
Abstract
Background:
Mitochondrial 17β-hydroxysteroid dehydrogenase type 10 (17β-HSD10) is necessary for brain cognitive function, but its studies were confounded by reports of Aβ-peptide binding alcohol dehydrogenase (ABAD), formerly endoplasmic reticulum-associated Aβ-peptide binding protein (ERAB), for two decades so long as ABAD serves as the alternative term of 17β-HSD10.
Objective:
To determine whether those ABAD reports are true or false, even if they were published in prestigious journals.
Methods:
6xHis-tagged 17β-HSD10 was prepared and characterized by well-established experimental procedures.
Results:
The N-terminal 6xHis tag did not significantly interfere with the dehydrogenase activities of 17β-HSD10, but the kinetic constants of its 3-hydroxyacyl-CoA dehydrogenase activity are drastically distinct from those of ABAD, and it was not involved in ketone body metabolism as previously reported for ABAD. Furthermore, it was impossible to measure its generalized alcohol dehydrogenase activities underlying the concept of ABAD because the experimental procedures described in ABAD reports violated basic chemical and/or biochemical principles. More incredibly, both authors and journals had not yet agreed to make any corrigenda of ABAD reports.
Conclusion:
Brain 17β-HSD10 plays a key role in neurosteroid metabolism and further studies in this area may lead to potential treatments of neurodegeneration including AD.
Keywords
INTRODUCTION
Most 17β-hydroxysteroid dehydrogenases are associated with the endoplasmic reticulum (ER) [1]. Among fourteen types of 17β-hydroxysteroid dehydrogenases, only types 8 and 10 are found in mitochondria [1–8]. The first-discovered function of human 17β-HSD10 is its ability to catalyze the redox conversion between L-3-hydroxyacyl-CoA and 3-ketoacyl-CoA with NAD+/NADH as coenzyme [3]— namely, its L-3-hydroxyacyl-CoA dehydrogenase activity (Fig. 1). Accordingly, 17β-HSD10 was initially called short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD) or L-3-hydroxyacyl-CoA dehydrogenase type 2 (HADII) [2–6]. Its catalytic activities are involved not only in neurosteroid metabolism [1–5, 9–18], but also in acyl thioester metabolism [2–4, 15–20].
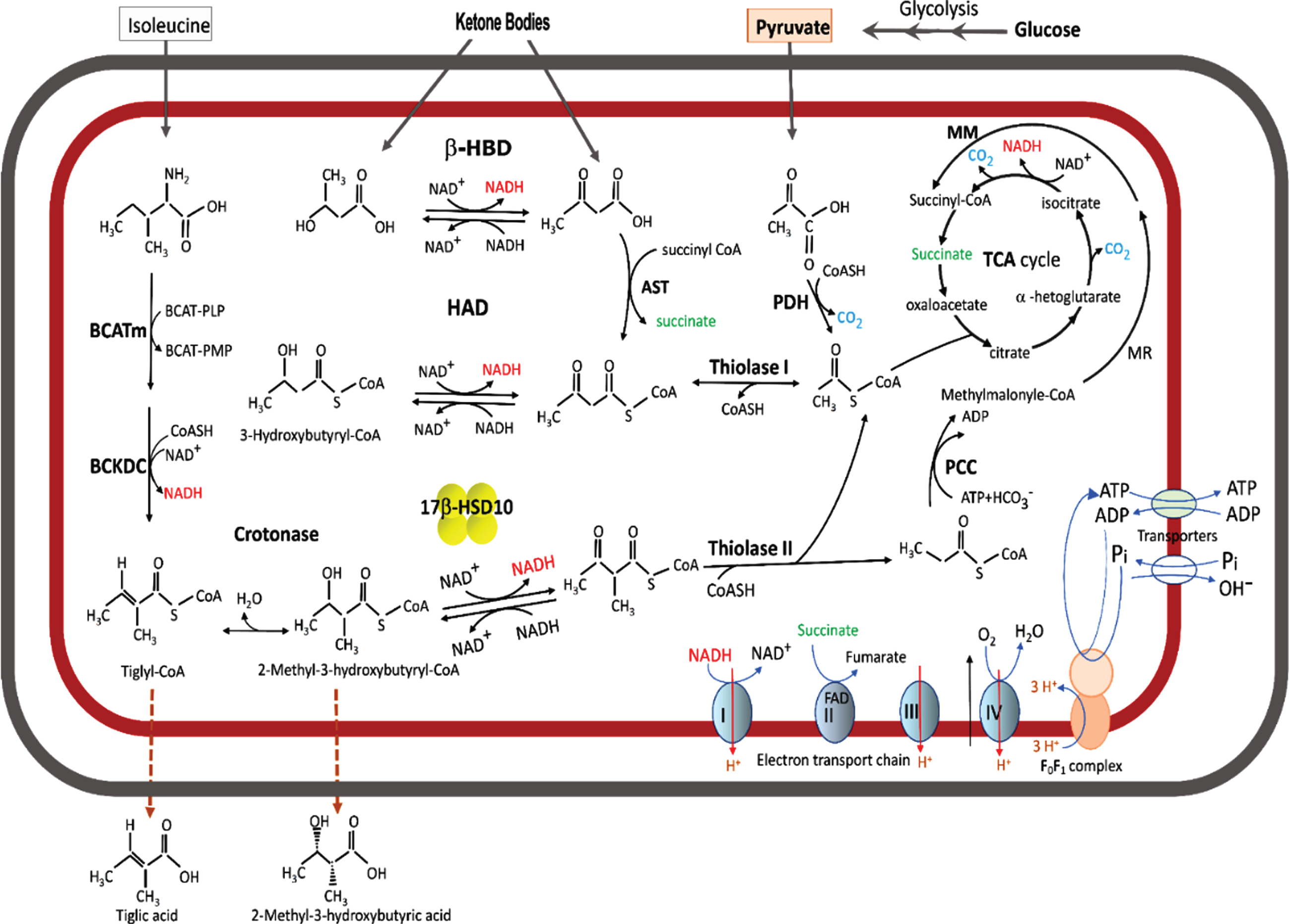
Acyl thioester metabolism and oxidative phosphorylation in brain mitochondria. The fatty acid β-oxidation pathway is shown only the last two steps. A cluster of four yellow balls represents 17β-HSD10. β-HBD rather than HAD or 17β-HSD10 plays a key role in ketone body metabolism. A missense mutation in the 17 β-HSD10 gene would block the isoleucine catabolic pathway to result in the accumulation of isoleucine metabolites such that tiglic acid and 2-methyl-3-hydroxybutyric acid would be released from the HSD10 deficienct-cell and their glucoronated derivatives be excreted in the urine of patient having an inborn error in isoleucine metabolism.
