
Abstract
Background:
There are more than 300 presenilin-1 (PSEN1) mutations identified but a thorough postmortem neuropathological assessment of the mutation carriers is seldom performed.
Objective:
To assess neuropathological changes (NC) in a 73-year-old subject with the novel PSEN1 G206R mutation suffering from cognitive decline in over 20 years. To compare these findings with an age- and gender-matched subject with sporadic Alzheimer’s disease (sAD).
Methods:
The brains were assessed macro- and microscopically and the proteinopathies were staged according to current recommendations.
Results:
The AD neuropathological change (ADNC) was more extensive in the mutation carrier, although both individuals reached a high level of ADNC. The transactive DNA binding protein 43 pathology was at the end-stage in the index subject, a finding not previously described in familial AD. This pathology was moderate in the sAD subject. The PSEN1 G206R subject displayed full-blown alpha-synuclein pathology, while this proteinopathy was absent in the sAD case. Additionally, the mutation carrier displayed pronounced neuroinflammation, not previously described in association with PSEN1 mutations.
Conclusion:
Our findings are exceptional, as the PSEN1 G206R subject displayed an end-stage pathology of every common proteinopathy. It is unclear whether the observed alterations are caused by the mutation or are related to a cross-seeding mechanisms. The pronounced neuroinflammation in the index patient can be reactive to the extensive NC or a contributing factor to the proteinopathies. Thorough postmortem neuropathological and genetic assessment of subjects with familial AD is warranted, for further understanding of a dementing illness.
INTRODUCTION
Mutations in the presenilin-1 (PSEN1) gene are the most common genetic cause of familial Alzheimer’s disease (AD) [1–3]. The mutations are autosomal dominant and mostly associated with an aggressive progression of the disease, with onset of symptoms before the age of 65 [1–4]. Up to date, there have been over 300 different mutations identified within PSEN1 [5].
Presenilin 1 (PS1) is a constituent of the γ-secretase enzyme complex involved in the cleavage of amyloid-β protein precursor (AβPP), yielding the amyloid-β (Aβ) fragment [6, 7]. The Aβ peptides, especially Aβ42, have an ability to accumulate into larger oligomers and fibrils that aggregate extracellularly, forming what is referred to as amyloid plaques, one of the hallmark lesions of AD neuropathological change (ADNC) [8, 9]. During the course of the disease, Aβ plaques are observed in predisposed neuroanatomical regions, initially in the neocortex but subsequently engaging also the limbic structures, brainstem and, at advanced stages, the cerebellum [10, 11]. Some reports have described that certain PSEN1 mutations feature unique characteristics of Aβ aggregates, so called “cotton wool plaques” [2, 13]. Recently, a new type of Aβ aggregates, “coarse-grained plaques” were described to be seen in early onset AD [14].
Hyperphosphorylated τ (HPτ) is the other constituent of ADNC. The formation of HPτ and the extent of HPτ pathology have been claimed to be influenced by PSEN1 mutations [8, 16]. One previously described case, with a PSEN1 mutation, displayed severe neurodegeneration of the type seen in frontotemporal lobar degeneration of the Pick’s disease subtype and thus lacking Aβ pathology [17]. The severity of the neuropathology of ADNC varies with the location of the mutation. Mutations beyond codon 200 on the PSEN1 gene are described as displaying a more severe neurofibrillary (NF) pathology and more cerebral amyloid angiopathy (CAA) than mutations located in the 5' end of the gene [16, 18]. Noteworthy, even within the same family, a particular mutation might exhibit significant neuropathological variations [19].
Concomitant pathologies are frequently seen in the setting of sporadic AD (sAD), and mixed pathologies can be seen in an aged population [20–25]. When the disease is caused by a genetic defect, the initiation of pathology and the symptoms are generally observed early on; thus, aging-related concomitant pathologies are usually sparse or even lacking [16, 27]. However, both alpha-synuclein (αS) and transactive DNA binding protein 43 (TDP43) have, in addition to ADNC pathology, been reported in brains of PSEN1 subjects [26, 27].
Five familial and de novo PSEN1 mutations within codon G206 have been identified and clinically characterized [1, 28–32]. Noteworthy, the neuropathological features accompanying PSEN1 mutations at this codon have not previously been studied in detail. Here, we present the clinical course and postmortem (PM) neuropathological findings in a 73-year-old female PSEN1 G206R mutation carrier with a premortem clinical diagnosis of AD. Furthermore, to illustrate the mutation specific pathological features, we have compared the pathology with that of an age and gender matched subject with sAD.
MATERIALS AND METHODS
Ethical statement
The Regional Ethical Committee in Uppsala, Sweden and the Swedish Ethical Review Authority had approved the study (Dnr 2005-244, 2005-11-02; Dnr 2021-04766, 2021-09-16).
Neuropathological examination
At autopsy, the brains were removed and weighed. The left hemisphere from the index PSEN1 G206R subject was freshly frozen, whereas the right part was placed in 4% buffered formalin (10% formaldehyde). The whole brain, from the control sAD case, was placed in the fixative. For the subject with PSEN1 G206R, the PM delay (PMD) was 4 h and for the sAD patient 120 h, and the total fixation time (FT) was 14 and 23 days, respectively. The brains were macroscopically assessed, after 7 and 8 days of fixation, and cut into 1 cm thick coronal slices. The gross alterations were registered, and routine tissue blocks, were sampled from 16 defined neuroanatomical regions, as previously described, shortly: frontal, temporal, parietal, occipital, motor cortices, gyrus cingula, anterior and posterior hippocampus, basal forebrain including amygdala and nucleus basalis of Meynert, striatum with insular cortex, thalamus, mesencephalon with substantia nigra, pons with locus coeruleus, medulla with dorsal motor nucleus of vagus, cerebellar vermis and dentate nucleus, and cerebellar cortex [33]. The samples were processed into paraffin blocks and cut into 7μm thick sections for hematoxylin-eosin (HE) and immunohistochemical (IHC) stainings. The IHC stainings were carried out on the Dako Autostainer Plus and Dako Omnis (DakoCytomation, Glostrum, Denmark) platform with Dako EnVision Flex detection system (DakoCytomation). Table 1 provides a summary of the antibodies and the pretreatment strategies used. The assessment of the slides was performed using light microscopy at x20 to x400 magnification. The neuropathological lesions were noted and the assessment of the proteinopathies, such as Aβ, HPτ, αS, and TDP43 followed the current recommendations and grading schemes [10, 34–37]. Photographs were taken using an Olympus BX45 microscope with Olympus UC30 camera.
