
Abstract
Citrate synthase is a key mitochondrial enzyme that utilizes acetyl-CoA and oxaloacetate to form citrate in the mitochondrial membrane, which participates in energy production in the TCA cycle and linked to the electron transport chain. Citrate transports through a citrate malate pump and synthesizes acetyl-CoA and acetylcholine (ACh) in neuronal cytoplasm. In a mature brain, acetyl-CoA is mainly utilized for ACh synthesis and is responsible for memory and cognition. Studies have shown low citrate synthase in different regions of brain in Alzheimer’s disease (AD) patients, which reduces mitochondrial citrate, cellular bioenergetics, neurocytoplasmic citrate, acetyl-CoA, and ACh synthesis. Reduced citrate mediated low energy favors amyloid-β (Aβ) aggregation. Citrate inhibits Aβ25–35 and Aβ1–40 aggregation in vitro. Hence, citrate can be a better therapeutic option for AD by improving cellular energy and ACh synthesis, and inhibiting Aβ aggregation, which prevents tau hyperphosphorylation and glycogen synthase kinase-3 beta. Therefore, we need clinical studies if citrate reverses Aβ deposition by balancing mitochondrial energy pathway and neurocytoplasmic ACh production. Furthermore, in AD’s silent phase pathophysiology, when neuronal cells are highly active, they shift ATP utilization from oxidative phosphorylation to glycolysis and prevent excessive generation of hydrogen peroxide and reactive oxygen species (oxidative stress) as neuroprotective action, which upregulates glucose transporter-3 (GLUT3) and pyruvate dehydrogenase kinase-3 (PDK3). PDK3 inhibits pyruvate dehydrogenase, which decreases mitochondrial-acetyl-CoA, citrate, and cellular bioenergetics, and decreases neurocytoplasmic citrate, acetyl-CoA, and ACh formation, thus initiating AD pathophysiology. Therefore, GLUT3 and PDK3 can be biomarkers for silent phase of AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease that affects the elderly population and results in dementia [1]. It is the sixth leading cause of death in elderly people. AD leads to declines in cognitive function like memory, language problems, planning, execution, and visuospatial problems and noncognitive psychiatric symptoms like depression, anxiety, agitation, hallucination, and delusion, which result in difficulties in daily activities of living [2]. Currently, 6.2 million people ≥65 years of age are living with AD, which may rise to 12.7 million by 2050 [3].
The underlying pathomechanism of AD starts years before the appearance of the clinical symptoms [4]. In AD, a decrease in acetylcholine (ACh) and its degradation by acetylcholinesterase or butyrylcholinesterase causes cognitive symptoms [5, 6]. Furthermore, senile plaques due to amyloid-beta (Aβ) deposition and neurofibrillary tangles due to tau hyperphosphorylation are two diagnostic parameters in AD [7], which cause N-methyl-D-aspartate (NMDA) induced excitotoxicity. Current treatment strategies such as acetylcholinesterase inhibitors and NMDA antagonists slow down the disease progression but do not cure the disease. So, we need to search for other pathways interlinked with the production of Aβ, its deposition, metabolism, clearance, and involvement of peripheral organs, which influence cognitive function and start the progression of AD [8]. In this review article, we investigate the role of the citrate synthase (CS) and its product citrate in AD and in pathophysiology of the prodromal phase of AD.
THE ROLE OF CITRATE SYNTHASE AND CITRATE IN CELLULAR BIOENERGETICS
Neuronal cells depend on glucose as their energy source. Glucose metabolizes to generate ATPs by glycolysis, tricarboxylic acid (TCA), and electron transport chain (ETC) pathways which involve glucokinase, CS, and cytochrome oxidase enzymes, respectively. CS is an important mitochondrial enzyme encoded in nuclear DNA. It is synthesized in the cytoplasmic ribosome and migrates to the mitochondrial matrix, where CS plays a key role in the TCA cycle in aerobic energy production by interconversion of metabolites.
Glycolysis produces pyruvate, which generates acetate. Acetate is carried with co-factor (acetyl-CoA) in the TCA cycle [9]. In neuronal cells, mitochondrial acetyl-CoA is utilized by CS [10–12]. In the TCA cycle, CS catalyzes the condensation of acetate residue of acetyl-CoA and oxaloacetate to form citrate [9].
This citrate in the TCA cycle is utilized to form 2NADH, 1FADH, and 1 ATP, which are further used in the ETC cycle to generate more ATPs using cytochrome oxidase enzymes (mitochondrial complex IV) by oxidative phosphorylation [13].
Low CS activity impairs the aerobic ATP synthesis pathway and decreases citrate or decreases cytoplasmic acetyl-CoA, which causes a low energy environment inside the cells [14]. A low-energy environment favors the aggregation of Aβ16–22 [15]. Several neurodegenerative conditions occur due to the inhibition of pyruvate dehydrogenase (PDH) that ultimately inhibits the acetyl-CoA formation in the mitochondria, which reduces the metabolic flux via the TCA cycle, decreases cellular bioenergetics, and inhibits the synthetic acetylation reaction in the neuronal cells sub-compartments.
Changes in the mitochondrial and neurocytoplasmic acetyl-CoA can significantly contribute to neurotoxic pathomechanisms and other neurodegenerative disorders [16]. Neuronal cells require a higher source of glucose energy than the non-excitable cell to strengthen the functions of neurotransmitters. Acetyl-CoA also serves as a direct substrate of energy for the brain neuronal cells [16]. ETC produces higher energy, and mitochondria perform these tasks if they are supplied with enough energy. At a low ATP state, mitochondria are unable to pump-out calcium from the inner mitochondrial membrane (IMM), and accumulation of Ca+2 causes mitochondrial dysfunction. Ca+2 overload in mitochondria promotes apoptosis by release of pro-apoptotic factors which lead to impaired mitochondrial membrane potential, decreased ATP production, increased reactive oxygen species (ROS) production, alteration of Ca+2 homeostasis, and mitochondrial dysfunction [17]. Aβ was also seen to cause mitochondrial and cytosolic Ca+2 overload in both in vivo and in vitro studies [18]. The fibroblasts of patients with AD show disrupted mitochondrial dynamics, cellular bioenergetics, and Ca+2 homeostasis [19, 20]. In AD, altered mitochondrial Ca+2 homeostasis causes defective mitophagy and accumulation of the damaged mitochondria leading to neurodegeneration, hence resulting in cell death by Ca+2 overload [21–24].
CS is the main rate limiting enzyme in the TCA cycle [25, 26]. Furthermore, CS is the quantitative marker of mitochondrial integrity, function, mass [27–29], and mitochondrial respiratory chain enzymes [30], as CS and citrate maintain the TCA, ETC energy cycle, and mitochondrial health. Lower levels of CS lead to decreased citrate, cytoplasmic acetyl-CoA, and ACh neurotransmitter formation, release, and re-synthesis [31].