17β-HSD10 is a homo-tetrameric protein with a molecular weight of 108 kDa and plays a key role in the pathogenesis of Alzheimer’s disease [2–5, 10–22], Parkinson’s disease [23], developmental disabilities [18–20, 24–34], as well as certain cancers [10, 19]. A 27 kDa peptide was discovered by using a portion of amyloid-β peptide (Aβ) protein precursor as a bait and nominated as an endoplasmic reticulum-associated Aβ-binding protein (ERAB) in a high-impact journal [21]. The primary structure of ERAB turned out to be almost identical to a subunit of SCHAD/17β-HSD10 [2, 3]. No one has ever purified such a 27 kDa protein, although the ERAB reportedly exhibited L-3-hydroxyacyl-CoA dehydrogenase (HAD) as well as generalized alcohol dehydrogenase activities [35]. This report was challenged almost immediately [4] (see Supplementary Material 1). Yet no corrigenda were published to date, because neither authors nor the journal that published the ERAB report agreed to revisit the issue (see Supplementary Material 2). Nevertheless, ERAB was renamed Aβ-peptide-binding alcohol dehydrogenase (ABAD), and more unreproducible data were published in the high-impact journal [36] (see Table 1 of [17]). The endoplasmic reticulum-associated ABAD reportedly plays an important role in ketone body metabolism, allegedly modulating nutritional stress such as cerebral ischemia in stroke [35, 36]. Thus, the lack of response to published challenges to the validity of the ERAB/ABAD reports [21, 36] has led to the dissemination throughout the scientific literature of ERAB/ABAD data, hypotheses, theories, and conclusions— without correction. This dissemination has continued to interfere with research studies on valid mechanisms of neurodegeneration [11].
Missense mutations in the HSD17B10/HADH2 gene [8] are known to generate 17β-HSD10 mutants, most of which cause progressive neurodegeneration— namely HSD10 deficiency [13–20, 24–34]. Although it was emphasized by ABAD studies [21, 36] that ABAD/ERAB mediates Aβ neurotoxicity to cause senile neurodegeneration, alterations of 17β-HSD10 activity itself would result in neurodegeneration because little Aβ was found in the cerebrospinal fluid of infantile neurodegeneration patient [37].
The HAD activity of 17β-HSD10 usually serves as an indicator for the catalytic function of this multifunctional protein [2–4, 30–33]. Because extremely distinct catalytic capabilities between ABAD and 17β-HSD10 were found by use of different experimental procedures for assaying HAD [2–4, 31–36], it is necessary to investigate which procedure(s) could best evaluate HAD activity.
Here we show that the experimental procedure described in [3], but not that in [35], can accurately determine HAD activity, and that no reliable or valid data can be obtained for an alleged “generalized alcohol dehydrogenase” activity [35, 36]. Further, 17β-HSD10 is not involved in ketone body metabolism; the data reported for Aβ inhibition of the dehydrogenase activity of ABAD (i.e., 17β-HSD10) are unreliable and invalid. Taken together, all the above establishes that no rationale exists to employ “ABAD” as an alternative name for 17β-HSD10, and the major role of 17β-HSD10 in AD pathogenesis is not due to “mediation of Aβ neurotoxicity,” as reported previously [21, 22].
