
Abstract
Oxidative stress is associated with the progression of Alzheimer’s disease (AD). Reactive oxygen species can modify lipids, DNA, RNA, and proteins in the brain. The products of their peroxidation and oxidation are readily detectable at incipient stages of disease. Based on these oxidation products, various biomarker-based strategies have been developed to identify oxidative stress levels in AD. Known oxidative stress-related biomarkers include lipid peroxidation products F2-isoprostanes, as well as malondialdehyde and 4-hydroxynonenal which both conjugate to specific amino acids to modify proteins, and DNA or RNA oxidation products 8-hydroxy-2’-deoxyguanosine (8-OHdG) and 8-hydroxyguanosine (8-OHG), respectively. The inducible enzyme heme oxygenase type 1 (HO-1) is found to be upregulated in response to oxidative stress-related events in the AD brain. While these global biomarkers for oxidative stress are associated with early-stage AD, they generally poorly differentiate from other neurodegenerative disorders that also coincide with oxidative stress. Redox proteomics approaches provided specificity of oxidative stress-associated biomarkers to AD pathology by the identification of oxidatively damaged pathology-specific proteins. In this review, we discuss the potential combined diagnostic value of these reported biomarkers in the context of AD and discuss eight oxidative stress-related mRNA biomarkers in AD that we newly identified using a transcriptomics approach. We review these genes in the context of their reported involvement in oxidative stress regulation and specificity for AD. Further research is warranted to establish the protein levels and their functionalities as well as the molecular mechanisms by which these potential biomarkers are involved in regulation of oxidative stress levels and their potential for determination of oxidative stress and disease status of AD patients.
INTRODUCTION
Alzheimer’s disease (AD) patients show hallmarks of oxidative stress early in the progress of disease. The excessive accumulation of reactive oxygen species (ROS) associated with oxidative stress leads to membrane damage, cytoskeletal alterations, and cell death [1, 2]. This review covers recent research on biomarkers that signify early-stage oxidative stress in AD. We aim to critically appraise the differentiation potential of these early-stage oxidative stress biomarkers for AD compared to other neurodegenerative disorders. We extend on these known biomarkers by employing transcriptomics-based analysis on an AD patient population to identify potential novel mRNA biomarkers that associate with oxidative stress and discuss their potential application in the field of neurodegeneration.
PRODUCTS OF OXIDATIVE STRESS AS BIOMARKER
A series of biomarkers has been clinically validated to allow for differential diagnosis of AD. Examples of those currently used in clinical applications are cerebrospinal fluid (CSF) levels of total tau, phosphorylated tau, and amyloid-β (Aβ) 1–42 [3]. Oxidative stress is an established early-stage phenomenon in AD pathology. However, none of the currently known AD-related biomarkers target conditions of oxidative stress. At the same time, two reviews have suggested the relevance of oxidative stress related biomarkers in AD [4, 5]. Peroxidation and oxidation of biomolecules, including lipids, proteins, DNA, and RNA, caused by oxidative stress generate products that can be detected in plasma, serum, or CSF. These products may act as biomarkers although their specificity and differentiation potential toward AD compared to other oxidative stress-related disorders may be questioned as oxidative stress is generally observed in many pathologies. Several biomarkers have been validated for use in clinical diagnostics and are commonly used as output measure for oxidative stress in experimental research and clinical trials. This section will discuss distinct molecular features of oxidative stress that show significant correlation with AD and their potential for use in diagnosis of AD. Table 1 provides an overview of previously identified biomarkers obtained from AD patient studies.