CITRATE SYNTHASE AND CITRATE IN ACETYLCHOLINE SYNTHESIS
Oxaloacetate and acetyl-CoA are impermeable through mitochondrial membranes, whereas pyruvate and citrate can cross the mitochondrial membranes [32]. Citrate moves from mitochondrial membrane to neuronal cytoplasm through citrate-malate antiporter and converted to acetyl-CoA by ATP citrate lyase [33–38].
In neuronal cytoplasm, acetyl-CoA combines with choline in the presence of choline acetyl transferase (CAT) to form ACh [39, 40]. ACh is the main neurotransmitter for memory and cognition, and it is found to be deficient in AD [41]. Therefore, citrate as a substrate forms acetyl-CoA, that is utilized for ACh synthesis [38, 43].
The mitochondrial acetyl-CoA is responsible for the level of neuro-cytoplasmic acetyl-CoA and ACh synthesis. The decrease in mitochondrial acetyl-CoA in neuronal cytoplasm results in many neurodegenerative diseases [16]. Pyruvate dehydrogenase complex (PDHC) and CAT correlated well in different brain regions, such as the caudate nucleus, amygdala, and putamen, but not in the frontal cortex or hippocampus. In patients with C-21 trisomy Down syndrome, the total activation and activities of PDHC and CAT were below normal, which increases the risk of AD. PDHC deficiency may also play a vital role in AD and Huntington’s disease because decreased PDHC activity correlates with the loss in cholinergic neurons and decreased cognitive functions due to less mitochondrial and neuronal cytosolic acetyl-CoA and less ACh synthesis [44–48].
Studies show that acetyl unit (acetyl-CoA) is transported as citrate for ACh synthesis and lipogenesis for brain tissues [35, 50]. In the immature brain, Acetyl-CoA is utilized in large amounts for the lipid synthesis that is necessary for structural element and myelin formation of neuronal tissue, while in the mature brain, acetyl-CoA is more utilized in the formation of ACh. Both glucose and ketone bodies are precursors for acetyl-CoA in suckling animal brains, while glucose is an exclusive substrate in the adult brain [38, 52]. Acetyl-CoA has a significant role in neurodegenerative disease for the survival and death of the cholinergic neurons [48] and also has a role in epigenetic regulation [53] (Fig. 1).
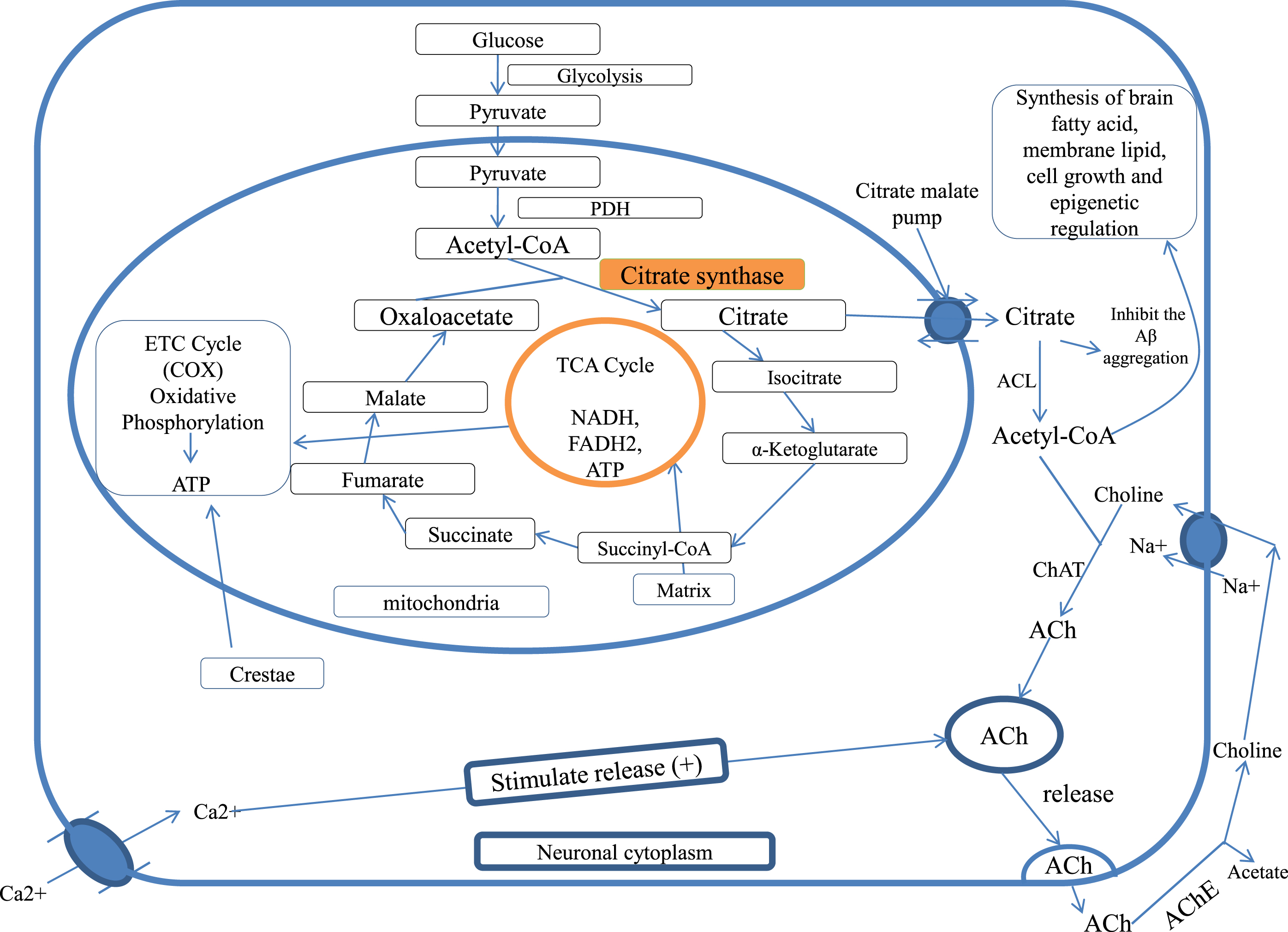
Citrate synthase and citrate in cellular bioenergetics and ACh formation, PDH, pyruvate dehydrogenase; TCA cycle, tricarboxylic acid cycle; CAT, choline acetyltransferase; ACL, ATP citrate lyase; ACh, acetylcholine; AChE, acetylcholinesterase; COX, cytochrome c oxidase.