MATERIALS AND METHODS
Materials
Acetoacetyl-CoA, (–)-2-octanol and enantiomers, NAD+, NADH and standard biochemicals were obtained from Sigma-Aldrich.
Enzyme preparation and assays
6xHis-tagged-HSD10 was prepared as previously reported [31]. Protein concentration was determined by the Bio-Rad reagent according to the manufacturer’s instruction. The L-3-hydroxyacyl-CoA dehydrogenase activity of 6xHis-tagged-HSD10 was determined in the backward direction with acetoacetyl-CoA as substrate and NADH as coenzyme according to two different experimental procedures Measurements of HAD activity were performed by a traditional method [38] using a continuously-recording spectrophotometer (Hitachi U-3010). Data were then analyzed by the Leonora computer program [39]; HAD activity was determined by another published method [35]. The assay for reduction of S-acetoacetyl-CoA employed 6xHis-tagged HSD10 (333 ng/ml), a range of S-acetoacetyl-CoA concentrations up to 0.36 mM and NADH (0.1 mM) in 97 mM potassium phosphate (pH 7.3). The reaction was run for a total of 2 h at 25°C “under steady-state condition (24), and the change in NADH absorbance at 340 nm was determined every 5 min” as described in [35]. The “(24)” in the quotation above is a well-known text on enzyme kinetics [40].
The principle of experimental procedure
Preparation of assay mixture to allegedly measure generalized alcohol dehydrogenase activities of human ABAD/ERAB†
†Concentrations of these prepared (–)-2-Octanol, (+)-2-Octanol, and (±)-2-Octanol solution supposed to be 43 mM, 84 mM and 85 mM, respectively, which are equal to previously reported Km value of ABAD/ERAB for these optical isomers shown in Table 1 of [35]. *Mixed with alcohol first and vortexed for 3 min, adding buffer and water vortex again, then incubated at 25°C for 72 h; /, Do not apply.