Immunohistochemical stains
Dako Autostainer Plus (Dako Cytomation) was used for α-synuclein, amyloid-β, Tau 8, HLA-DR, and Iba1, and Dako OMNIS was used for GFAP. ac, autoclave; FA, formic acid; TE, Tris-EDTA buffer pH 9.0; CB, citrate buffer pH 6.0.
Genetic analyses
Genomic DNA from the index patient was extracted from the cerebellar tissue sample using DNeasy® Blood & Tissue kit (Qiagen, Germany) and analyzed by whole exome sequencing (INVIEW Core Exome, Eurofins, Germany). Sequenced regions were aligned to the human reference genome (assembly hg38), and SNP and InDel calling were performed. Mutation was confirmed by Sanger sequencing. In addition to the sample from the mutation carrier, a sample without mutation was included in the analysis. For the Sanger sequencing, PSEN1 exon 7 sequence containing the mutation site was amplified by PCR. Reactions were performed in a Phusion DNA polymerase buffer in a final volume of 25μl containing 80 ng genomic DNA, 0.5μM of each primer (TGTTTGGGAGCCATCACATTAT and GGGGCATTCCTGTGACAAAC) and 1.25 U Phusion DNA polymerase (Thermo Scientific, USA). The following amplification protocol was used: denaturation for 45 s at 98°C, followed by 34 cycles of 98°C for 10 s, 62°C for 30 s and 72°C for 20 s; this was followed by a final extension at 72°C for 5 min. Amplification products were examined on a 1% agarose gel and purified by PureLink kit (Invitrogen, USA). Two sequencing reactions were carried out for each sample using forward (GTACATCTTTTAAAATCTGTG) and reverse (AACAAATTATCAGTCTTGGG) sequencing primers, respectively. Sanger sequencing was performed at the Eurofins sequencing center (Eurofins, Germany). The novelty of the mutation was ascertained by searching the NCBI SNP database dbSNP (https://www.ncbi.nlm.nih.gov/snp), the Genome Aggregation Database gnomAD (https://gnomad.broadinstitute.org/) and the Alzheimer’s disease mutation database (https://www.alzforum.org/mutations).
RESULTS
Clinical presentation
The patient with the PSEN1 G206R mutation had a long prodromal stage over several years with vague symptoms, including anxiety, stress intolerance, impaired attention, and short-term memory. Due to her cognitive dysfunction, she went into early retirement at the age of 54 years. Four years later, she scored 24 out of 30 on the Mini-Mental State Examination (MMSE) scale, which was administered by her general practitioner [38]. Two years later, at the age of 60, she was referred to the Memory Clinic at Uppsala University Hospital. She was tense and refused to complete more than a few cognitive tests. Her episodic memory was markedly impaired with frequent repetitions of her statements, and her calculation skills were impaired. Copying the pentagons, Verbal Fluency Test, Clock Drawing Test, and the Trail Making Test A yielded normal scores, but she failed on the Trail Making Test B. She showed limited self-awareness of her deficits, and her family reported irritability and anxiety as major problems. A CT scan of her brain displayed a mild cortical atrophy, and the patient declined lumbar puncture. Two years later, her MMSE score was 21 points. Also, her memory impairment and mood swings were more aggravated, according to her family. An 18-fluoro-deoxyglucose positron emission tomography was performed, which showed markedly reduced glucose uptake in the parietal lobes and the posterior temporal lobe, as well as moderately reduced uptake in the frontal lobes, especially on the right side. Four years later, she could not manage any household chores, and anxiety and depression continued to be the major problems. At the age of 67, she was transferred to a dementia group living facility, and the patient died six years later, at 73 years of age.
The patient’s mother had also been diagnosed with early onset AD, with similar symptoms, but no PM examination was carried out (Fig. 1A).
The control patient with sAD was a female who died at the age of 71 years. She had a clinical diagnosis of AD without any known heredity.