CITRATE SYNTHASE AND CITRATE IN ALZHEIMER’S DISEASE
Neuronal cells require higher amounts of overall energy for functioning neurotransmitters due to their continuous depolarization and repolarization. Acetyl-CoA is the direct source of energy for the brain cells. Neuronal cells mainly utilize the energy by oxidative phosphorylation [16].
The reduced activity of cytochrome c oxidase (COX) is found in neurodegenerative and non–degenerative AD tissue due to mitochondrial mutation [54]. COX maximizes mitochondrial energy production; therefore, when COX activity is reduced, it results in a lower energy production, which favors Aβ16 - 22 aggregation [15]. Aβ deposition in AD causes cytotoxicity by increasing oxidative stress, lipid peroxidation of the plasma membrane, ion motive ATPase impairment, GPCR uncoupling, glutamate uptake, and altering of calcium homeostasis intracellularly [55, 56].
Furthermore, Aβ generates tau protein kinase1 (TPK1) [57, 58]. TPK1 hyperphosphorylates tau [59] and inhibits PDH [60], which inhibits acetyl-CoA and citrate in mitochondria resulting in a decrease of ACh synthesis and causes memory and cognition impairment [15]. Aβ interacts with mitochondrial cyclophilin D and causes cell death [61]. Acetyl-CoA is the direct energy source for the brain cells [16]. Low CS activity impairs ATP synthesis aerobically [14] and decreases acetyl-CoA. Low energy favors Aβ16 - 22 aggregation [15] that further induces TPK1, which causes tau hyperphosphorylation and PDH inhibition. Thus, the same cycle continues with the inhibition of acetyl-CoA and ACh synthesis. Screening of patients with AD showed an overall reduction in brain COX activities. Hence, mitochondrial dysfunction in the cerebral or extracerebral contributes to AD pathology [54]. Therefore, instead of Aβ causing AD, low CS activity and citrate mediated low ACh production and bioenergetics could be the main reason for underlying AD symptoms.
Human teratocarcinoma cell line (NT2 cell) treated with Aβ25 - 35 and Aβ1 - 42 differentiates into neurons, secretes amyloid, and exhibits an excitotoxic response [62, 63]. Aβ1 - 42 is more toxic than Aβ25 - 35. Aβ25 - 35 leads to ATP depletion, reduces membrane potential of mitochondria, and inhibits mitochondrial respiratory chain complexes (complex 1-NADH ubiquinone oxidoreductase, complex 2/3-succinate cytochrome-c oxidoreductase, and complex 4-cytochrome–c oxidase) enzymes [56]. Pre-incubation of NT2 cells with antioxidants like vitamin E, melatonin, idebenone, and glutathione for 22 h prevents Aβ25 - 35 toxicity [64].
Interestingly, a lack of mitochondrial DNA prevents oxidative phosphorylation in NT2 cells. Furthermore, NT2 cells remained unaffected by Aβ1 - 42 or Aβ25 - 35. This proved that Aβ causes mitochondrial dysfunction and oxidative stress [56], promoting neuronal death and degeneration [65]. This shows that mitochondrial respiratory chain malfunction causes Aβ toxicity.
CITRATE SYNTHASE AND CITRATE IN TAU HYPERPHOSPHORYLATION
AD is caused by extracellular deposition of Aβ plaques and intracellular neurofibrillary tangles by abnormal paired helical filaments (PHF), leading to neuronal cell damage [57]. The abnormal PHF is composed by hyperphosphorylation of tau [58, 66] and in small portions by ubiquitin [67]. Tau is a microtubule-associated protein that provides strength and stability to axonal microtubules in neurons by tubulin-binding property and helps in intraneuronal transportation [68].
In AD, Aβ induces the synthesis of glycogen synthase kinase-3 beta [60] and tau protein kinase 1 and 2 (TPK1 and TPK2) in AD [58]. TPK1 hyperphosphorylates tau protein. It converts tau to PHF and forms neurofibrillary tangles, while TPK2 does not participate in PHF formation [59]. TPK1 was found to be positive in neurodegenerative Aβ plaques and neuropil threads in Down syndrome brains [69]. TPK1 also phosphorylates PDH and reduces the conversion of pyruvate to acetyl-CoA, which is needed for ACh synthesis [60], whereas TPK2 does not inactivate PDH [70]. In rat hippocampal cells, Aβ-mediated TPK1 activation causes inhibition of PDH and leads to accumulation of pyruvate, impaired glucose metabolism, decreased acetyl-CoA production, low CS activity, and citrate and ACh synthesis, resulting in AD [71].
Aβ AND IMPAIRMENT OF GLUCOSE TRANSPORTERS
The decrease in glucose uptake is observed in the silent phase of AD prior to neurodegeneration in the study of genetically at-risk AD patients [72–74]. By using brain imaging methods, neurotic plaques are detected in the brain regions where glucose uptake is impaired [75–77]. The mitochondrial enzyme activities of ketoglutarate dehydrogenase and PDHC also decreased [78]. In the impairment of neuronal glucose uptake, ATP production in mitochondria is reduced and increased neuroexcitotoxicity due to calcium overload is seen [79, 80]. In the brain, glucose is taken up by the special transporter GLUT3 in neuronal cells and by GLUT1 in endothelial cells, and the levels of these glucose transporters are reduced in the AD brain [81]. Aβ, the proteolytic fragment of the amyloid-β protein precursor (AβPP), is majorly responsible for neurotic plaques in AD [82]. Aβ toxicity leads to membrane peroxidation [83] and membrane transporter signaling impairment, along with impairment of ion-motive ATPases [84], ion homeostasis disruption [85], apoptosis [86], glutamate transporter [87], and muscarinic cholinergic receptor binding signaling [88]. The level of lipids [89], proteins by oxidative stress [90], and the endproducts of glycation are associated with neurofibrillary tangles [91], an aldehydic product of lipid peroxidation (4-hydroxynonenal), disruption in ion-motive ATPases, and calcium homeostasis [92]. According to data, Aβ also impairs the glucose uptake reduction in ATP levels in cultured cortical and hippocampal neurons mediated by 4-hydroxynonenal to GLUT3 conjugation and lipid peroxidation. So Aβ may result in a decrease in glucose uptake and neurodegeneration in AD [93]. Glucose transporter is impaired by Aβ deposition that ultimately affects the intra-mitochondrial acetyl–CoA synthesis and low substrate availability for CS that is responsible for low levels of citrate and neuronal cytoplasmic acetyl–CoA, which affects the synthesis of ACh.