The molar extinction coefficient used for calculating rates was 6220 M–1cm–1. A unit of activity is defined as the amount of enzyme that catalyzes the conversion of 1μmol of substrate to product per minute.
RESULTS
Comparison of enzyme activity data secured by use of experimental procedure b versus experimental procedure a
When 0.36 mM acetoacetyl-CoA and 0.1 mM NADH were used as substrate and coenzyme in the enzyme assay, the reaction ended rapidly following addition of the 6xHis-tagged-HSD10 (333 ng/ml). As shown in Fig. 2, the absorbance at 340 nm (black dots) decreased with time gradually but evidenced no further decline at about 15 min (see a blue dash line). After 20 min the absorbance change at 340 nm with time becomes barely measurable spectrophotometrically, even if the reaction time had been intentionally extended further until 120 min [35]. Since the total ΔA340 was only –0.42 in this measurement (see the blue dashed line in Fig. 2), the HAD specific activity determined by use of experimental procedure
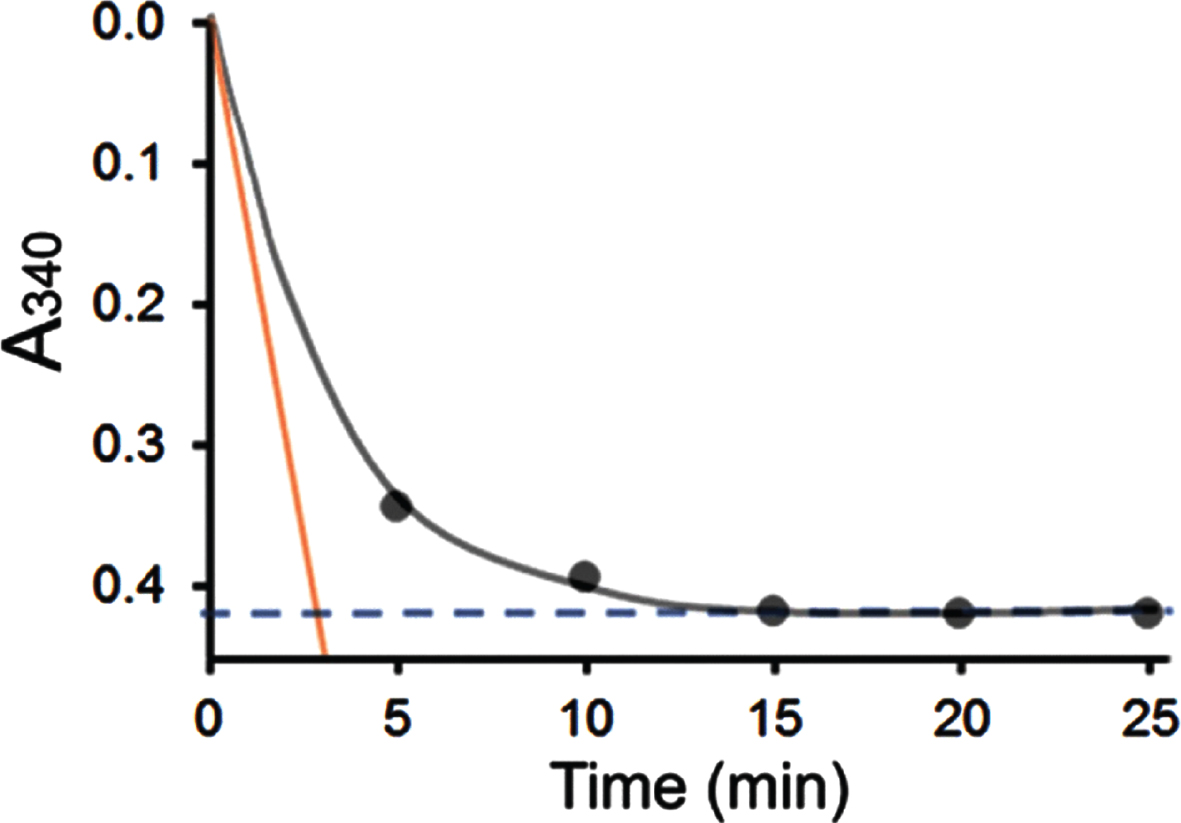
Comparison of the HAD activity of 6xHis-tagged HSD10 (human) (333 ng/ml) measured using different experimental procedure
In contrast, the absorbance change with time was recorded as a curve by use of a continuously recording spectrophotometer according to experimental procedure
Impacts of addition of 6xHis tag on the HAD activity of 17β-HSD10
The HAD activity of 6xHis-tagged-HSD10 was measured according to experimental procedure
Comparison of 3-hydroxyacyl-CoA dehydrogenase activity of human 17β-HSD10, 6xHis-tagged HSD10 and ABAD/ERAB
†Results of the present study by use of the experimental procedure
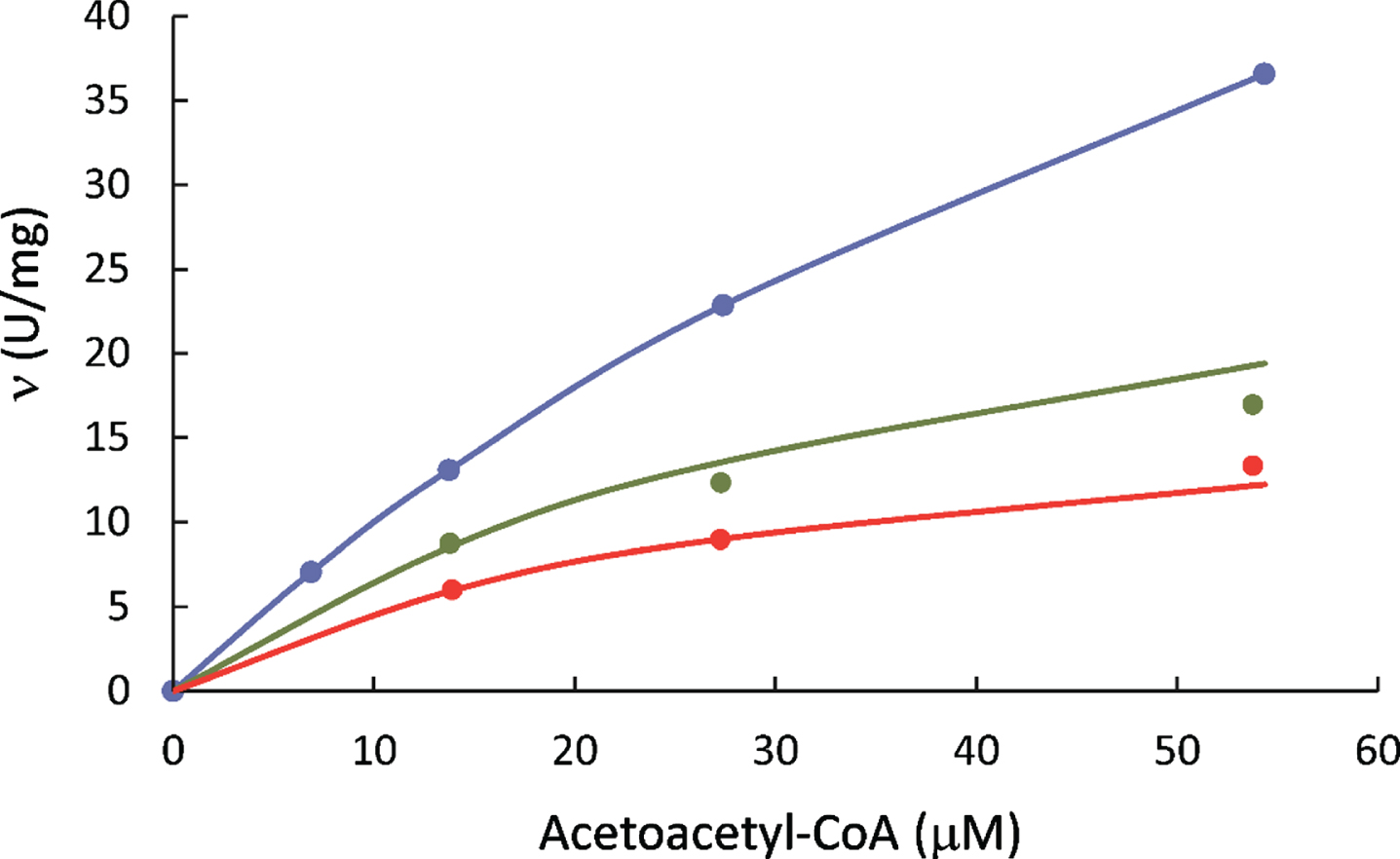
L-3-Hydroxyacyl-CoA dehydrogenase activities of 6xHis-tagged HSD10 (human) measured in the presence of 118.6μM NADH (blue), 23.7μM NADH (green), and 11.9μM NADH (red), respectively.