Summary of previously identified oxidative stress-related biomarkers in AD
F2-isoprostanes
Increased levels of lipid peroxidation are observed in postmortem AD brains and regional variation of lipid peroxidation closely corresponds to the regional distribution of AD pathology in the AD brain. Generation of isoprostane, a prostaglandin isomer, is mediated via a multi-step process involving various unstable intermediates such as bicycloendoperoxides by non-enzymatic peroxidation of polyunsaturated fatty acids (PUFAs), particularly arachidonic acid, which are a major component of cellular membrane phospholipids [6, 7] (Fig. 1). Measurement of oxidative stress by probing in vivo lipid peroxidation is an attractive approach as these biomarkers are an excreted metabolically stable end product [8]. Protocols for establishing cellular levels of F2-isoprostanes, a class of isoprostanes, have been published and are based on ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS), or gas chromatography-mass spectrometry (GC-MS) [8 –11] using an analytical C18 column [8] or a fused silica capillary column [9]. Using GC-MS, brain specimens of 19 AD patients were subjected to quantification of F2-isoprostane regioisomers iPF2α-III and iPF2α-VI. AD patient CSF, temporal and frontal cortical regions, but not the cerebellar cortex, were found to contain higher levels of iPF2α-III and iPF2α-VI [6], as opposed to Parkinson’s disease patients and patients suffering from schizophrenia [6]. In another study, lateral ventricular CSF levels of F2-isoprostanes were found to be significantly elevated in 23 AD patients compared to age-matched controls [12]. In this study, F2-isoprostane levels in the ventricular fluid showed a significant negative correlation with brain weight and positively correlated with degree of atrophy in the cerebral cortex, and Braak staging which is a method to classify the degree of AD pathology based on brain regional distribution of neurofibrillary tangles. While no significant correlation was found between F2-isoprostane levels and APOE genotype, another study showed that levels of 8,12,-iso-iPF2α-VI, a member of prostaglandin F2α isomers, correlated with the number of APOE ɛ4 alleles [13]. APOE codes for the protein apolipoprotein E (apoE) which is a cholesterol carrier but this protein was also demonstrated to display antioxidant activity [14], both in vivo and in vitro [16, 17]. Individuals that carry APOE polymorphic allele ɛ4 were shown to be at increased risk of developing AD compared to those individuals that carry alleles ɛ2 and ɛ3 [18]. Damage in the brain mediated by oxidative stress was found to correlate with the number of cysteine residues in the different apoE isoforms [19]: ApoE ɛ2 (2 cysteines) < ApoE ɛ3 (1 cysteine) < ApoE ɛ4 (no cysteines) [20, 21]. Other apoE isoform-dependent contributing factors to the extent of oxidative stress measured in AD patients include the role of apoE in low density lipoprotein receptor-related protein (LRP)-1 [22] and LRP-4-mediated [23] Aβ uptake [24], and apoE ɛ4-associated downregulation of thioredoxin-1 as have been extensively reviewed before [25]. This study reported that levels of 8,12,-iso-iPF2α-VI were elevated in CSF of AD but also mild cognitive impairment (MCI) patients suggesting that oxidation of PUFAs occurs early in AD pathogenesis [13]. CSF isoprostane levels were demonstrated in some studies to predict progress of MCI into AD [13] although reports are contradicting. F2-isoprostane levels in AD and MCI patient-derived urine were also increased compared to healthy controls [13], although low numbers of subjects were included in this study [13], and various other reports were unable to reproduce this observation [26]. Further, a correlation between plasma or urine F2-isoprostane levels with brain oxidative status has not been observed [26]. This lack of correlation limits application of F2-isoprostane as a biomarker to access oxidative stress status in patients in a little or non-invasive manner.