Fluorodeoxyglucose-positron emission tomography (PET) studies in asymptomatic middle-aged APOEɛ4 allele carriers show the reduction in glucose utilization and changes in the mitochondrial bioenergetics [94]. APOEɛ4, an isoform of apolipoprotein E, forms a peptide of mitochondrial targeting sequence that inhibits COX and results in mitochondrial dysfunction and neurotoxicity [95–97]. Activities of the mitochondrial COX and CS were also reduced in the platelets of APOEɛ4 carriers versus non-carriers [98].
ABNORMAL ENERGY METABOLISM IN ALZHEIMER’S DISEASE
Poly ADP ribose polymerase-1 (PARP-1) activation affects the protein levels of cytochrome oxidase-IV and causes a decline in functional bioenergetics, rate of oxygen consumption, and membrane potential that results in deficits in cellular bioenergetics and also increases the expression of pyruvate kinases and modulates the glycolytic pathway through PARP-1. PARP-1 overactivation is associated with neurodegenerative diseases [99]. In addition, the level of full-length AβPP and its β-secretase cleavage product C99 were reported to cause impairment in energy metabolism and mitochondrial functions in the mice [100–102].
Moreover, low levels of peroxisome proliferator-activated receptor-gamma co-activator (PGC-1α) might also result in mitochondrial dysfunction. There is convincing evidence of a link between AD and PGC-1α [103]. Postmortem brain tissue samples of patients with AD showed a decrease in PGC-1α levels [104]. Additionally, AβPPswe/PS1dE9 mice were also reported to show decreased PGC-1α levels [105]. PGC-1α is one of the important regulators of carbohydrates and proteins and initiates mitochondrial respiration and biogenesis. The proteins and the mRNA level of PGC-1α were significantly reduced in neurodegenerative diseases [106].
The low levels of PGC-1α may cause mitochondrial dysfunction [107] and impairment of mitochondrial biogenesis [108]. In different AD mouse models, CS activities were significantly decreased in the mitochondria of brain cells, which indicated a decrease in mitochondrial mass and reduction in the levels of PGC-1α, cytochrome c-oxidase (complex IV) activity, ATP, and mitochondrial membrane protein [109–111].
Mitochondrial function is improved by restoring the levels of ATP, COX, and mitochondrial membrane potential, mitochondrial mass, enhancement of CS activity, and the levels of oxidative phosphorylation. Physiological CS activity and citrate level helps in enhancing acetyl-CoA-mediated cellular bioenergetics and ACh synthesis [112].
REDUCTION IN COX AND ENERGY ALTER AβPP TO THE AMYLOIDOGENIC PATHWAY
The presence of distinct features outside of the AD brain and inside the many tissues suggested that AD is not a brain-limited event or process. Aβ oligomerization and aggregation processes in the brain can alter AβPP homeostasis and energy metabolism in multiple tissues. Gabuzda et al. reported that the inhibition of COX shifts the process of AβPP to the amyloidogenic pathway and results in Aβ deposition [113]. Mitochondrial ROS also shifts the AβPP process towards Aβ [114], and different cell culture studies suggest that interference with cellular bioenergetics shifts the AβPP process to the amyloidogenic pathway [115, 116]. The triple transgenic female mice model of reduced COX activity showed an increase in glycolytic activity prior to amyloidosis [117]. CS knock down cells also result in impairment of respiratory activity and significantly decrease ATP production [118] (Fig. 2).
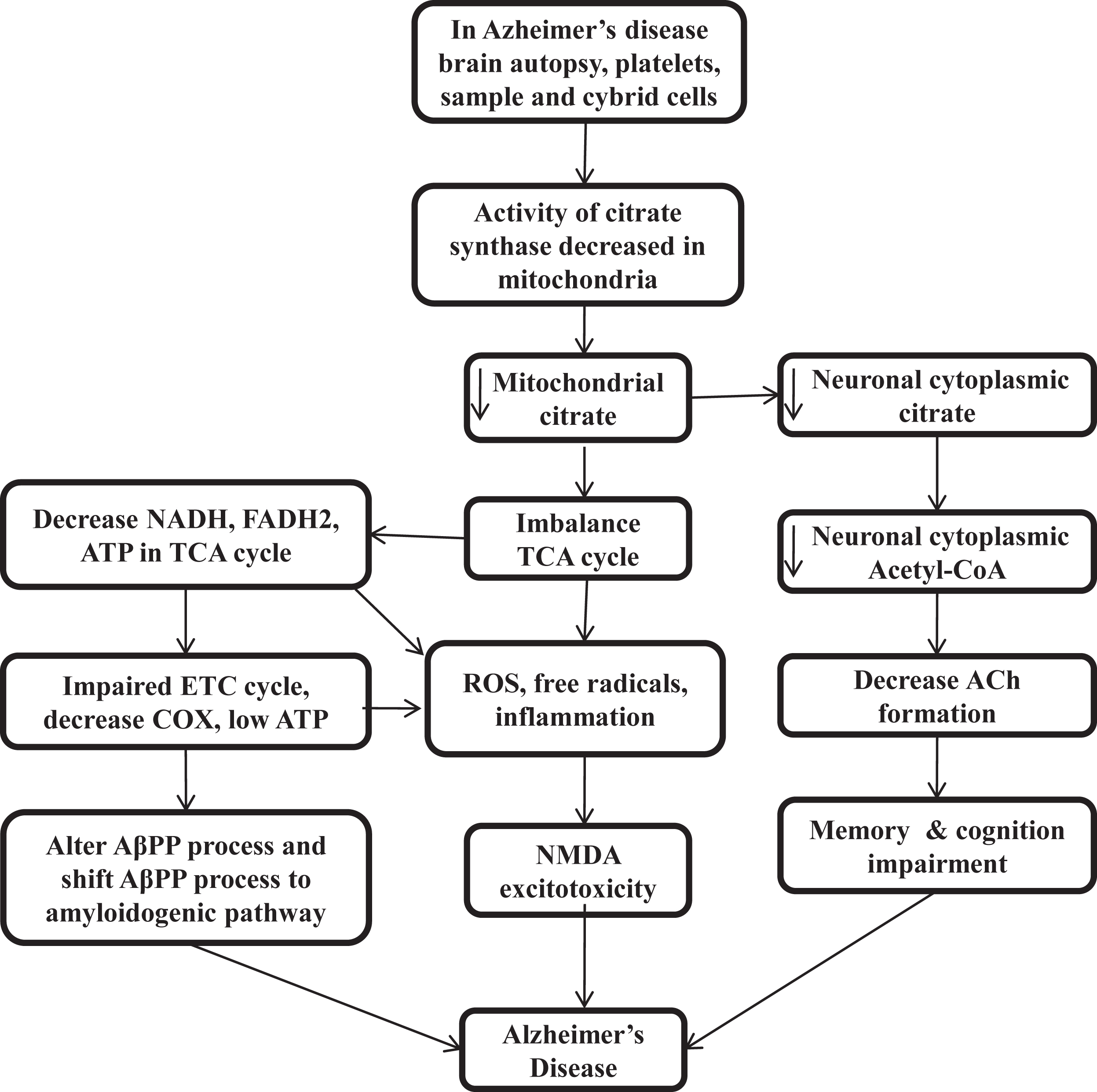
Low citrate synthase and citrate impairs cellular bioenergetics, acetylcholine synthesis, and loss of cognitive function in Alzheimer’s disease
CITRATE SYNTHASE ACTIVITY IN THE CLINICAL STUDIES OF ALZHEIMER’S DISEASE PATIENTS
In patients with AD, low CS activity was found in brain homogenate [119, 120], platelets [121, 122], prefrontal cortex [123, 124], temporal cortex [125], parietal cortex [126, 127], cybrid cells [128], and APOE4 carrier [98]. Patients with low CS levels show low citrate production, low cellular bioenergetics, reduced acetyl-CoA, and decreased ACh formation. Then low energy and mitochondrial ROS alter the AβPP process and shift it to amyloidogenic pathways [113–116]. At low energy, proteins have the tendency to self-aggregation, while at physiological ATP level, it prevents self-aggregation. Low cellular energy induces free radicals, ROS, and NMDA excitotoxicity [129]. The low level of PDH and Acetyl-CoA also results in many neurodegenerative diseases [44–47] that decreases the level of citrate in mitochondria and in neuronal cytoplasm and decrease the ACh synthesis and impairs cellular bioenergetics homeostasis or calcium homeostasis. Therefore, maintaining the level of CS and citrate by external sources can be a promising therapeutic target for curing AD by maintaining the cellular energy homeostasis, acetyl-CoA, and ACh production and lipogenesis of brain tissue. Interestingly, citrate, as a surfactant, also inhibits Aβ aggregation [130–133] (Fig. 2).