For investigating the alteration of its catalytic activity by a mutation or residual activity in the presence of an inhibitor, HAD data of 6xHis-tagged HSD10 obtained in the absence of inhibitor was taken as 100%.
Comparison of the v versus [s] curve of 6xHis-tagged-HSD10 and the reported v versus [s] curve for ABAD
When the enzyme activity was measured by use of the experimental procedure
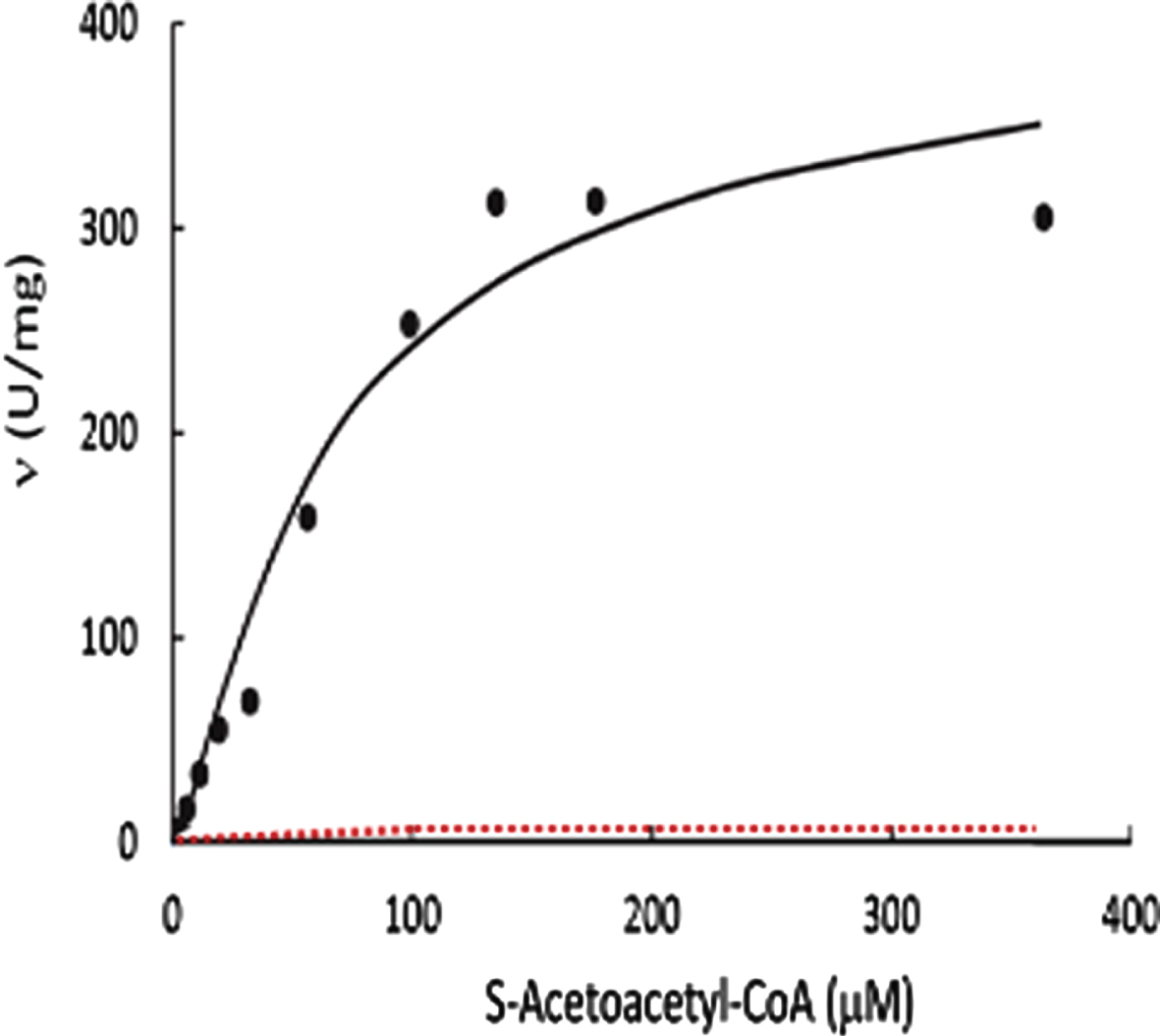
Measurement of generalized alcohol dehydrogenase activities of 6xHis-tagged HSD10 by use of experimental procedure c
When the alcohol dehydrogenase assay mixture was prepared according to the experimental procedure

Interface between alcohol and phosphate buffer in generalized alcohol dehydrogenase assay mixtures. The assay mixture prepared according to experimental procedure
Comparison of alcohol dehydrogenase activity of human 17β-HSD10, 6xHis-tagged HSD10, and ABAD/ERAB
†Data from the present study by use of the experimental procedure
Michaelis constants of ABAD, 17β-HSD10, and 6xHis-tagged HSD10
K m is a pseudo-equilibrium constant expressing the relationship between the actual steady-state concentrations of enzyme, substrate, and the enzyme •substrate complex [40]. A comparison of K m values of 17β-HSD10, 6xHis-tagged HSD10, and ABAD/ ERAB for their 3-hydroxyacyl-CoA dehydrogenase and alcohol dehydrogenase functions are displayed in Tables 2 and 3, respectively. Comparing data listed in the first and second column of the Table 2, it was illustrated that adding a 6xHis tag to the N-terminal of 17β-HSD10 does not cause any significant alteration of its binding to its substrates and its catalytic capability for the redox reaction of L-3-hydroxyacyl-CoAs.