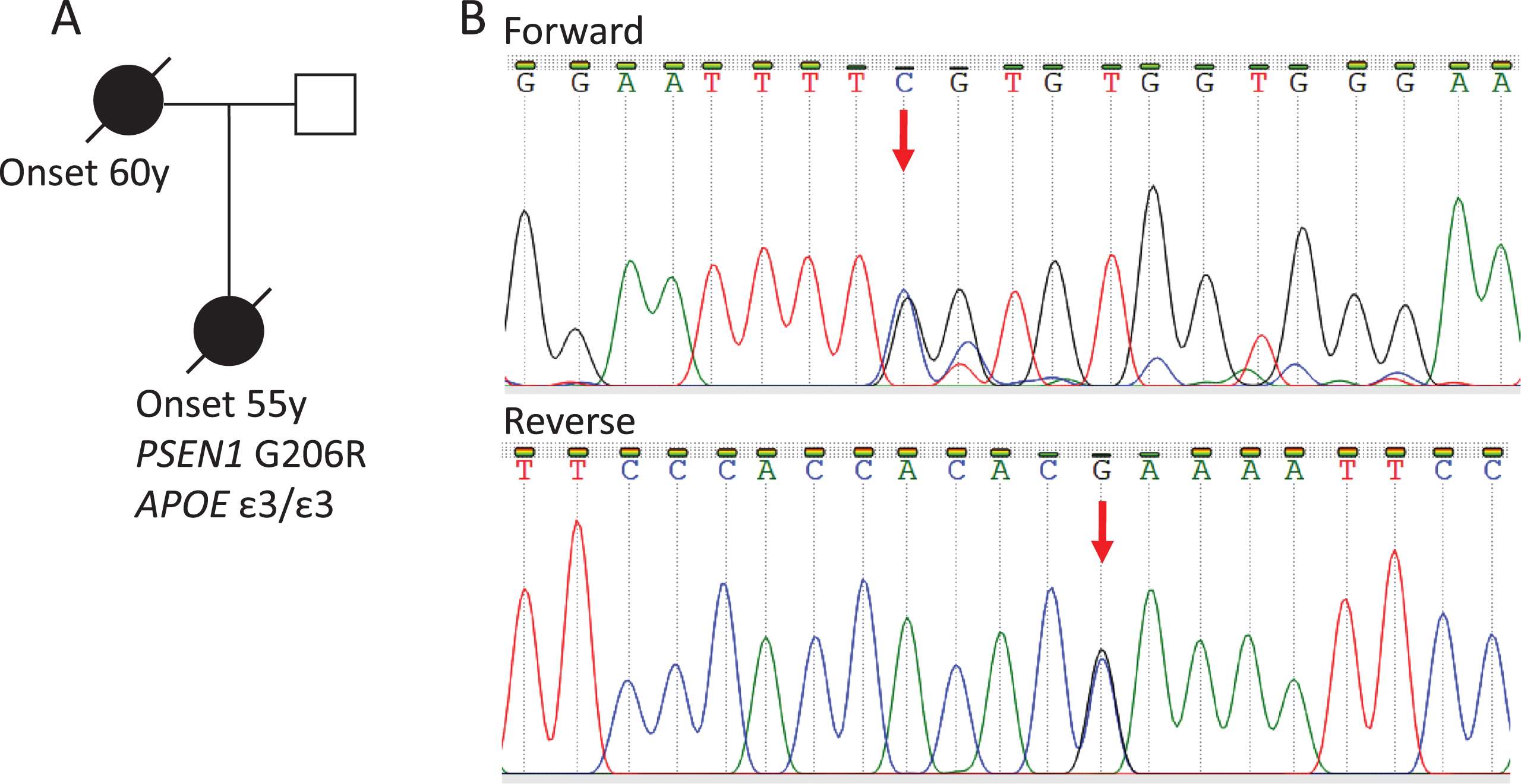
Pedigree of the PSEN1 G206R mutation family (A). Sanger sequencing results of the patient sample. Mutation position is indicated by an arrow (B).
Identification of the PSEN1 G206R mutation
The PSEN1 G206R mutation was identified by whole exome sequencing. Besides PSEN1, PSEN2, APP, and APOE were also analyzed, but no other likely pathogenic mutations were found. The patient was found to have the APOE ɛ3/3 genotype. The presence of the mutation was also confirmed by Sanger sequencing (Fig. 1B). This mutation causes a codon change from GGT to CGT, resulting in an amino acid change from glycine to arginine. It is located at the same chromosomal position as the previously described PSEN1 G206 S mutation (rs63750569). However, no G206R mutation was reported in the searched databases.
Neuropathological findings
The PSEN1 G206R patient: The brain weight was 945 g, displaying severe atrophy of the limbic structures and the cerebral cortex and thus, severe hydrocephalus. The inferior horn of the lateral ventricle had lost its crescent shape, and was seen as a large cyst, rimmed with a sparse extent of brain tissue. Grossly, it was difficult to identify the amygdala and the different anatomic structures of the hippocampus. Three sections were obtained from this region: one section from the basal forebrain (BFB) taken coronally at the level of the mammillary body incorporating the remains of the amygdala, followed by a second section incorporating remains of the anterior hippocampus and finally, a third section incorporating remains of the posterior hippocampus. In the HE stained BFB slides, the optic tract, supraoptic nucleus, paraventricular nucleus as well as mammillary body were identified, thus functioning as roadmaps. The nucleus basalis of Meynert (nbM) displayed severe neuronal loss and gliosis. Remains of the amygdala stretched along the enlarged ventricular space inwards and was delineated by the leptomeninges outwards. In the next section, with remains of anterior hippocampus, neither the transentorhinal cortex nor Cornu Ammonis (CA) structures were identified. In the section incorporating remains of posterior hippocampus, at the level of lateral geniculate body, the CA region was identified with a defined granular cell layer. CA1 region, remnants of entorhinal and temporo-occipital cortices were atrophic, displaying severe neuronal loss and gliosis. Hirano bodies were not observed within the few remaining neurons of the hippocampus. Granulovacuolar degeneration was not observed within the limbic structures or neocortex. The silhouettes of rounded structures, “plaques”, were visible in HE stained sections in the most affected limbic structures. These structures were 75–100μm in size, and the cellular component could be seen within them; thus, the term “cotton wool” plaque was not applicable. The remaining neuroanatomical regions, such as the thalamus and striatum, were identified macro- and microscopically. The neocortex displayed cell loss and diffuse gliosis. On the level of mesencephalon, in the substantia nigra (SN), the neuronal loss was most prominent laterally, and pale bodies were observed in some of the pigmented neurons, whereas Lewy bodies (LB) were scarce. Corticospinal tracts were unaffected. In the pons, the Locus Coeruleus (LC) was relatively well preserved. On the level of medulla, the pyramids and nucleus hypoglossus were unaffected, whereas a slight neuronal loss was observed in the dorsal motor nucleus of vagus. Cerebellum was histologically unaffected.