CITRATE IN PREVENTION OF AβAGGREGATION
The Aβ molecule is an amphiphilic peptide in nature and has N-terminal hydrophilic and C-terminal hydrophobic regions. Aβ form self-assembled aggregates of various morphologies like fibrils, protofibrils, filaments, dimers, and oligomers [134–138] due to intermolecular electrostatic and hydrophobic interaction [139–143]. Studies show that in different forms of Aβ, like monomeric, oligomeric, and fibrillar Aβ, the soluble forms of Aβ oligomers are more toxic in comparison to fibrillar and monomeric forms [144]. Further, inter-molecular electrostatic and hydrophobic interaction results in the aggregation of Aβ [139–143]. The aggregation of Aβ depends on the hydrophobicity of Aβ [133], so the molecule which inhibits the formation of Aβ aggregation could be a therapeutic option. The aggregation of Aβ was found to be inhibited by small number of surfactants that bind to the hydrophobic region of Aβ, which is responsible for its self-assembly [145–148]. Here citrate has an important role due to its high anionic charge density and small size [130]. Citrate has been reported to inhibit the aggregation of Aβ25 - 35 [131, 132] and aggregation of Aβ1 - 40in vitro [133].
Furthermore, cerebrospinal fluid (CSF) levels of citrate were significantly related to CSF total proteins levels and changes in CSF citrate can be a diagnostic parameter in patients with AD and other neurologic disorders [149]. Citrate also dissolves Aβ1 - 40 and β-2-macroglobulin, presumed to chelate calcium [150].
MITOCHONDRIAL FUNCTION IN THE NORMAL NEURONAL CELLS
Neuronal cells depend on the mitochondria at the synapse to get ATP and the concentration of buffer Ca++ ion to generate membrane potential for neurotransmission. Therefore, the amounts of the mitochondria are higher in the synaptic area as compared to the other parts of neuronal cells [151–153]. The correct transport of mitochondria to the synapse is an indicator of its efficient function. The synaptic and non-synaptic mitochondria are produced in the neuronal soma and transported to the other required area of the neuronal cells. This transport of mitochondria with axon towards synapse occurs through microtubules, motor proteins like dynein and kinesin, and outer mitochondrial protein Rho GTPase. The metabolic demand and Ca++ concentration at the synaptic level influence this axonal transport of the mitochondria [154–157].
Oxidative phosphorylation of mitochondrial respiratory chain complex causes two side effects; first, the generation of IMM-potential necessary for the import of nuclear-encoded proteins of the mitochondria and shows the health of cell and mitochondria itself. Second, electron leakage from the respiratory chain complex forms ROS, so ROS is the by-product of mitochondrial bioenergetics pathways [158]. But under normal physiological conditions, this ROS production is counter balanced by the mitochondrial antioxidant system. In neurodegenerative conditions, this equilibrium fails and leads to molecular damage in the native and surrounding areas. ROS targets the protein, carbohydrates, lipids, nucleic acids, and all the macromolecules of the cell [159].
In the brain, the hippocampus and cortex region is more affected by ROS due to their higher consumption of oxygen and dependence on the energy produced by mitochondria. Low levels of antioxidants and high levels of polyunsaturated fat trigger the vulnerability of the brain to ROS [160].
Mitochondria have a tubular network throughout the cytosol. The structural and morphological regulation of this mitochondrial network is made by the fusion and fission reaction [161]. Fission occurs when the mitochondrial tubule divides into the fragments and is controlled by proteins like dynamin-1-likeprotein (Drp1) with mitochondrial fission 1 protein (Fis1) and mitochondrial dynamics protein (MID49). Fusion occurs when two or more mitochondria are fused into one by the joint action of protein optic atrophy protein1 (OPA1) and Mitofusion1 protein (Mfn1 & 2). Through this fission and fusion mitochondrial biogenesis reaction, cells increase their mitochondrial mass [162].
PGC-1α is one of the key regulators of the mitochondrial biogenesis [163], which activates the chain of transcription factors and regulates the transcription, and replication of mitochondrial DNA [164] along with NFR-1, NFR-2 (Nuclear respiratory factor 1 & 2) and TFAM (Mitochondrial transcription factor A) and control the nuclear gene-encoded with mitochondrial proteins [163].
The cooperation between mitochondria and endoplasmic reticulum (ER) by forming a contact site mediates the intracellular buffer of calcium [165] that allows the uptake of calcium from cytosol and ion exchange between these two organelles [166]. Ca++ is an important regulator of mitochondrial metabolic enzymes [167], and mitochondria have two calcium transporter: 1) mitochondrial calcium uniporter, having high selectivity for calcium ions in the IMM [168], and 2) voltage dependent an ion channel (VDAC) in the outer mitochondrial membrane that regulates calcium release from the mitochondria [165].