DISCUSSION
17β-HSD10 is reportedly essential for (a) degrading isoleucine and promoting fatty acid oxidation [12–19, 30–33]; (b) serving as a cardiolipin phospholipase [41] involved in the metabolism of cardiolipin (an important constituent of the mitochondrial inner membrane); and (c) binding with RNA (quinine-9-)methyltransferase domain containing 1 and KIAA0391 (protein-only RNase P catalytic subunit) to form a holoenzyme (mitochondrial RNase P) necessary for mtRNA maturation [42].
The three-dimensional structure of 17β-HSD10 was reported to be identical to that of ABAD [43], and the latter was also reported to exhibit L-3-hydroxyacyl-CoA dehydrogenase activity [35, 36]. To our knowledge, three types of mitochondrial L-3-hydroxyacyl-CoA dehydrogenase exist due to their distinct substrate chain length preference [33]. 17β-HSD10 belongs to the short-chain dehydrogenase family [2–5, 32–34].
Despite that reliable HAD assays had already been well-established [38, 45], a novel method to determine HAD activity was created by Yan et al. [35], which is identified as experimental procedure
As shown in Fig. 4, the v versus [S] curve obtained by using experimental procedure
As shown in Fig. 2, about two thirds of the NADH in the assay system was oxidized to NAD+ using experimental procedure
According to a published bell-shaped pH curve of SCHAD/17β-HSD10 [2], the difference in HAD activity due to a pH difference between 7.0 and 7.3 is insignificant as both pH values are close to the optimal pH (see Fig. 2 of [2]). Therefore, the pH difference between experimental procedures
Although K m values of ABAD/ERAB for medium-chain alcohols had been reported in Table 1 of [35], we establish here that all those data are unreliable. For example, the concentration of a saturated solution of (–)-2-octanol is about 8.6 mM [46], which is only one fifth of the K m value reported for ABAD/ERAB (see Table 3). Assuming the reported K m value of 43 mM for (–)-2-octanol (see Table 1 of [35]) is correct, the maximal velocity of ABAD will be attained until 86 mM (–)-2-octanol is used as substrate in an assay. Since the addition of 1% DMSO is certainly unable to solubilize (–)-2-octanol to form a sufficiently high concentration in the enzyme assay mixture (see Fig. 5), it was concluded that data published in Fig. 2B of [35] is questionable (see Supplementary Material 2).
The reported specific HAD activity of ABAD/ERAB determined in the absence of Aβ140— an alleged inhibitor— was considered as 100%. Its real value determined in the present study (1.7±0.0 U/mg) was found to be even smaller than the previously reported ABAD/ERAB HAD specific activity under the inhibition of 5μM Aβ140 (see Fig. 6). Thus, data reported for inhibition of ABAD/ERAB by Aβ140 [35] could not have been derived from any valid reproducible experiments. With respect to the inhibition of Aβ peptides to L-3-HAD activity of ABAD/ ERAB it had been reported that, in addition to Aβ140, Aβ2535 could also inhibit 17β-HSD10 effectively [47].