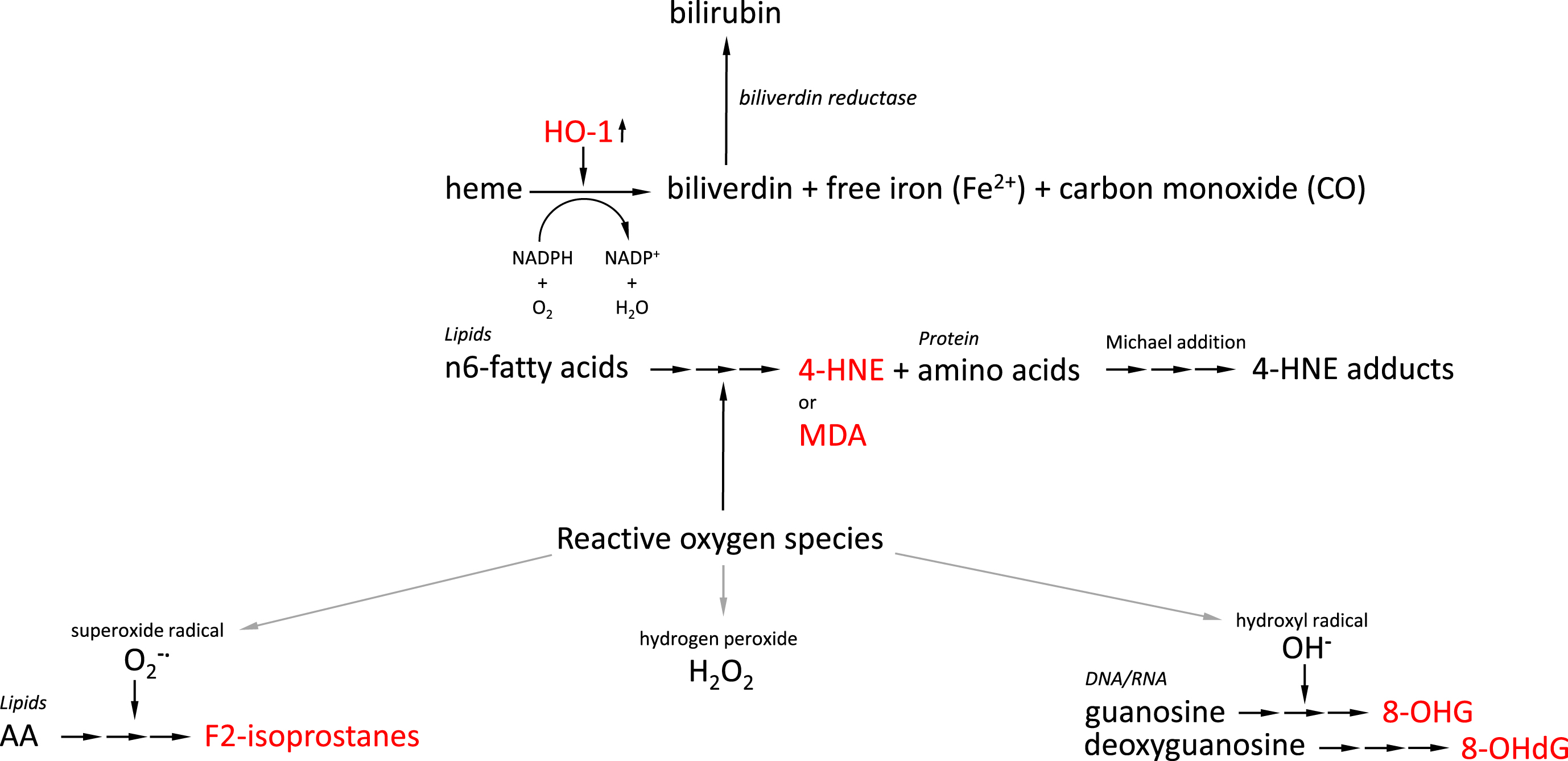
Overview of generation of AD-related oxidative stress-induced biomarkers. Interrupted arrows indicate multi-step conversion processes. AD-related oxidative stress-induced biomarkers are indicated in red. AA, arachidonic acid; HO-1, heme oxygenase 1; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; 8-OHG, 8-hydroxyguanosine; 8-OHdG, 8-hydroxy-2’-deoxygyguanosine.
3-nitrotyrosine (3-NT)
Tyrosine is an amino acid that may be subject to nitration by conjugation with a nitrite-NO2 group [27]. This chemical reaction can alter protein conformation and function [28]. Agents known to induce nitration of tyrosine residues include peroxynitrite (ONOO-), nitrogen dioxide, and peroxidases, each associated with oxidative stress, to produce 3-nitrotyrosine (3-NT). While levels of free 3-NT are elevated in CSF [29, 30], and hippocampus [31] of AD patients [32], this compound has been suggested a potential biomarker for neurodegeneration [33]. The stability of 3-NT lends itself very well for measurement of 3-NT levels in biological samples and the use of 3-NT as a biomarker of oxidative stress in neurodegeneration [34] as well as different methods for its detection [35] have been reviewed. Apart from the detection of increased levels of tyrosine nitration in the hippocampus [36] and inferior parietal lobe [37] of AD patients, also MCI patients [38] display elevated levels of protein nitration in the hippocampus and inferior parietal lobe indicating that nitrosative stress occurs early in AD pathogenesis. Also in AD model systems, like rat hippocampal neurons treated with Aβ1–42, in combination with glucose to mimic hyperglycemic conditions of diabetes, a significant induction of 3-NT formation and toxic response were observed [39]. Apart from in neurodegenerative processes, also in other pathologies co-occurring with oxidative stress, such as cancer and diabetes, increased levels of 3-NT have been reported questioning the specificity of 3-NT as biomarker for AD. However, proteins targeted by nitration seem to vary between the different neurodegenerative pathologies. For example, two AD-related proteins found to be targeted by nitration are tau and Aβ [40, 41]. Specifically the N-terminal tyrosine regions of tau were reported to be subject to nitration inhibiting tau polymerization [41] while nitration of Aβ tyrosines was observed to induce Aβ aggregation [40]. Ready formed tau-derived neurofibrillary tangles significantly colocalize with antibodies recognizing 3-NT-modified proteins in the postmortem AD brain [42, 43]. Apart from modulating tau aggregation, tyrosine modification also ameliorates the normal phosphorylation of tau that critically regulates association of tau with the microtubular network, affecting biological functionality of this protein. Thus, even though 3-NT levels may be generally increased in neurodegenerative diseases, targeted proteins seem to be more defined as a function of a specific pathology. For example, while in AD tau and Aβ are nitrated at higher levels, in Parkinson’s disease nitration of α-synuclein is observed [44] and in amyotrophic lateral sclerosis (ALS) the enzyme superoxide dismutase (SOD) is targeted [45], while in prion disease primarily prion protein is affected [46].