The proteinopathies, visualized by IHC, involving the hippocampal formation, the amygdala and the neocortex are visualized in detail in Fig. 2.
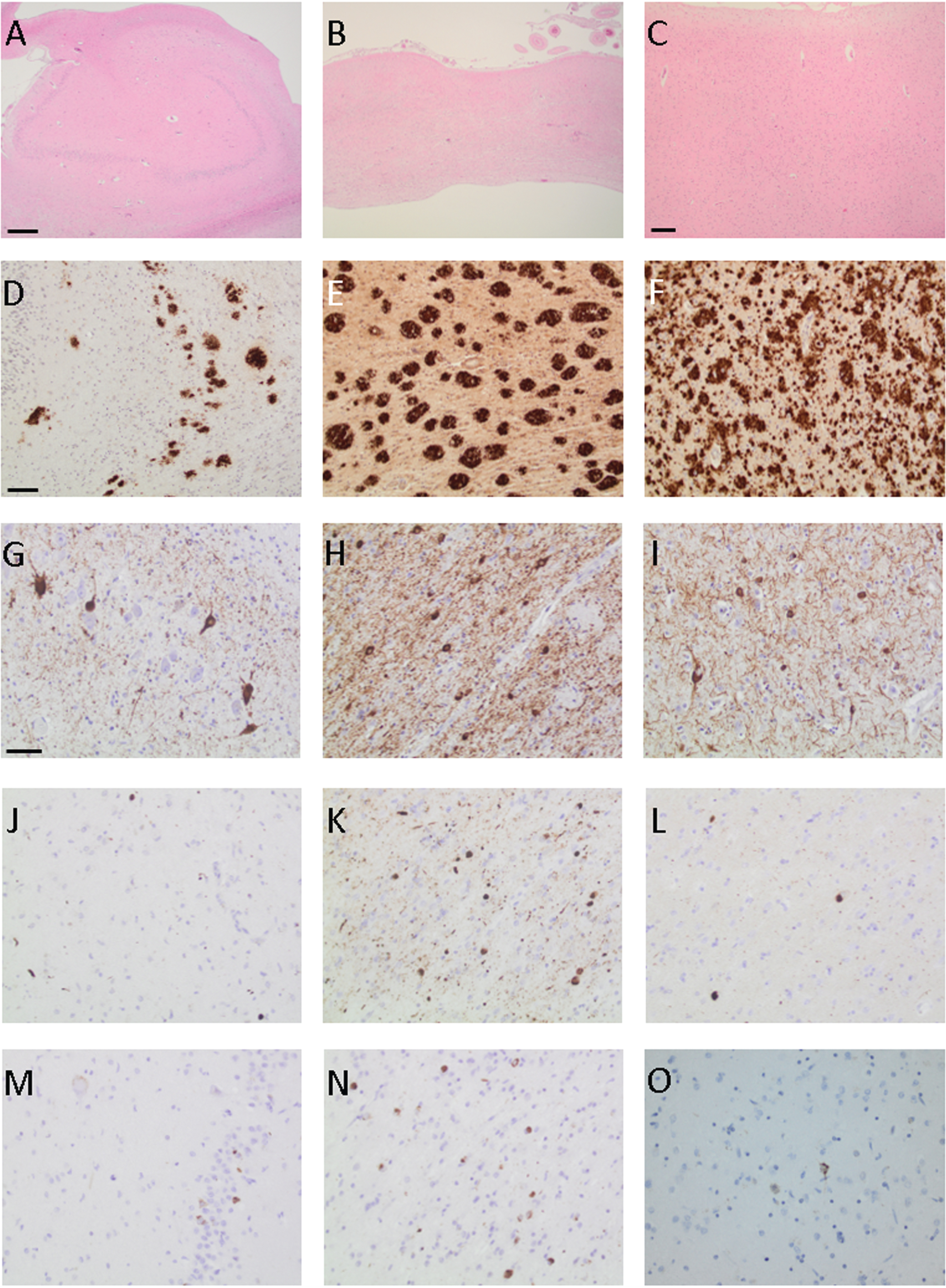
Photomicrographs of brain samples from a PSEN1 G206R mutation carrier from the hippocampus (A, D, G, J, and M), remnants of amygdala (B, E, H, K, and N) and neocortex (C, F, I, L and O) when applying hematoxylin-eosin staining (A–C) and antibodies towards total amyloid β (D–F), hyperphosphorylated τ (G–I), α-synuclein (J–L), and transactive DNA binding protein 43 (M–O). Bar in A-B 500μm, C 200μm, D–F 100μm, and G–O 50μm.
The IHC displayed extensive Aβ pathology with multiple extracellular aggregates within the cortex, visualized with all three Aβ markers (Figs. 2 and 3). The staining was less intense and less pronounced using Aβ40. There were smaller and larger protein aggregates; focally, small aggregates merged into larger groups. Some previously described “coarse-grained plaques” measuring 80–100 μm in size were seen. In line with previous literature these aggregates expressed Aβ6F/3D, pyroglutaminated Aβ (N3pE) and phosphorylated Aβ at serine 8 Aβ (1E4E11) [14]. Diffuse Aβ aggregates were observed within the subcortical white matter. Numerous Aβ aggregates were observed in the striatum and thalamus. In mesencephalon, the pathology was most pronounced in the tectum and tegmentum, except for the area of the red nucleus. In the cerebellum, particularly in the molecular layer, the Aβ pathology was seen as streaks of various thickness. Overall, the distribution of Aβ pathology fulfilled the ADNC criteria for Thal phase 5 [10].
CAA type 2 was noted, affecting mainly the meningeal vasculature [39].