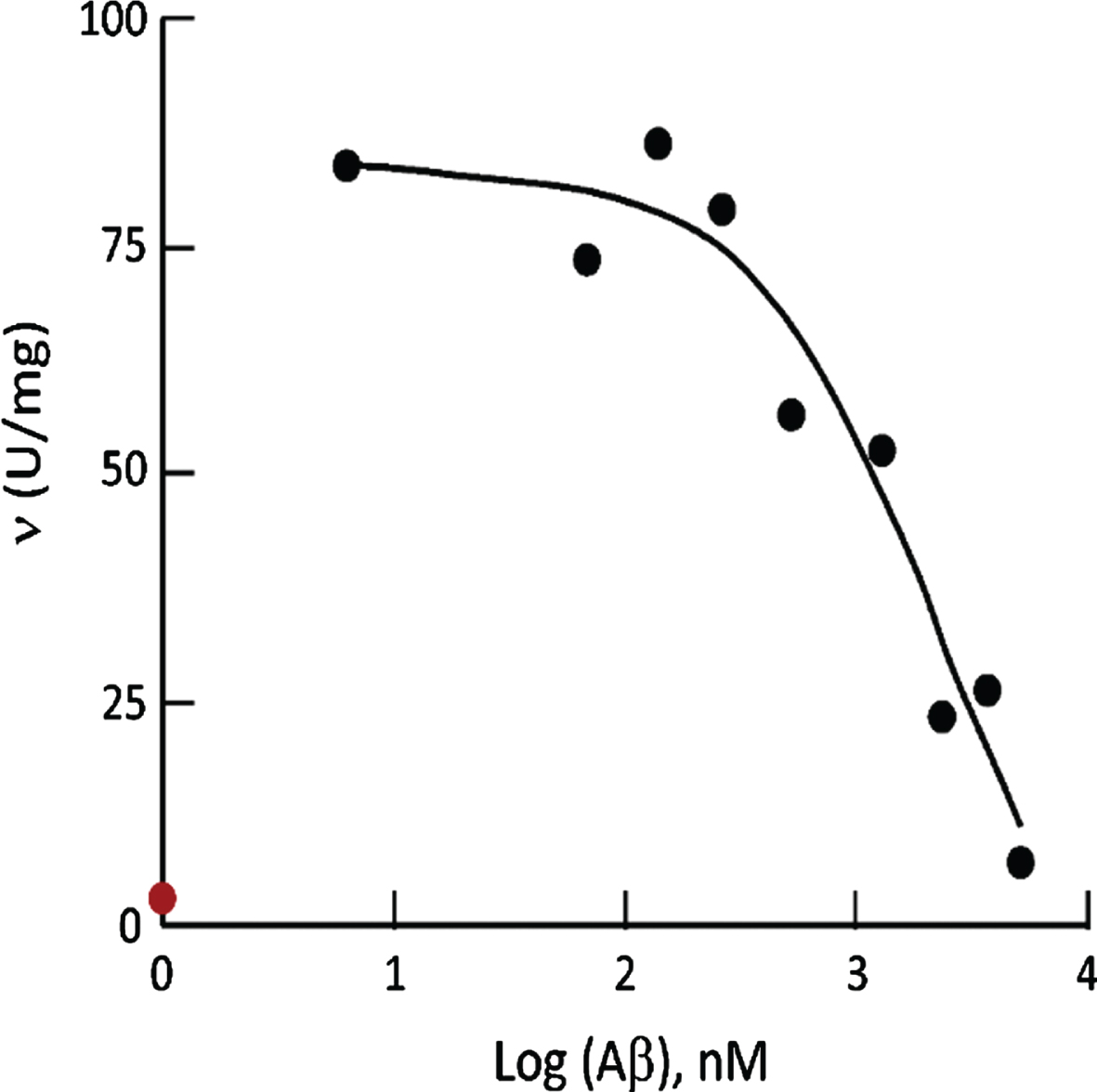
Comparison of the HAD activity of 6xHis-tagged human HSD10 in the absence of Aβ (red dot) to that of ABAD/ERAB under Aβ140 inhibition (black dots). The specific HAD activity of 0.67μg/ml 6xHis-tagged human HSD10 was measured using 0.18 mM acetoacetyl-CoA and 0.1 mM NADH as the substrate and coenzyme, respectively, according to experimental procedure
The reduction of S-acetoacetyl-CoA by NADH is an ordered Bi-Bi reaction [40]. The inhibition constant of Aβ140 should be determined according to [40], which had been cited as the kinetic reference book by [35, 36]. The Aβ140 inhibition data was reportedly fitted to a one-site model for competitive inhibition [35], but we were told by the author of that enzyme kinetic book [40] that there is no one-site model for competitive inhibition mentioned in his book and he does not know what the novel inhibition model means (Segel IH, personal communication) (see Supplementary Material 2).
More surprisingly, ABAD/ERAB was reported to exhibit not only L-3-HAD activity but also D-3-hydroxyacyl-CoA dehydrogenase activity to support the astonishing assertion that ABAD/ERAB plays a critical role in ketone body metabolism [35, 36]. It was also concluded [36] that this enzyme is essential for supplying energy to brain cells under nutritional stress. In contrast, it had already been reported [3] that this human brain dehydrogenase was found to metabolize only L-3-hydroxyacyl-CoA, but not the D-isomer. To settle this debate once and for all, it was demonstrated here that none of the dehydrogenase activities reported in [35, 36] are reliable, notwithstanding that it had been reported in a prestigious journal [36] that “we asked whether it might also catalyze the oxidation of the ketone body D-3-hydroxybutyrate. This was indeed the case, and oxidation proceeded with Km of ∼4.5 mM and Vmax of ∼4 mU/mg.”