8-hydroxy-2’-deoxyguanosine (8-OHdG) and 8-hydroxyguanosine (8-OHG)
Widely used biomarkers to assess levels of oxidative stress are 8-OHdG and 8-OHG. These are ROS-induced oxidation products of DNA and RNA bases, respectively, formed by hydroxyl radical (•OH)-mediated damage to deoxyguanosine and guanosine [47, 48]. Deoxyguanosine and guanosine are both subject to high mutagenic potential [49] (Fig. 1). While an age-related increase in oxidatively damaged DNA can be observed [50], increased levels of 8-OHdG have been detected in AD postmortem brain tissue including cerebral cortex and cerebellum, as well peripheral cells compared to healthy age-related individuals [51]. The accumulation of Aβ1–42 peptide, one of the key hallmarks of AD pathology, but not Aβ1–40, was observed to be inversely related with the concentration of intraneuronal 8-OHG [52], although regional significance of these observations does not entirely overlap. While this observation argues for an antioxidant effect of Aβ, Aβ has also been observed to induce oxidative stress in other conditions [53]. CSF levels of 8-OHdG were found to be increased up to five times in CSF of AD patients compared to healthy age-matched controls [54 –56] combined with a deficient DNA repair mechanism [55]. Interestingly, while mitochondrial DNA in AD patients in the parietal cortex, which also shows an AD-related reduction in glucose metabolism [57], showed significant oxidative damage compared to healthy age-matched controls, nuclear levels of oxidized DNA were not affected [58]. Mitochondrial localization of oxidized RNA, particularly in neurons, was also reported in a study that used antibody-based immunoreactivity in combination with RNase and DNase treatment to show that cytoplasmic immunoreactivity to oxidized products was significantly reduced in the presence of RNase but not DNase [59]. The high reactivity of •OH in combination with its slow diffusion rate allows it to react more extensively with the unprotected single stranded RNA in its vicinity. A striking number of mRNA species that were identified to be oxidized in AD brains were found to be transcripts related to previously identified AD genes such as mitogen-activated protein kinase 1 (MAPKI), dismutases, and enolase [60] and their oxidation hampers their translation into functional proteins. Also, using immunoprecipitation, significant levels of mRNA oxidation were observed in the frontal cortex of AD patients compared to age-matched groups [61]. While nuclear DNA is protected against oxidative damage by histones and other extensive repair mechanisms, this is not the case for mitochondrial DNA rendering this DNA more susceptible for the consequences of oxidative stress. Moreover, the mitochondrion houses the machinery to generate adenosine triphosphate (ATP) from oxidative phosphorylation which is a source of free radical formation. Mitochondrial abnormalities in terms of morphology and dysfunction are frequently observed in AD patients [62 –65]. Also MCI patients transitioning to prodromal stages of AD are generally characterized by mitochondrial dysfunction, which was found to correlate well with neuronal RNA damage determined by 8-OHG-immunoreactivity assays [58, 66]. Lymphocyte levels of 8-OHdG were significantly higher in AD patient samples compared to a control group and these higher levels were shown to correlate well with low plasma antioxidants, including vitamin E, vitamin A, and carotenoids (lycopene, lutein, α-carotene, and β-carotene) [49]. Apart from AD, also other neurodegenerative disorders, including Parkinson’s disease [67], ALS [68], and dementia with Lewy bodies [69] are characterized by oxidation of neuronal RNA. Thus, while blood-derived 8-OHdG and 8-OHG levels may provide information on oxidative stress levels in AD [49], this biomarker should be used in combination with AD-specific biomarkers to establish progression of the disease.