VDAC works with IMM adenine nucleotide transporter and in the matrix with cyclophilin D after forming membrane permeability transition pore (mPTP) [169]. The opening of mPTP results in cell death by the activation of apoptosis [170], and the amount of calcium is regulated by the mitochondria for the neuro transmission and the execution of synaptic functions [171, 172].
Cellular homeostasis and mitochondrial functions are maintained by the quality control system of mitochondria. At the same time, the endogenous proteases and chaperons regulate the folding state and mitochondrial protein activity [173], and selective autophagy removes the damaged mitochondria, termed mitophagy [174]. That is primarily regulated by the signaling system of protein Pink1 (PTEN induced kinase1) and ubiquitin ligase parkin, which activate after losing the mitochondrial membrane potential [175]. Pink1accumulation at the damaged mitochondrial outer membrane recruits the ubiquitin ligase Parkin, which might label the mitochondria to the process of autophagy and engulfment of mitochondria by fusion with the lysosomes, and the material of mitochondria is digested [176].
MITOCHONDRIAL FUNCTION IN ALZHEIMER’S DISEASE
The Aβ oligomer is responsible for primary neurotoxicity [177] by numerous mechanisms like the production of ROS, inflammatory response, and altered Ca+2 homeostasis [178]. The accumulation of Aβ and mitochondrial dysfunction results in ROS production in AD [179]. In the human AD brain, different metabolic pathways and enzymes involved in glycolysis and ETC oxidative phosphorylation are affected at the transcriptional level and reduced their activity [180, 181]. In ETC, reduced oxidative phosphorylation protein expression of complex-1andcomplex-IVinpR5/APP/PS2 (triple transgenic mice model) leads to progressive reduction in cerebral aerobic glucose metabolism [182]. The bioenergetics deficiency of mitochondria is the main sign of AD pathophysiology in the AD triple transgenic female mouse model by decreasing the activity of PDH E1α and COX 4 and protein expression [117]. In mitochondrial deficiency, the cell may shift its energy requirements from mitochondria to glycolysis and other metabolic pathways. In the conversion of phosphoenolpyruvate to pyruvate, the pyruvate kinase is the main catalytic enzyme and rate-limiting step in glycolysis for energy production [183]. PKM2 slows the mitochondrial oxidative phosphorylation even in the oxygen presence like Warburg effects [184].
The variation in the energy pathway reduces glucose consumption in the AD disease brain [185]. According to PET studies in AD, glucose metabolism was found to be 20–30% lower in the area of memory processing like the hippocampus, parietal lobe, temporal lobe, and posterior cingulated area than in a healthy brain [186]. That shows the variation in metabolic signs occurs early than the appearance of changes in histopathological signs and symptoms [185]. In several animals models and human AD patients, mitochondrial functions are severely compromised [107] in terms of morphology [187] and numbers of mitochondria [188], oxidative phosphorylation, mitochondrial membrane potential, buffer calcium concentration, production of ROS [189], mutation and oxidation of mitochondrial DNA [190], contact sites of mitochondria and ER [191], biogenesis of mitochondria and transport to the neuronal axon [192], and mitophagy [172]. Dysfunction in these processes results in synaptic loss and other consequences in the brain [192].
The activity of mitochondrial enzymes involved in energy production like PDHC, citrate synthase isocitrate dehydrogenase, αKGDH (α-ketoglutarate dehydrogenase), complex-IVCOX, and ATP synthase were decreased in AD, while the activity of complex-II succinate dehydrogenase and the malate dehydrogenase were increased [120, 121]. That compromises the mitochondrial membrane potential and production of ATP [193].
The imbalance between the generation of ROS and antioxidants was found in the blood, CSF, and in the brain of AD patients [194]. Oxidative stress caused by ROS is considered a main contributing factor in the pathogenesis of AD [195] and mild cognitive impairment (MCI) in the early stage of AD by increasing protein oxidation and lipid peroxidation and decreasing the levels of antioxidants in the brain and the peripheral regions [196].
The sample of AD patients and transgenic mice models (mAPP mice) shows the association of mitochondrial dysfunction with oxidative stress by impaired oxidative phosphorylation, axonal transport of mitochondria, increased oxidative stress, and mPTP [197]. The triple transgenic mice model of AD shows the compromised bioenergetics, and increased oxidative stress level appears early than the development of Aβ plaque [107, 117]. The lymphocytes sample of peripheral blood of AD, AD brain, and MCI patients found the oxidation of the ATP synthase and the enzyme of oxidative phosphorylation [198] represents the reduction in the activities of ATP synthase, and the levels of the ATP in AD. Reduction in plasma antioxidant level and aconitase (ACO2) activity in the peripheral lymphocytes of MCI and AD patients also proves the correlation between mitochondrial dysfunction and oxidative stress [199].
In human-induced pluripotent stem cells (iPSCs) model of AD also shows that oxidative stress and mitochondrial dysfunction are the main causative factor in AD pathology by increasing ROS, respiratory chain complexes, and vulnerability to stressors in neurons and astrocytes of AD-iPSCs [200].
Mitochondrial dynamic processes of fusion and fission were imbalanced in AD patients, resulting in the distribution of compromised mitochondria in neurons [188] and fragmented mitochondria in fibroblast and the brain of AD patients [201]. In the hippocampus of AD, the mitochondrial protein expression is abnormal and finds an increase in fission protein Fis1 along with the downregulation of the Drp1, and the fusion proteins OPAI, Mfn1, and Mfn2 [202]. The same is also found in the cybrid cells model with shorter and bleb-like mitochondria compared to the control [203]. And the further increase in phosphorylation of Drp1 S-nitrosylation at the site of Ser-616 promotes the fission of mitochondria [204] which is higher in the AD brain than in control [202]. Protein Drp1 interacts with hyperphosphorylated tau and Aβ in brain homogenates of the AD patients [205]. A recent study of a sample of the healthy control group and AD subject shows the link between the polymorphism in AD and the MFN2 gene, suggests that polymorphism in the regulatory process of mitochondrial fusion might have a role in the pathogenesis of the AD [206]. Protein mfn2 work as a cord between ER and mitochondrial membrane [207]. Presenilin 2 (PS2) is influenced by mfn2 and its mutation is linked to familial AD in ER-mitochondrial contact site modulation [208]. Different AD experimental models of Aβ peptide treatment and over expression of AβPP are characterized as fragmentation of mitochondria and due to variation in protein levels of fusion and fission process leads to abnormal distribution of mitochondria along with neurons [209–211].
Another important mitochondrial function is bioenergetics which is impaired in AD and decreases the mitochondrial number in the hippocampus of the human AD brain or in other cell culture models due to compromised mitochondrial bioenergetics [188, 209]. The level of mitochondrial regulatory proteins like NRF1, NRF2, PGC1α, and TFAM was decreased in the hippocampus overexpressing cellular models of APP Swedish mutation [104, 212]. In other mouse models of AD, i.e., humans carrying mutant transgenes of the Presenilin-1 (PS1) and APP, the markers of mitochondrial bioenergetics were also found to decline in the hippocampus, and here melatonin shows some beneficial effects [213].