Because most of the cell’s NAD+ is sequestered in mitochondria, it is difficult to understand how such an “ER-associated NAD+-dependent dehydrogenase” (namely, ABAD) (see Fig. 2C of [36]) could indeed play a physiological role in ketone body metabolism. Furthermore, most well-trained life scientists are aware that mitochondrial β-hydroxybutyrate dehydrogenase, β-HBD [48], rather than 17β-HSD10, is involved in ketone body metabolism (see Fig. 1). Although data about D-3-hydroxybutyrate supporting the growth of COS cells were reported previously (see Fig. 3 of [36]), this could not serve as support for the assertion that ABAD is involved in ketone body metabolism because β-HBD had not yet been knocked out in the COS cell line used by Yan et al. [36].
The localization of 17β-HSD10/SCHAD in mitochondria was first demonstrated by Fig. 1 of [2] in 1999, and then was further proven by identifying its mitochondrial targeting signal [5, 49] and endorsed by Prof. C. de Duve (see Supplementary Material 1 of [17]). Unfortunately, even this did not lead to a corrigendum of endoplasmic-reticulum associated ABAD/ERAB reports [21, 36]. Instead, a mitochondrial ABAD article appeared in the journal Science in 2004 [22] without citing or addressing any mitochondrial SCHAD/17β-HSD10 studies [2–5, 49].
The catalytic capabilities reported for ABAD/ERAB [35, 36] were incredibly higher than those of 17β-HSD10 or 6xHis-tagged HSD10, especially for the oxidation of various alcohols by NAD+ (see Table 3). But the truth is that it is impossible to accurately measure such an alcohol dehydrogenase assay as previously reported [35] (see Fig. 5). Furthermore, the K m values of (±)-2-octanol and (+)-2-octanol were reported to be as incredibly high as 85μM and 84μM, respectively [35]. As is well known, (±)-2-octanol is a racemic mixture that consists of 50% of (–)-2-octanol and 50% of (+)-2-octanol. Because the oxidation of (–)-2-octanol and (+)-2-octanol by NAD+ under the catalysis of an NAD+-dependent dehydrogenase generated the same product, a serious scientist should be aware of that it is impossible for the K m value of (±)-2-octanol to be greater than that of (+)-2-octanol because the K m value for (–)-2-octanol shown in the same report was just 43μM (see Table 1 of [35]).
Although a more sophisticated HAD assay had already been available [38, 45], it is not known why Yan et al. created a rather peculiar method and then published their method in a high-impact journal [35, 36]. Erroneous data revealed here cannot simply be attributed to the incredible methodology employed as described in the published work (Fig. 2). A tremendous amount of incredible data has also been found in those ABAD/ERAB reports (see Figs. 4–6). If ABAD/ERAB are accepted as alternative names for 17β-HSD10 (OMIM300256), the unsolved reproducibility issues in the published ABAD/ERAB studies cannot be overlooked any longer (see Supplementary Material 2).
A series of ABAD/ERAB reports [11, 36] not only conflict with 17β-HSD10 research [2–5, 49–51] but also encourage and support an erroneous trend to designate the HSD17B10/HADH2 gene [8] with various confusing names allegedly related to erroneously reported function(s) of this multifunctional protein. For example, fifteen alternative names have been listed in the Gene ID: 3028 including ABAD; CAMR; ERAB; etc. This confusion originated from the ABAD/ERAB publications [21, 36], and persist to the present [11, 52]. This uncorrected phenomenon resulted in wasted research resources and directly interfered with studies on 17β-HSD10 and HSD17B10 (HADH2) gene-related disorders such as Alzheimer’s disease [17] and developmental disabilities, e.g., infantile neurodegeneration [20]. Because 17β-HSD10 is involved in the etiology of human diseases including prostate cancer [10–34, 49–54], this interference is not a trivial problem (see Supplementary Material 2) and deserves a public and official correction of ABAD/ERAB reports in the scientific record [55]. Erroneous data supporting ABAD/ERAB should be exposed and corrected so that the data, and hypotheses, theories, and conclusions reliant thereon no longer confuse the scientific community (see Supplementary Material 2) and adversely affect patients and their treating physicians.
Conclusion
This report not only provides new information for the mitochondrial multifunctional protein 17β-HSD10 important in the pathogenesis of neurodegenerative disorders, including Alzheimer’s disease and developmental disabilities such as infantile neurodegeneration, but also clarifies confusion concerning its role in human health and disease.
Footnotes
ACKNOWLEDGMENTS
ABBREVIATIONS
acetoacetylsuccinyl-CoA transferase; branched-chain amino acid transaminase; branched-chain amino acid transaminase pyridoxal 5'-phosphate; branched-chain α-keto acid dehydrogenase complex; methylmalonyl-CoA mutase; methylmalonyl-CoA racemase; propionyl-CoA carboxylase; pyruvate dehydrogenase; tricarboxylic acid cycle.