Carbonylated proteins
The highly reactive electrophile 4-hydroxynonenal (4-HNE) is formed by peroxidation of n-6 fatty acids under conditions of oxidative stress which can then target the amino acids lysine, arginine, proline, threonine, cysteine, or histidine to generate protein carbonyl derivatives including aldehydes and ketones [70] (Fig. 1). Formed 4-HNE is normally degraded by a set of enzymes that effectively eliminate HNE, including glutathione S-transferases, aldo-keto reductases, and aldehyde dehydrogenases [71], as well as proteasomal degradation [72] or metabolism of HNE [71]. Aging, and neurodegeneration in a more pronounced manner, results in less efficient HNE elimination leading to accumulation of HNE, subsequently leading to HNE-mediated modification of cellular components [73]. A number of proteins were found to be readily carbonylated, in response to oxidative stress conditions, in the hippocampus of MCI subjects [2]. In the AD brain it was shown that glutamine synthase is extensively carbonylated compared to healthy brain [74]. Carbonylation of glutamine synthase lead to partial inactivation of the enzyme which was used as read-out to demonstrate that activity of this enzyme was selective for the frontal pole compared to the occipital pole [75]. A decrease in activity of glutamine synthase, and also of creatine kinase, was further observed in a different study but activities of both enzymes were found to be globally decreased throughout the entire brain without any brain-region specific effects [76]. However, brain regional differences in protein carbonyl levels were reported in a different study by comparing hippocampus, inferior parietal lobule and cerebellum, where hippocampal carbonyl content was observed to be significantly higher in AD patients compared to healthy age-matched controls [76]. Another protein that is found to be modified in an Aβ42-mediated manner by 4-HNE is glial glutamate transporter GLT1, also known as Excitatory Amino Acid Transporter-2, or EAAT2, leading to loss of transporter activity in rat cortical synaptosomes [77]. This transporter importantly regulates the concentration of synaptic glutamate which critically avoids excitotoxic events [78]. These findings translate well into the observations in AD patient brains showing brain region-specific differential EAAT2 expression and activity [79], and dysregulation of glutamate homeostasis in transgenic mouse models [80, 81]. Other carbonylated proteins that were identified using redox proteomic analysis in the inferior parietal lobule of six MCI and four early-stage AD subjects were MAPKI, heat shock protein 70 (Hsp70), carbonic anhydrase II, and syntaxin binding protein I [82]. In addition to this set of proteins, early-stage AD carbonylated proteins in this brain region include fructose bisphosphate aldolase C, glial fibrillary protein, and phosphoglycerate mutase 1. Other targets for carbonylation by HNE include the cysteine residues of apoE ɛ2 and ɛ3. Interestingly, while apoE ɛ4 is considered a major genetic risk factor for developing AD, this apoE isoform differs from apoE ɛ2 and ɛ3 by the lack of cysteine residues. It has been suggested before that this difference between the isoforms may contribute to the increased risk of oxidative stress in AD observed in APOE4 bearing individuals [21 , 83]. Carbonylation of these proteins coincided with a loss of enzymatic activity of carbonic anhydrase II [82]. Specific targeted carbonylation was observed in a 12-month old Senescence Accelerated Mouse-prone 8 model, SAMP8, of α-enolase, collapsing response mediator protein-2, creatine kinase, lactate dehydrogenase, and α-spectrin [84]. Levels of carbonylated proteins were observed to correlate with the degradation of L-tryptophan by the kynurenine pathway [85] which is implicated in systemic immune activation. This observation is of interest as inflammatory response and oxidative stress are both commonly observed in AD patient brains but their interaction at a molecular mechanistic level is little understood. An enzyme-linked immunosorbent assay (ELISA)-based study on clearance of oxidative stress markers showed that, in patients undergoing coronary heart surgery, protein carbonyl levels were cleared from the serum at a slower rate than lipid peroxidation products suggesting that the detection of protein carbonyls provides a relatively stable read-out for oxidative stress status [86]. As many other diseases have demonstrated increased levels of carbonylated proteins, the differential power of this oxidative stress-related marker to AD may be limited. As carbonyl levels were found to correlate well with the detection of neurofibrillary tau tangles [87] and severity of histopathological alterations [76] the reported brain-region specific differences may be used to establish the extent of oxidative stress as factor to map progress of AD.