Another factor, mitophagy, was able to prevent cognitive deterioration, deposition of Aβ, tau hyperphosphorylation, and reverse memory impairment in many AD models [214]. But in AD, mitophagy is affected and causes neuronal dysfunctions by accumulations of mitochondrial damage. This may be due to fusion impairment between the lysosomes and the autophagosomes [215].
In AD, the somatic mutation found in mitochondrial DNA is higher than in the healthy brain, which triggers ROS production and promotes the amyloidogenic cascades and other neuropathological consequences [216].
The histopathological markers of AD, Aβ and tau, interact with mitochondria non-specifically [182] and adversely affect the mitochondrial axonal transport from soma to the synapse of the neurons. Overexpression Aβ mouse model of AD has damaged mitochondria, impaired mitochondrial axonal transport, decreased mitochondrial membrane potential, and inhibition of respiratory chain complexes with lower ATP production [217]. The AβPP inside the mitochondria interacts with the Aβ peptide and with the mitochondrial matrix component [218, 219]. Aβ impaired the import of mitochondrial nuclear-encoded proteins by the process of mitochondrial co-aggregation [220].
The tauopathies in AD also impaired the functions of mitochondria and their axonal transport at the presynaptic terminals [221]. In AD, hyperphosphorylated tau interacts with VDAC1 and leads to mitochondrial dysfunction [205]. Hyperphosphorylated tau decreases the activity of complex-1 and ATP production, increases ROS, lowers the mitochondrial membrane potential, stimulates the mitochondrial fission process, and the fragmentation of mitochondria in the postmortem brains of murine models and AD patients [222, 223], and oxidative stress stimulates tau hyperphosphorylation [224].
The outer membrane translocase homolog unit Tomm40 is a channel at the outer mitochondrial membrane responsible for the import of nuclear-encoded protein [225]. Aβ affected this import machinery and caused mitochondrial dysfunction in AD [226, 220]. TOMM40 has a closed gene cluster with the APOE gene at chromosome-19 and APOE is the most significant risk factor in sporadic genetic late-onset AD (LOAD) linked with high-risk isoform ɛ4. A variable-length and the polymorphism of deoxythymidine homo polymer of TOMM40 gene intron 6 shows genetic risk factors for LOAD [227–229]. In the Caucasian ethnic group, there are three variants of TOMM40 polymorphism. One of the variants, rs10524523 in APOE3/4 carriers, lowered the onset of LOAD by 7 years [230] which impaired the volume of the grey matter and cognition in the brain of AD [231]. Some other groups also influence TOMM40 “523” variant on APOE and TOMM40 gene transcription [232].
OXIDATIVE STRESS MEDIATED INFLUENCES ON CS AND CITRATE AND BEGINNING OF THE SILENT PHASE PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
An accumulation of Aβ and hyperphosphorylation of tau proteins might not be the primary cause of neuronal degeneration in LOAD. Additionally, some other factors might be responsible for it [233, 234]. Abnormal cellular bioenergetics contributes to normal aging and pathogenesis of LOAD [235, 236]. The hippocampal culture or neuronal cells get oxygen consumption and energy utilization by oxidative phosphorylation to power the neuronal cells activities [237]. But, when neuronal cells are highly synaptically active or high in metabolic activity, the O2 consumption is increased multifold, which generate H2O2 and ROS in higher amounts. Therefore, the neuronal cell shifts ATP utilization from oxidative phosphorylation to aerobic glycolysis (pentose sugar pathway) to prevent this ROS and free radicals that damage the neuronal cell and start inflammatory pathway. This shift is called the Warburg effect. The loss of CS is directly linked with the Warburg effect. CS knockout cells severely decrease the respiratory activity and showed a marked decrease in ATP production but a huge increase in glycolytic metabolism [11]. This shift of neuronal cell energy utilization from the glycolysis pathway decreases ROS and decreases the activity of mitochondria [238–240]. Actually, it is the mechanism of acquired neuroprotection [241]. This transition helps in decreasing the accumulation of ROS. But, at the same time in order to prevent ROS and free radicals, the shift of energy utilization from glycolysis pathway by the hippocampal neurons that are highly synaptically activated, upregulate both GLUT3 (neuronal glucose transporter) for increasing glucose uptake and the pyruvate dehydrogenase kinase-3 (PDK3) enzyme [242–244]. In the hippocampus, PDHC activity is found to be highest [44], and PDK3 phosphorylates the PDH in mitochondria and inhibits the conversion of glycolysis endproduct pyruvate to acetyl-CoA or ultimately inhibit acetyl-CoA and citrate. Then less acetyl-CoA results in less substrate for CS and ultimately less citrate and further less acetyl-CoA (serve as direct energy source to neuronal cell and also lipogenesis for brain tissue) decreases ACh synthesis in neuronal cytoplasm [245, 246], which starts the silent phase pathogenesis of AD.
The shift in energy utilization from the oxidative phosphorylation pathway to glycolysis causes a decrease in ROS production by decreasing mitochondrial activity as the reduction in oxidative phosphorylation and reduction in the inducing effects on oxidative phosphorylation by calcium in the matrix of mitochondria [247], represses the transcriptional mitochondrial calcium uniporter via calcium-Npas4induction [248], and reduces the harmful conversion of mitochondrial energy at the cost of low ATP yield by glucose metabolism or glycolysis [247]. A recent study also shows that the soluble form of Aβ aggregates is found in the silent phase of AD [249].
Oxidative stress-mediated damage if found in an early phase of AD and it decreases as the disease progresses, and increased deposition of Aβ is associated with reduced oxidative damage [250]. In humans, studies find enlargement of the resting synapse as a result of compensatory response that allows the system to perform well and counter balance this initial damage [251, 252]. So that in humans, the progression of AD from an early phase to symptomatic phase takes a long time [253]. The upregulation of GLUT3 and PDK3 can be biomarkers for the silent phase of AD. While GLUT3 transporter is reduced in the brain of AD patients, results show decreased glucose metabolism [81], which again hamper overall brain functions, cellular bioenergetics, ACh synthesis in AD, and cause loss of memory and cognitive functions. It also reduces acetyl-CoA mediated lipogenesis and shrinks the brain tissue over the time (Fig. 3).
