be reported as there is a lack of strong morphological evidence.
Axonal degeneration
Axonal degeneration is another important pathological event of advanced AD [136]. Recent studies have found that necroptosis is a key mechanism in axonal degeneration following excitotoxicity [137]. Activation of necroptosis, mitochondrial dysfunction, and axonal degeneration has been frequently observed in neurons [136]. Moreover, the pharmacological inhibition of RIPK1 has been found to prevent key steps in the axonal degeneration cascade, including mitochondrial depolarization, opening of the permeability transition pore, and Ca2 + dysregulation in the axon. In accordance with this finding, the same effect was observed for RIPK3 and MLKL knockdown [118].
THERAPEUTIC MEASURES TARGETING THE AD BRAIN NECROPTOSIS PATHWAY
Pharmacological intervention of necroptosis in AD animal models (Table 1)
Data from in vivo experiments have indicated that several compounds inhibit necroptosis and improve cognitive impairment in AD models. These compounds include necroptosis (RIPK1/RIPK3/MLKL) inhibitors and other drugs.
Pharmacological intervention of necroptosis in AD animal models
IV, intravenous injection; PO, oral; ICV, lateral ventricle injection; Bcl-2, B-cell lymphoma-2; p-RIPK3, phosphorylated receptor-interacting serine-threonine kinase 3; mGluR2/5, metabotropic glutamate receptors2/5; LC3II, microtubule-associated proteins 3II; NF-κB, nuclear factor-κB; pRIPK1, phosphorylated receptor-interacting serine-threonine kinase 1; ACh, acetylcholine; Bax, pro-apoptotic gene; PARP2, poly adenosine diphosphate ribose polymerase; Bmf1, BCL2 modifying factor 1; Rab25, member RAS oncogene family 25; IL-6, interleukin 6; IFNβ, interferon beta; CCL5, C-C motif chemokine ligand 5; TMEM119, transmembrane protein 119; MLKL, mixed lineage kinase domain like pseudokinase; vECs, venous ECs; capECs, capillary ECs; VCAM1, vascular cell adhesion molecule 1; ICAM1, intercellular cell adhesion molecule-1; BACE1, β-site amyloid cleavage enzyme 1; GSK-3β, glycogen synthase kinase 3 beta; PSD95, postsynaptic density-95; GluR1, Glutamate receptor 1; ADAM10, ADAM metallopeptidase domain 10; JNK, c-Jun N-terminal kinase; p-JNK, phosphorylated amino-terminal protein kinase; STAT3, signal transducer and activator of transcription 3; p-STAT3 (Ser 727), phosphorylated signal transducer and activator of transcription; t-STAT3, total STAT3; FGF2, Fibroblast growth factor 2; BDNF, brain-derived neurotrophic factor; TrKB, Tyrosine Kinase receptor B; AKT, Protein Kinase B, PKB; TBK1, TANK-binding kinase 1; CIG, Cornel Iridoid Glycoside; EGb761, Gingko biloba extract 761; CE, Coeloglossum viride var. bracteatum extract; Mouse Transmembrane protein 119.
RIPK1 inhibitors: Nec-1 is a classic inhibitor in targeting RIPK1 kinase [138]. To date, Nec-1 has been shown to have an ameliorating effect on multiple AD/cognitive deficit-related animal models, including APP/PS1 mice [11], aluminum induced-AD mice [139], aluminum-zebrafish AD models [140], and prediabetic rats [141]. However, the specificity of Nec-1 in inhibiting RIPK1 kinase has been widely debated [142]. This may be due to its inhibitory effect on indoleamine-2,3-dioxygenase (IDO) [142]. IDO has been found to be upregulated in inflammation and plays a major immunomodulatory role [143]; hence, the dual response to RIPK1 and IDO may have important in vivo effects. Nec-1s is a more specific RIPK1 inhibitor that lacks the IDO-targeting effect [142]. The corresponding findings suggested that Nec-1s improves cognitive deficits in SAMP8 mice [144], TauP301s mice [12], and APP/PS1 mice [19]. RIPK3 inhibitors: GSK’872 is a RIPK3 inhibitor that binds to the RIPK3 kinase domain with high affinity and inhibits kinase activity, which has also been shown to ameliorate cognitive decline in an AβO-induced AD model [10]. Although GSK872 has limited research data in terms of AD animal models, its potential in improving cognitive deficits from in vitro experiments can be appreciated [8, 106]. For example, GSK’872 was added to human iPSC-derived glutamatergic neurons and M1 macrophages induced by TNF-α, SMAC mimic, and ZVAD-fmk (TSZ, a commonly used in vitro method to induce necroptosis), which significantly reduced cytotoxicity and increased neuronal survival [8, 106]. MLKL inhibitors: NSA is a specific inhibitor of necroptosis that targets MLKL. The study of Motawi and colleagues showed that NSA reduced the pathological changes associated with cognitive decline in an AlCl3-induced AD rat model given oral administration of AlCl3 (17 mg/kg/day) for 6 consecutive weeks [20]. In future studies, the therapeutic effect of NSA must be validated in further AD models. Recently, GSK’074 and TAK-632 have been identified as dual inhibitors of RIPK1 and RIPK3, both of which exhibited extremely low cytotoxicity [145–147]. Although dual inhibitors have advantages in blocking RIPK1-dependent and -independent forms of necroptosis, how effective these dual inhibitors are in AD must be clarified.