The HPτ staining revealed severe NF pathology, i.e., tangles and neurites, within all predisposed regions, corresponding with ADNC Braak stage VI [8, 34]. In the medulla oblongata, HPτ was seen in the dorsal vagal nucleus and the reticular formation, on the level of pons in the LC and Raphe nucleus. Scattered tangles and neurites were seen in the SN as well as in the central grey. In the basal ganglia, the claustrum was severely affected by HPτ, in the nucleus caudatus and putamen numerous neurites and a few tangles were present. Only a single tangle was present in the globus pallidus. In the thalamus, mostly the medial nuclei were affected. In the BFB, there was a severe NF pathology within the remains of the amygdala, nbM, and paraventricular nucleus. Within the posterior hippocampus CA1-CA4 region few remaining pyramidal neurons with HPτ were seen whereas granular layer was severely affected. Severe NF pathology was seen in the transentorhinal region and in the neocortex, reaching the main visual cortex area within the occipital lobe. One single tangle was detected within the nucleus dentatus of the cerebellum.
The TDP43 pathology was widespread, seen in all predilected areas, reaching the neocortex, a pathology consistent with stage 6, according to Josephs or Limbic-predominant age-related TDP43 encephalopathy (LATE)-NC stage 3 [36, 37]. Scattered neurons containing intracytoplasmatic TDP43 protein were seen within the amygdala, nbM, and paraventricular nuclei. Within the posterior hippocampus CA1-4 regions, a few remaining pyramidal neurons displayed TDP43 positivity. TDP43 pathology was also noted in the region of the fascia dentata, subiculum, and transentorhinal cortex. Within the neocortex and insula, a few scattered neurons with TDP43 pathology were observed. A few TDP43 deposits were seen within the striatum and in the thalamus. Sparse TDP43 deposition was seen within the SN and tectum. At the medulla level, sparse TDP43 pathology was detected within the dorsal vagal nucleus, nucleus hypoglossus, and the inferior olivary nuclei.
The distribution of αS was seen in all the predicted neuroanatomical areas, confirming Braak stage 6 [35]. Pronounced pathology, with intracellular aggregates, LB, and Lewy neurites (LN), was observed in the medulla oblongata, dorsal vagal nucleus, reticular formation, and olivary nuclei. Within pons, αS pathology was seen in the LC, raphe nucleus, and basis pontis. The αS pathology was abundant within the SN. In the hippocampus, αS was seen in the remaining few pyramidal neurons of CA1-CA4 region with scattered LN; in the transentorhinal cortex, abundant LN and scattered LB were seen. In the BFB, there was pronounced αS pathology in the area of the amygdala and nbM. The gyrus cingulate was severely affected. In the parietal neocortex, both scattered LB and LN were seen.
Diffuse gliosis verified by GFAP stain was prominent in areas with neuronal loss. In the neocortex, in particular, the gliosis was accentuated in areas associated with the Aβ aggregates. The HLA-DR and Iba1 staining visualized abundant microglial activation within both grey and white matter (Fig. 3). The microgliosis was diffuse, and microglial noduli were not identified. Sparse perivascular T-lymphocyte infiltrates were observed, which was confirmed by the CD3 immunostaining. In addition, a few lymphocytes were seen within the parenchyma.
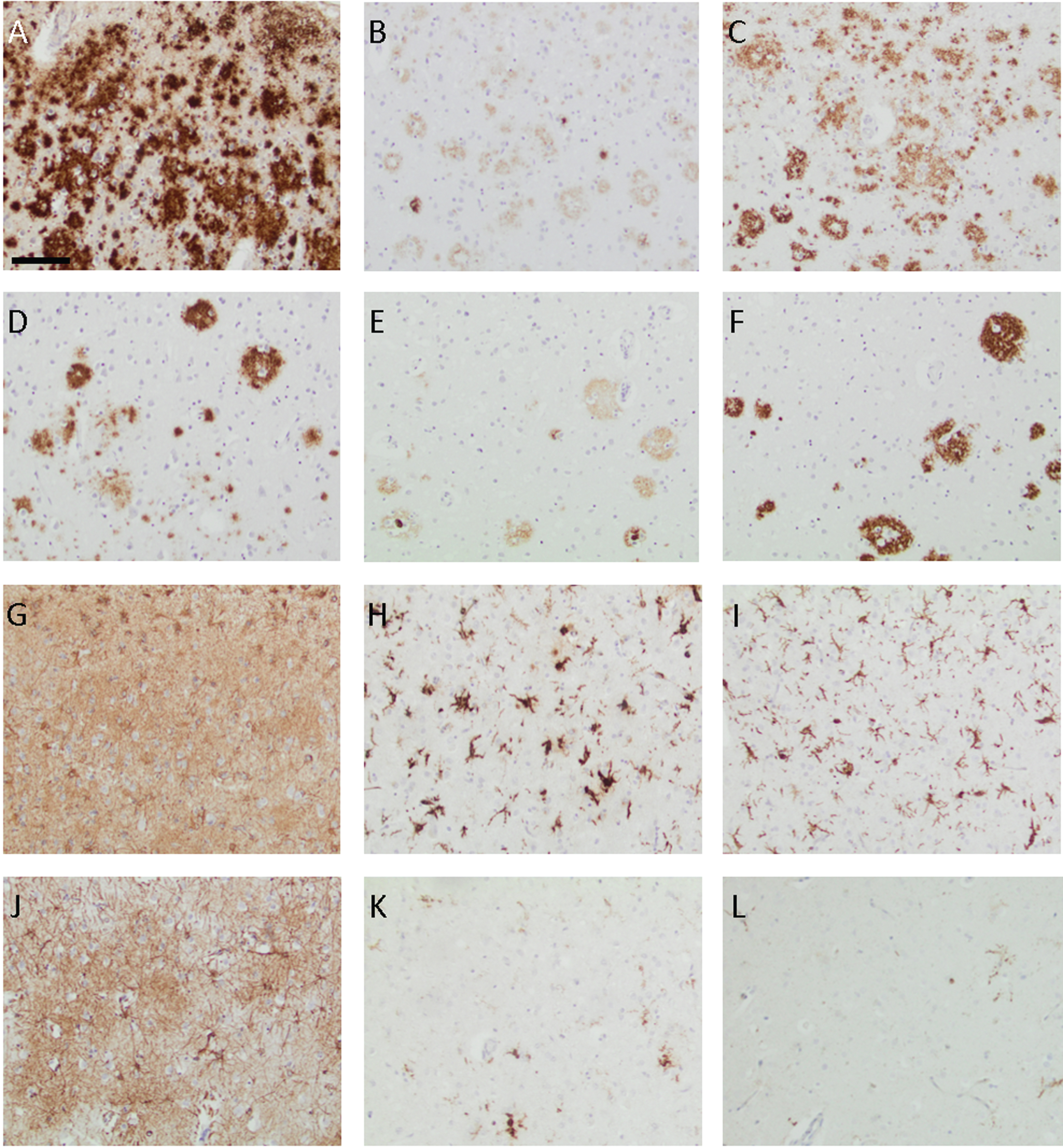
Photomicrographs of the neocortex from the PSEN1 G206R mutation carrier (A–C and G–I) and the subject with sporadic Alzheimer’s disease (D–F and J–L) when applying antibodies to the total amyloid-β (Aβ) (A and D), Aβ40 (B and E), Aβ42 (C and F), glial fibrillary acidic protein (G and J), human leucocytic antigen-DR (H and K), and ionized calcium-binding adaptor molecule 1 (I–L). Bar in A-L 100μm.