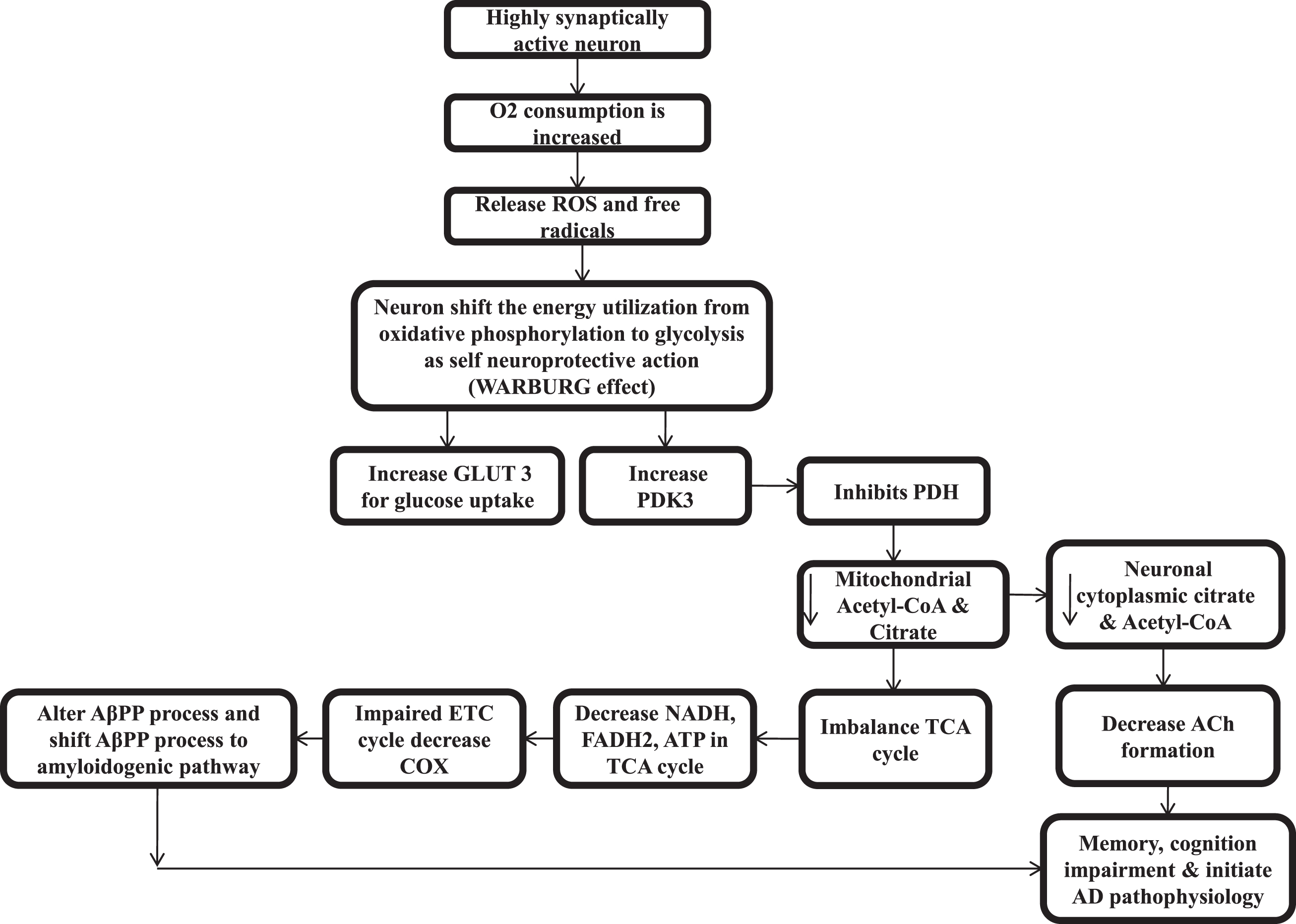
Pathophysiology of silent phase of Alzheimer’s disease.
In recent reports, these above-mentioned factors, like changes in energy metabolism, etc., result in normal aging and are responsible for the pathogenesis of LOAD. The cells of LOAD also show impaired mitochondrial metabolic and redox potential, decreased metabolism of NAD+, altered activity of the TCA cycle, and shifting of the energy production towards glycolysis [254].
CONCLUSION
According to this review, mitochondrial CS activity and citrate are important factors in AD. It suggests that AD is a metabolic disorder more than a neurodegenerative disease. In clinical studies of AD patients, mitochondrial enzyme CS activity is found to be lower in different regions of brain tissue, platelet samples, and cybrid cells. Decreased CS reduces citrate, a substrate for cellular bioenergetics in TCA cycle, and acetyl-CoA, ACh synthesis in cytosol. Decreased citrate, cellular bioenergetics, and neurocytoplasmic acetyl-CoA reduce ACh synthesis and results in memory and cognition impairments. Furthermore, reduced acetyl-CoA also decreases lipogenesis that leads to shrinking of the brain tissues. A lower energy environment in the cell favors Aβ aggregation. Therefore, citrate can be a more effective treatment for AD because it maintains cellular energy, improves ACh synthesis physiologically, lipogenesis, and inhibition Aβ aggregation. Citrate can also be utilized in the treatment of type 2 diabetes.
In AD’s silent phase, higher oxidative stress, free radicals, and ROS upregulate GLUT3 and PDK3. GLUT3 promotes more glucose uptake, whereas PDK3 inhibits PDH and inhibit acetyl-CoA, which is the substrate of CS, and same pathway of CS related to citrate, cellular bioenergetics, and ACh synthesis is inhibited. So GLUT3 and PDK3 can be biomarkers for the silent phase of AD. For further development, inhibition of PDK3 can play an important role in the silent phase of AD; behavioral therapy of breathing exercises and intermittent fasting can also prevent this oxidative stress and improve the mitochondrial quality control system in initial stages. Oxidative stress can be overcome by decreasing the exposure of environmental pollutants having oxidizing properties, by increasing the levels of endogenous and exogenous antioxidants, and by stabilizing the production and efficiency of mitochondrial energy. Importantly, citrate maintains mitochondrial energy production as well as having antioxidant properties.
In the future, an animal model can be develop by reducing the CS and citrate for the silent phase of AD. Current treatments like acetylcholinesterase inhibitors and NMDA antagonists only slow down the disease progression. However citrate can be a new and more effective therapeutic option by improving mitochondrial health, cellular bioenergetics, acetyl-CoA, and ACh synthesis, than the current treatments of AD.
Footnotes
ACKNOWLEDGMENTS
We thank the whole Department of Pharmacology, PGIMER, Chandigarh, Dr. Prem Prakash Khosla, Abhishek Chhimpa, Dr, Harmanjit Singh, Dr. Parvinder, Dr. Meenakshi Raju, Dr. Shikha Raheja, Dr Sudeep Bhardwaj, Dr. Ashutosh Agarwal and Dr. Muskan Malhotra.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.