Other related studies have demonstrated that O-GlcNAcase (OGA) inhibitors can increase O-GlcNAcylation [17, 148]. Interestingly, O-GlcNAcylation of RIPK3 has been shown to inhibit the phosphorylation of RIPK3 as well as its interaction with RIPK1, thereby further inhibiting the activation of necroptosis [16, 149]. Notably, OGA inhibitors have also been found to ameliorate cognitive impairment in Tau/APP [16] and 5xFAD mice model [17] through this effect.
Regarding other drug treatments, CIG has been reported to increase the expression of synapse-associated proteins by inhibiting necroptosis and ultimately improve cognition in aged SAMP8 mice [15]. Gingko biloba extract 761 has also been shown to improve cognitive impairment in a Aβ1 - 42-induced AD rat model by attenuating RIPK1-mediated mitochondrial dysfunction and ROS [150]. Coeloglossum viride var. bracteatum extract (CE) is a plant that grows on snowy plateaus at an average altitude of 4000 meters and possesses antioxidant and anti-inflammatory properties. CE ameliorates cognitive impairment in Aβ25 - 35 induced AD mice by modulating RIPK1-driven inflammation and necroptosis [151].
Potential gene targets of necroptosis in AD animal model (Table 2)
Recently, the potential gene targets of necroptosis in AD have been gradually revealed, and data from in vivo studies have demonstrated its ability in improving cognitive impairment in the AD animal models [10, 152]. For example, knockout of RIPK1D138N kinase in APP/PS1 mice has been found to ameliorate cognitive impairment by reducing microglia-associated inflammation [19]. Furthermore, knockout of MLKL improved cognition as well as learning and memory in Aβo-induced AD mice models [10]. Murine N-acetyltransferase 1 (mNat1) has been shown to play an important role in preventing insulin resistance-mediated endothelial necroptosis, with its deficiency leading ECs vulnerable to necroptosis [13]. The selective restoration of mNat1 expression in ECs can inhibit EC necroptosis and ameliorate BBB damage and cognitive impairment in APP/PS1 mice [13]. UVRAG serves as a key regulator in several steps of autophagic flux. Interestingly, UVRAG overexpression has been found to inhibit necroptosis by regulating autophagy, ultimately alleviating cognitive impairment in APP/PS1 mice [152]. Previous studies have also identified p62/SQSTM1, which is a molecule responsible for the clearance of aggregated proteins, in which RIPK1 directly interacts to activate necroptosis [163]. Xu et al. showed that p62 is highly expressed in hippocampal CA1 cells of APP/PS1 mice as well as in the cortical neurons of AD patients. In contrast, p62 knockdown in APP/PS1 mice has been found to inhibit necroptosis and alleviate cognitive impairment [152]. In addition, various genes play important roles in AD necroptosis, such as: A20 [13, 154], TRAF2 [155], and low-density lipoprotein receptor-related protein 1 [13]. Given that information on these genes is admittedly limited in in vitro studies, further research is needed in order to demonstrate their potential as a target for inhibiting necroptosis in AD.
Potential genetic targets of necroptosis in AD animal models
EC, Endothelial cells; VCAM1, vascular cell adhesion molecule 1; ICAM1, intercellular cell adhesion molecule-1; LRP1, low-density lipoprotein receptor-related protein 1; mNat1, murine N-acetyltransferase 1; TSZ, TNF-α, SMAC mimetic, ZVAD-fmk; UVRAG, ultraviolet irradiation resistance-associated gene; LC3, microtubule-associated proteins, the ratio of LC3II/I can estimate the level of autophagy; P62, Sequestosome 1; shRNA, short hairpin RNA.
CLINICAL TRIALS TARGETING AD NECROPTOSIS
Drug developers are now working to move drug candidates into preclinical studies or clinical trials by targeting necroptosis. Currently, RIPK1 inhibitors, RIPK3 inhibitors, MLKL inhibitors, and OGA inhibitors related to necroptosis have been gradually suggested for the treatment of neurodegenerative diseases such as AD. Therefore, clinical trials regarding these inhibitors were searched for using the Informa Pharma Intelligence database (https://citeline.informa.com/), where five RIPK1 inhibitors and four OGA inhibitors were found for AD (Table 3). However, it is disappointing that no RIPK3 and MLKL inhibitors have entered clinical trials so far.