The subject with sAD: The brain weight was 1075 g displaying atrophy of the limbic structures and hydrocephalus. All anatomical regions could be identified macroscopically. The HE stained slides displayed neuronal loss and gliosis within the amygdala, hippocampus, and nbM. Vascular changes,
Extensive Aβ pathology, corresponding to Thal phase 5, was detected [10]. Multiple strongly Aβ- and Aβ42- positive aggregates were seen while the labelling was weak/moderate, applying Aβ40 staining (Fig. 3). In the grey matter, the aggregates were small to moderate in size. Diffuse Aβ aggregates were observed within the subcortical white matter. Numerous small Aβ aggregates were seen within the basal ganglia. In the cerebellum, scattered small Aβ aggregates were noted, and only focally confluent diffuse aggregates were detected.
Aβ40 aggregates were focally notable within vessel walls of larger meningeal arteries, CAA type 2, a finding not noted in other Aβ IHC stainings [39].
The NF pathology was widespread, corresponding to Braak stage V [8, 34]. In the brainstem, the pathology was sparse, with a few scattered tangles and neurites within dorsal vagal nucleus, reticular formation, LC, and raphe nuclei. A few neurites were observed in the SN and NF pathology was mainly seen in the tectum and the central grey. The HPτ pathology was sparse in the striatum, while some neuritic pathology was seen in the globus pallidus. A few scattered tangles were seen in the thalamus. In the BFB, the amygdala, nbM, and supraopticus were the most affected regions. In the hippocampus, the pathology affected the pyramidal cells of the Ammons horn, and scattered neuritic plaques were identified. Only occasional cells within the granular cell layer contained HPτ. Transentorhinal cortex was severely affected. The pathology was widespread in the neocortex, reaching the peristriate area of the occipital lobe. No NF pathology was seen within the cerebellum.
The intracytoplasmic TDP43 deposits were seen in scattered neurons in the amygdala. A few pyramidal neurons within the Ammons horn displayed TDP43 pathology, but no pathology was observed within the granular cell layer. Other neuroanatomical areas remained unaffected by the TDP43 pathology, corresponding with Joseph’s stage 2 and LATE-NC stage 2 [36, 37].
αS pathology was not present.
GFAP positive gliosis was seen within both the grey and white matter and was accentuated in areas associated with Aβ aggregates. Pronounced subpial gliosis was seen. Sparse HLA-DR and Iba1 positive microgliosis was seen both in the grey and white matter (Fig. 3). Focally, small aggregates of microglia were observed in the cortex associated with Aβ aggregates.
In summary, both subjects displayed severe atrophy of the cerebral hemispheres and limbic structures. The atrophy was more pronounced in the PSEN1 G206R mutation carrier. Both subjects displayed a high grade of ADNC and a presence of CAA type 2. Both cases fulfilled the criteria for Thal phase 5, whereas the density of Aβ pathology was higher in the index patient. The index case fulfilled Braak stage VI, whereas the sAD fulfilled Braak stage V criteria for HPτ. The NF pathology was more severe and more extensive in the index patient engaging the granular cell layer of the hippocampus and the striatum. Furthermore, TDP43 pathology was seen in both cases, being moderate in the sAD case (Josephs 2/LATE-NC 2) and full-blown in the PSEN1 G206R subject (Josephs 6/LATE-NC 3). The mutation carrier displayed severe αS pathology (Braak 6), not seen in the sAD brain. Both brains displayed diffuse astrogliosis within the parenchyma as well as activated microglia, which were sparse in the sAD patient and abundant in the PSEN1 G206R subject.
DISCUSSION
Here, we present clinical and PM neuropathological findings seen in a 73-year-old subject with the novel PSEN1 G206R mutation. This case is unique, as all common protein alterations, considered causative regarding defined neurodegenerative diseases, are seen. Moreover, according to international consensus guidelines, these alterations fulfil the end-stage criteria for each proteinopathy [10, 34–37].