Heme oxygenase type 1 (HO-1)
HO-1, also known as heat shock protein-32 (HSP-32), is an oxidative stress-inducible enzyme that acts as cellular adaptation mechanism to stress and catalyzes the oxidation of heme into biliverdin, free iron and carbon monoxide. Biliverdin is then further converted into bilirubin, a structure reported for its hydroxyl radical, superoxide anion, and singlet oxygen scavenging activity, catalyzed by the enzyme biliverdin reductase-A [88] (Fig. 1). Modification of hippocampal biliverdin reductase-A by 3-NT and 4-HNE, as observed in MCI and AD, leads to loss of function of this enzyme [89], which, in turn, is associated with impairment of autophagy [90] and increased insulin resistance in the brain [91]. Low levels of constitutive expression of HO-1 in the healthy brain selectively by neuroglia and specific types of neurons are regulated by a set of different transcription factors, including nuclear factor erythroid 2–related factor 2 (Nrf2), as well as stimuli including ROS [92, 93]. Considering these observations, HO-1 upregulation has been regarded as a cellular defense mechanism against oxidative stress. In line with this, neuronal and neuroglial expression of HO-1 in the hippocampus and cerebral cortex is increased with age in a Nrf2-dependent manner [94]. For example, in a study of the parieto-occipital lobe and the hippocampus derived from 31 individuals that died from mechanical asphyxia, the number of HO-1 positive cells were found to be positively correlated with aging [94]. Specifically postmortem brain cortical samples derived from the middle frontal gyrus showed upregulation of mitochondrial, but not astrocytic, HO-1 in AD patients compared to non-demented age-matched control individuals [95]. At the same time, two other studies, using Drosophila melanogaster [96] and rats [97] as aging models, showed that activation of Nrf2 in response to exposure to acute stress becomes inhibited with age. Some other studies have shown that with aging, HO-1 levels increased. Observations from AD and MCI patient brains have been inconsistent. For example, one study shows that already in the MCI stage, glial expression levels of HO-1 in temporal cortex and hippocampus significantly exceed those of non-demented patients [98] suggesting that HO-1 expression is an early-stage event in AD pathology. Amyloid precursor protein (APP) transgenic mice, a model for AD, similarly show increased HO-1 expression levels [99, 100]. Intriguingly, while HO-1 protein levels are generally observed to be upregulated in AD patient brains, mRNA levels of HO-1 were found to be reduced in probable AD patients, compared with healthy elderly controls. This was found to coincide with decreased plasma HO-1 protein, lymphocyte mRNA levels and CSF protein levels [98]. Suppressed mRNA levels of HO-1, detected by quantitative reverse transcription PCR (RT-qPCR), in lymphocytes was confirmed in a small-scale study involving twelve AD patients [101]. Further, HO-1 was found to colocalize with astrocytes, neurofibrillary tangles, and senile plaques in the human postmortem AD brain [102]. Colocalization of HO-1 with deposits of Aβ and tau proteins in AD has been reported on a few occasions [103 –105]. For example, in a five familial AD mutations (5xFAD) mouse model HO-1 overexpressing microglia in the hippocampus and cortex were found to be localized in close proximity to deposits of Aβ [95]. These observations raised the question whether HO-1 levels can be used as a biomarker to assess AD-related oxidative stress levels which has been reviewed before [106]. However, while HO-1 protein levels are upregulated in AD, it was observed that APP, which is processed into Aβ peptide, can interact with HO to reduce its activity in cerebral cortical cultures from APP mutant AD model mice rendering cells more vulnerable to hemin-induced toxicity [107]. Apart from AD, also other pathologies have been related to HO-1 upregulation, including Parkinson’s disease showing high HO-1 expression levels in dopaminergic neurons and nigral astroglia [108]. Apart from affecting HO-1 expression levels, HO-1 is also found to be subject to ROS-mediated modifications. For example, hippocampal HO-1 in AD patients shows significant conjugation with HNE and carbonyls compared to healthy age-matched individuals. On the other hand, in MCI patients, only HNE-bound HO-1 was found to be significantly increased in the hippocampus [109].