Clinical trials for AD
The above data comes from Informa Pharma Intelligence.
Clinical trials targeting RIPK1
DNL104 commenced trials in AD/ALS in September 2016, though further studies have been discontinued due to post-dose hepatotoxicity that was observed in six subjects (37.5%) in the multiple escalating dose group. SAR443060 (DNL747) is a selective, orally bioavailable, and central nervous system (CNS)-penetrant inhibitor of RIPK1. In two early-stage clinical trials (NCT03757325 and NCT03757351) in healthy subjects and in patients with AD or ALS, DNL747 has been shown to be distributed in cerebrospinal fluid after oral administration and decreased pRIPK1 in human peripheral blood mononuclear cells. However, the development of DNL747 has been discontinued due to long-term nonclinical toxicology findings [156]. Another BBB-penetrating RIPK1 inhibitor, SAR443820 (DNL788), demonstrated potent interactions with its target at well-tolerated doses in Phase 1 clinical trials (NCT04982991), which may have potential in certain neurological diseases, such as ALS, MS, and AD. It is now planned for Phase II clinical trials for both ALS and MS (NCT05237284). Importantly, the US FDA has granted Fast Track designation to SAR443820 for the treatment of ALS. SIR-2446 is an oral RIPK1 inhibitor being developed by Sironax for the treatment of AD and MS, which is currently in Phase I clinical trials. VRN-04-1 (VRN-04) is another oral RIPK1 inhibitor developed by Voronoi for the treatment of various autoimmune diseases and neuroinflammation diseases, such as AD, PD, ALS, and MS, which in preclinical trials.
Clinical trials targeting OGA
ASN120290 (ASN-561 or ASN90, developed by Asceneuron) is used for the treatment of progressive supranuclear palsy, AD, and tauopathies, which has already completed testing in three Phase I studies in healthy young and elderly subjects. Preclinical data has shown that daily oral administration of ASN120290 prevents the development of Tau tangles, functional deficits in motor behavior and breathing, and increased survival [157]. BIIB-113 (developed by Biogen) is currently in a Phase I clinical trial for AD to evaluate the safety and tolerability of single- and multiple-ascending oral BIIB113 in healthy participants (NCT05195008). ASN-51 (developed by Asceneuron) has also entered Phase I clinical trials for AD. The safety, tolerability, pharmacokinetics, and pharmacodynamics of ASN51 have been evaluated in healthy subjects and AD subjects (NCT04759365). LY-3372689 (developed by Eli Lilly and Company) for the treatment of AD has completed safety, tolerability, and pharmacokinetic evaluations in 23 healthy subjects (NCT03819270, NCT04106206, NCT03944031, and NCT04392271). It was well tolerated, with no serious adverse events reported and is currently undergoing recruitment for a Phase II clinical trial (NCT05063539).
CONCLUSION AND FUTURE DIRECTIONS
Due to the rapidly aging population, novel disease-modifying therapies and treatment strategies are urgently needed for AD. In fact, for decades, researchers have characterized several aspects of AD pathology as separate components that may now be associated with susceptibility to necroptosis. The existing evidence has demonstrated that necroptosis is involved in the neuropathology of AD [8–10, 158]. For these reasons, there is indeed a broad correlation between AD neuropathology and the necroptosis pathway. Therefore, the role of necroptosis in AD is multifaceted, and inhibiting the activation of necroptosis may serve as a multi-targeted therapeutic approach for AD.
However, several outstanding questions regarding AD necroptosis remain to be further addressed. 1) The above studies show a strong correlation between necroptosis and AD pathological changes. However, as AD is a slowly progressive disease, it is necessary to clarify the degree of necroptosis in AD animal models at different pathological stages and to find the optimal treatment time. 2) Nec-1 targets the dissociation of Aβ aggregates; however, current evidence only supports a role in the pre-AD stage. Thus, more studies are warranted to investigate whether other dual inhibitors have binding sites for Aβ, and whether the treatment is better than Nec-1. 3) The hypothesis of GVDs as the morphological counterpart of AD necrosome needs to be further validated in vivo. Furthermore, the mechanism by which TDP-43 activates GVD-mediated necroptosis in AD should be elucidated. 4) Synapse loss is considered to be the main morphological counterpart of cognitive deficits and memory impairment in AD. Although the inhibition of necroptosis signaling improves the expression of synapse-related proteins, more robust morphological evidence is required. In conclusion, this review put forward evidence in regard to the involvement of necroptosis in AD progression according to AD neuropathology. However, the role of necroptosis in AD may only be confirmed when further necroptosis-based interventions successfully complete clinical trials. Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0809r2).
Footnotes
ACKNOWLEDGMENTS
This work is supported by grants from the Wu Jieping medical foundation clinical research project (320.6750.2022-08-3). The authors would like to thank Zibo Yimore Translation CO. LTD for providing English proofreading services for this paper.