To date, more than 300 different PSEN1 mutations have been identified [5]. Five mutations, both familial and de novo were previously described at the G206 position [1, 28–32]. One study identified G206A as one of the most common mutations in the coding region of PSEN1. All mutations located at codon G206 seem to be highly pathogenic, causing early onset dementia with a clinical diagnosis of AD whereas only two reports include neuropathological assessment confirming ADNC [40, 41]. The G206A mutation has been shown to double Aβ42 production compared to wild type PSEN1 in cell line studies [30]. PS1 is one of the proteins in the γ-secretase enzyme complex involved in cleavage of AβPP to Aβ. The atomic structure analysis of human γ-secretase is complex with a transmembrane AβPP fragment showing that mutations at the G206 residue likely affect the local conformation and eventually contribute to substrate binding [42]. When mutated, the proteolytic site of γ-secretase is altered, producing the amyloidogenic Aβ42, which is more prone to aggregate promoting more severe Aβ pathology [6, 7].
We compared the neuropathological findings in the PM brain of a PSEN1 G206R mutation carrier to a subject with sAD of the same gender and age range.
Severe atrophy of the neocortical and limbic structures was observed in both subjects. The mutation carrier displayed a more severe atrophy, especially within the limbic structures, where only posterior areas of the hippocampus were identified with an almost total loss of pyramidal neurons and with the amygdala being rudimentary and displaced. All neuroanatomical areas could be identified in the sAD patient. This is in line with previous literature, as more pronounced atrophy and cell loss are described in subjects with autosomal dominant AD (adAD). Such an aggressive phenotype can be caused by dysfunction of the γ-secretase enzyme complex, not only in relation to AβPP cleavage but also to other pathways involved in apoptosis, abnormal protein aggregation, cell cycle events and oxidative stress [2, 44].
The dominating NC in subjects with various PSEN1 mutations are multiple extracellular aggregates of Aβ, especially Aβ42 [2, 46]. This is in line with what was seen in the index subject, displaying more extensive Aβ pathology than the age- and gender-matched sAD case. Noteworthy, both index and sAD cases fulfilled the regional distribution of Aβ corresponding to Thal phase 5 [10]. Several publications, assessing the neuropathology in PSEN1 mutation cases, described a specific morphology of the Aβ aggregates, seen in HE stained sections and measuring up to 150μm, so called “cotton wool” plaques. When applying IHC, these structures displayed accentuated Aβ staining at the rim of the plaque, with no associated NF pathology [2, 13]. This plaque has been shown to be not specific for PSEN1 mutations, as similar structures have been described in the Arctic APP mutation, as well as in sAD [47, 48]. We could identify plaques with routine HE staining, in both the neocortical and limbic areas in our index subject, but the morphology of these lesions was not consistent with the “cotton wool” plaques. No plaques were visualized in HE stained sections in the sAD patient. Recently, new type of Aβ aggregates were described, “coarse-grained plaques”, to be seen in early onset AD [14]. We identified these large plaques in the brain of our mutation carrier by its morphology and proteomics and these structures were lacking in the sAD subject.
CAA was sparse in both of our subjects, mainly seen in the meningeal vessels. This is in line with previous studies on adAD, the most extensive CAA has been recognized in cases with APP mutations [2]. Mutations located beyond codon 200 of PSEN1 have been described to cause a more severe CAA than mutations located before codon 200, which thus is inconsistent with our results [16, 18].
The findings regarding the NF pathology are contradictory in the literature. Some studies describe no difference between PSEN1 and sAD cases, whereas some describe an increase in HPτ pathology in PSEN1 subjects [2, 50]. The opposing results are related to the location of the mutation within the PSEN1 gene, with mutations beyond codon 200 describing a more advanced NF pathology than mutations occurring before codon 200 [16, 50]. Another conflicting outcome is the variation of neuropathology described in different subjects within the same pedigree, carrying the same mutation [15, 19]. Both subjects assessed here displayed a high level of HPτ pathology. The PSEN1 mutation carrier displayed a more severe and extensive HPτ pathology when compared with the sAD subject. The index subject displayed neuritic HPτ pathology in the striatum and severe NF pathology in the claustrum, contrary to the sAD case. Contrary to the subject with PSEN1 E280A mutation, our index patient did not reveal any cerebellar NF pathology [50].
Our PSEN1 G206R mutation carrier displayed extensive αS pathology, corresponding with full-blown Lewy body disease, previously described in a few PSEN1 mutation carriers [26, 51–53]. The sAD patient lacked this proteinopathy. The lack of αS in our sAD case is related to selection bias, as it is acknowledged that concomitant αS pathology is frequently observed in sAD [20, 24]. In most individuals with PSEN1 mutations, the αS pathology has been described as being limited to the limbic structures, amygdala in particular, whereas involvement of neocortex is unusual [26, 54–56]. Other regions predilected for αS pathology, i.e., brainstem and mesencephalon, have been reported by some as being sparsely affected in the population with adAD [26, 56]. Contrary to the above, our index patient displayed a substantial number of LBs and LNs in the brainstem nuclei and mesencephalon. This finding is difficult to assert in relation to previous publications due to various selections of neuroanatomical regions analyzed [16]. Noteworthy, animal studies, have suggested an interaction between PS1 and αS in adAD that might explain this observation [57].
During the last decade, the significance of TDP43 pathology in AD has been acknowledged [58]. Several studies report a prevalence of over 50% of concomitant TDP43 pathology in sAD and a grading scheme has been established [24, 59]. In 2019 a new neuropathological entity, LATE, was defined affecting preferentially the older population and the LATE-NC is commonly seen concomitantly with ADNC [37]. There are only a few publications describing whether TDP43 pathology is observed in adAD cases [19, 60]. Our sAD patient with ADNC displayed a low-moderate grade of concomitant TDP43 pathology, corresponding with Joseph’s stage 2 and LATE-NC stage 2. The index subject displayed fulminant TDP43 pathology, Joseph’s stage 6 and LATE-NC stage 3, involving the limbic structures, striatum, and the neocortex [36, 37]. A widespread TDP43 pathology has not been previously described in AD caused by PSEN1 mutations or in adAD. Moreover, the extent of the pathology, found in our PSEN1 G206R case, is seldom seen in sAD cases at this age range [19, 60].