Malondialdehyde-lysine epitopes
Malondialdehyde (MDA) is an aldehyde generated upon peroxidation of lipids. It is produced when the double bonds of the PUFAs that are constituents of the cellular membrane are attacked by hydroxyl free radicals [110, 111] (Fig. 1). MDA promotes cross-linking of proteins and DNA causing structural modifications and mutations. In aging and under oxidative stress MDA accumulates and reacts with basic amino acid residues such as lysine, histidine, and arginine. These reactions lead to the generation of aldehyde adducts also known as advanced lipid peroxidation end products (ALEs) [112]. One prominent target of MDA to generate ALEs are the lysine residues of apoE [113], a lipid carrier that has been associated with risk of late onset and sporadic AD in a genotype-dependent manner [114, 115]. ApoE and its cargo are recognized by apoE receptors that are expressed in most cells. Neuronal apoE and its receptors critically regulate synaptic functioning by affecting dendritic spine density [116], and the function of the glutamate receptor [117], and apoE plays an essential role in clearance of Aβ [118]. Collectively, these observations suggest that in the AD brain synaptic signaling will be prone to modifications by MDA in an oxidative stress environment [119, 120]. MDA was also observed to modify proteins by reacting with lysine residues forming Nɛ-(malondialdehyde)-lysine [121]. This product is found at significantly higher levels in cortical neurons from brain tissue samples from AD patients in comparison to control samples when measured by GC-MS. In vitro treatment of primary cortical rat neurons with MDA resulted in protein cross-linking, observed by gel electrophoresis, that coincided with activation of apoptotic signaling pathways [122]. While brain levels of MDA were clearly increased according to previous studies, in a case-control study, plasma levels of total MDA in AD patients were also observed to be significantly increased, and inversely correlated with levels of retinol and α-tocopherol, compared to healthy controls [123]. In this 5-year interval study of AD patients, the MDA levels of blood samples in severe cognitive disorders measured with a Mini-Mental State Examination (MMSE), were observed to be significantly increased, coinciding with a decrease in the levels SOD and glutathione peroxidase (GSH-Px) in erythrocytes [123]. MDA is a recognized biomarker of oxidative stress in many diseases other than AD such as cardiovascular disorders, cancer, chronic inflammatory diseases, and neurodegeneration in general [124]. Free MDA and MDA bound to thiobarbituric acid can be measured in blood and various tissues [112]. Plasma MDA levels were found to be significantly increased in diabetes patients that undergo hemodialysis, particularly those patients suffering from cardiovascular complications [124] showing that raised MDA levels are non-specific to the AD brain. Even though MDA levels are often employed to study levels of oxidative stress in various types of systems, including clinical assays, and cell biological applications, the use of MDA as a clinical biomarker is further limited by virtue of its short half-life as it is rapidly metabolized and interacts with proteins [111] to form adducts [125]. MDA levels can also be measured in urine samples through aldehydic products, which are lysine-adducts, but this source is generally considered of low clinical significance [112] as nutritional lipid intake and exercise are known to affect levels of such products [126].
Oxidative stress as AD specific biomarker
The occurrence of oxidative stress at prodromal and early stages of AD [1 , 83] raised the possibility that the oxidized biomolecules that are generated in response to oxidative stress may act as biomarker indicative of early disease onset. A model to characterize specific biomarkers related to AD has been developed [127, 128], which includes increased levels of Aβ1–42 and tau in CSF, along with their structural imaging by magnetic resonance imaging (MRI), amyloid positron emission tomography (PET) and its hypo-metabolism in fluorodeoxyglucose (FDG)-PET [128], but excludes the use of oxidative stress biomarkers. The model has demonstrated its value by indicating the dynamics of the development of the various markers in relation to progress of AD. Even though oxidative stress levels can be measured in AD patients by means of the measurement of oxidized molecules or remaining antioxidant capacity, inclusion of a generic oxidative stress biomarker, including MDA, 4-HNE, indices of nucleotide oxidation, and enzymatic and nonenzymatic antioxidant capacity, may not enhance the objective measure of the pathogenic process of AD by the existing model. Published data, as discussed in the paragraphs in this section, demonstrate that, even though oxidative stress is a highly relevant pathogenic parameter in AD and somewhat correlates with disease progress and can also be determined at early stages of disease, such global indices do not seem to provide sufficient distinguishing power from other aging related diseases that are also often accompanied by increased levels of oxidative stress. However, viewing AD as multifactorial complex disease involving neurodegenerative processes, deposition of proteins Aβ and tau, inflammation, and failing mitochondrial and autophagosomal functioning, it is plausible to assume that inclusion of oxidative stress as a biomarker in early AD may result in more accurate definition of an individual’s needs in terms of therapeutic opportunities.