Our PSEN1 G206R subject displayed extensive concomitant pathologies, previously seen in only a few cases, mainly in the oldest individuals. Mixed neurodegenerative pathologies are currently acknowledged to be seen in the aged, but the level of pathology as seen here is unique in the setting of both sAD and adAD [20–25, 61]. Some studies have suggested that concomitant proteinopathies might be the result of a cross-seeding mechanism, i.e., one altered protein promotes alteration of other proteins, which can lead to mixed pathologies [62–64]. The above seem to occur in a prion like manner as recent studies describe that the wild type of prion protein seems to mediate the propagation of Aβ, HPτ, and αS pathology in cell-lines and animal models as well as in the human brain [65, 66]. Another suggested mechanism is a synergistic propagation of the pathology as described between TDP43 and HPτ in an animal model as well as in humans [67, 68]. The cross-seeding mechanism is certainly in line with the observations of concomitant TDP43 in some 30–70% and concomitant αS in up to 50 % in the setting of sAD [20–24, 63]. PSEN1 mutations is known to cause extensive Aβ pathology and promotes HPτ pathology and further PS1 is described to interact with αS in adAD. Thus cross-seeding mechanism through prion mediated pathways or synergistic exacerbation of the proteinopathies could be causative to the exceptional pathology as seen in our mutation carrier [17, 67]. Noteworthy, this is a singular case and the pathology as seen here has not been previously associated with any PSEN1 mutations so far published. Whether the concomitant proteinopathies in our index patient are driven by the ADNC, or whether they are caused by the PSEN1 G206R mutation, is still in need of investigation. Another issue that should not be ignored is the relatively long duration of the disease in our index patient, 19 years, which might also contribute to the extent of the pathology.
Here we implemented IHC in order to assess astrocytes and microglia cells. Previous studies of glial and microglial markers, using the same antibodies and pretreatment strategies as here, displayed that both HLA-DR and Iba1 were affected when PMD exceeded 168 h (PMD 4 respectively 120 h in our cases) whereas FT longer than 53 days affected the staining outcome of HLA-DR (FT 14 respectively 23 days). GFAP immunostaining was not affected by PMD or FT [69]. Thus, we feel confident that our results presented here are reliable and not influenced by the pre-analytical variables.
Activated astrocytes and microglia, have been associated with ADNC [70–73]. In sAD, the astrocytes are concentrated around the Aβ aggregates next to the microglia, which also seem to be closely associated with NF pathology [72–74]. The inflammatory cells in the setting of adAD and PSEN1 have been sparsely studied [2, 76]. “Cotton wool” plaques were described as lacking accompanying inflammatory cells, contrary to the Aβ aggregates seen in sAD [12]. In line with the literature, both our subjects displayed astrogliosis accentuated in association with the Aβ aggregates [72]. The microgliosis was mild in the sAD case, but abundant and diffuse, in all neocortical areas, of the PSEN1 mutation carrier. Diffuse microglial activation has been described in sAD and in some PSEN1 mutation cases and has been reported as more pronounced in females, which is supported by our findings [19, 76]. The prominent inflammatory process in the brain of our mutation carrier could be caused by the severe extent of all proteinopathies, as both astrocytes and microglia are described as being activated not only by Aβ and HPτ but also by αS and TDP43 [72, 77]. The relationship between protein alterations in neurodegenerative diseases and inflammation is intriguing, as neuroinflammation is proposed to promote and aggravate the disease process [72, 77]. Noteworthy, immune function alters with aging, and it is to be discovered whether neuroinflammation is merely a response to neurodegeneration or triggers further the degenerative process [78–80].
In summary, we identified a novel mutation in an early onset AD subject, PSEN1 G206R and performed a PM assessment of the neuropathological alterations. Previously, five PSEN1 mutations within the G206 codon have been identified, all with clinical dementia of AD-type but only two with verified ADNC by neuropathological examination. The PSEN1 G206R mutation, in line with the other mutations within the same codon, promotes ADNC but also seems to be prone to mediate other proteinopathies. The brain of our mutation carrier displayed the end stage of ADNC with multiple Aβ aggregates and severe NF pathology. In addition, an extensive and widespread αS pathology and TDP43 pathology was observed. This is unique, as every proteinopathy described here, by themselves, causes cognitive decline and ultimately dementia. Concomitant pathologies in the setting of aging and neurodegeneration are not uncommon, but the extent of each in our index subject, was surprisingly high [20–24, 61]. ADNC has been the prominent feature in the literature when assessing brain tissues from subjects harboring PSEN1 mutations whereas concomitant αS or TDP43 pathology has as previously described been sparse [16, 27]. One plausible cause that has been proposed for concomitant pathologies is the cross-seeding mechanism through prion like manner or synergistic protein propagation [62, 65–68]. In addition, diffuse gliosis and abundant microgliosis were observed in the index subject. Based on current knowledge neuroinflammation can be initiated by altered proteins and create an inflammatory environment that promotes further neurodegeneration.
The complex pathology in our mutation carrier is intriguing and raises several questions related to the cause, i.e., genetics, disease duration, cross-seeding, or inflammation or all of the above. In order to increase our understanding of the devastating neurodegeneration, that affect a large number of aged individuals, it would be of interest to carry out PM neuropathological investigation and not only accept assessment on the level of genetic analysis.
Footnotes
ACKNOWLEDGMENTS
The authors thank Meena Stromqvist for her critical reading of the manuscript and Svetlana Popova and Dijana Cerjan for their skillful laboratory work.
This study was funded by local grants from Uppsala University Hospital and by Hans Gabriel and Alice Trolle-Wachtmeister Foundation.