Recent advances in the field of redox proteomics do provide clear disease-specific oxidative stress indices that may allow for differential diagnosis. Redox proteomics allows for detection of individual oxidatively modified proteins based on mobility and isoelectric point [129, 130]. A review on the use of redox proteomics in the field of neurodegeneration has been published before [131]. Using a redox proteomics approach various proteins have been identified to be differentially and specifically damaged by oxidative modification in AD. For example, inferior parietal lobule samples from AD patients showed markedly increased levels of carbonylated creatine kinase BB, glutamine synthase and ubiquitin carboxy-terminal hydrolyse L-1, dihydropyrimidinase related protein-2, α-enolase, and heat shock cognate-71 when subjected to 2D oxyblot followed by mass spectrometric analysis [74, 132]. Other reviews on the application of redox proteomics to detect oxidatively damaged proteins specific for AD in various in vitro and in vivo models have been published before [133 –136], so we would like to refer to those for more details. Inclusion of such AD-specific oxidized protein indices in a biomarker panel may hold promise for accurate differential diagnosis and staging of AD.
Postmortem interval affects detection of oxidative damage
Postmortem interval (PMI) can be defined as the length of time that lapsed between an individual’s death and analysis of tissue. Postmortem degradation of proteins occurs and is an actual parameter used to determine time of death [137, 138]. Such degradation critically affects observations made concerning (oxidized) protein levels using postmortem patient material. While proteins are thus known to be degraded, also mRNA integrity and thus detection can be substantially reduced, up to 50%48 hours postmortem, as a function of PMI. Vulnerability to mRNA degradation over time was reported to depend on nucleotide sequence motifs [139]. Therefore, shorter PMIs are generally considered to provide more reliable information on oxidative stress-related proteins and mRNA levels in vivo in AD.
TRANSCRIPTOMIC ANALYSIS TO IDENTIFY NEW BIOMARKERS
To identify potential new biomarkers that can report on early-stage AD, we performed transcriptomics analysis on a publicly available dataset to identify previously unreported but oxidative stress related genes in AD patient brains. mRNA expression levels were assessed in the publicly available transcriptome datasets of brain samples from patients with various stages of AD (GSE1297; reference PMID: 14769913) as obtained from the National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo). Patients were categorized into four groups: healthy individuals (n = 9), incipient AD patient (n = 7), moderate AD patients (n = 8), and severe AD patients (n = 7), and data was analyzed using the online database GEO2R.
The heatmap provided in Fig. 2 shows that expression levels of some genes become differentially regulated only at later, or severe stages of disease. For example, DRD4 is a dopamine D4 receptor that has been reported to induce apomorphine-mediated protection against oxidative-stress-induced cell death [140]. In the light of their potential ability to collect information on an individual’s status of disease progress upon diagnosis, in this paragraph we will address only those markers that show up- or downregulation in incipient AD. One important remark to make in our approach is that mRNA values do not necessarily reflect protein levels. It is known that mRNA and protein expression levels as well as protein functionality correlate well for some proteins while for other proteins mRNA levels provide limited to no predicting power [141, 142]. Direct mRNA translation to proteins is regulated at its various stages, initiation, elongation, and termination by intricate mechanisms which have been elaborately reviewed before [143, 144]. While mRNA levels may serve as biomarkers, insight into protein levels and their functionalities reflect biological action and provide insight into oxidative stress pathways in neurodegeneration and AD in specific. Levels and activities of the proteins identified using our transcriptomic approach should therefore be further evaluated.
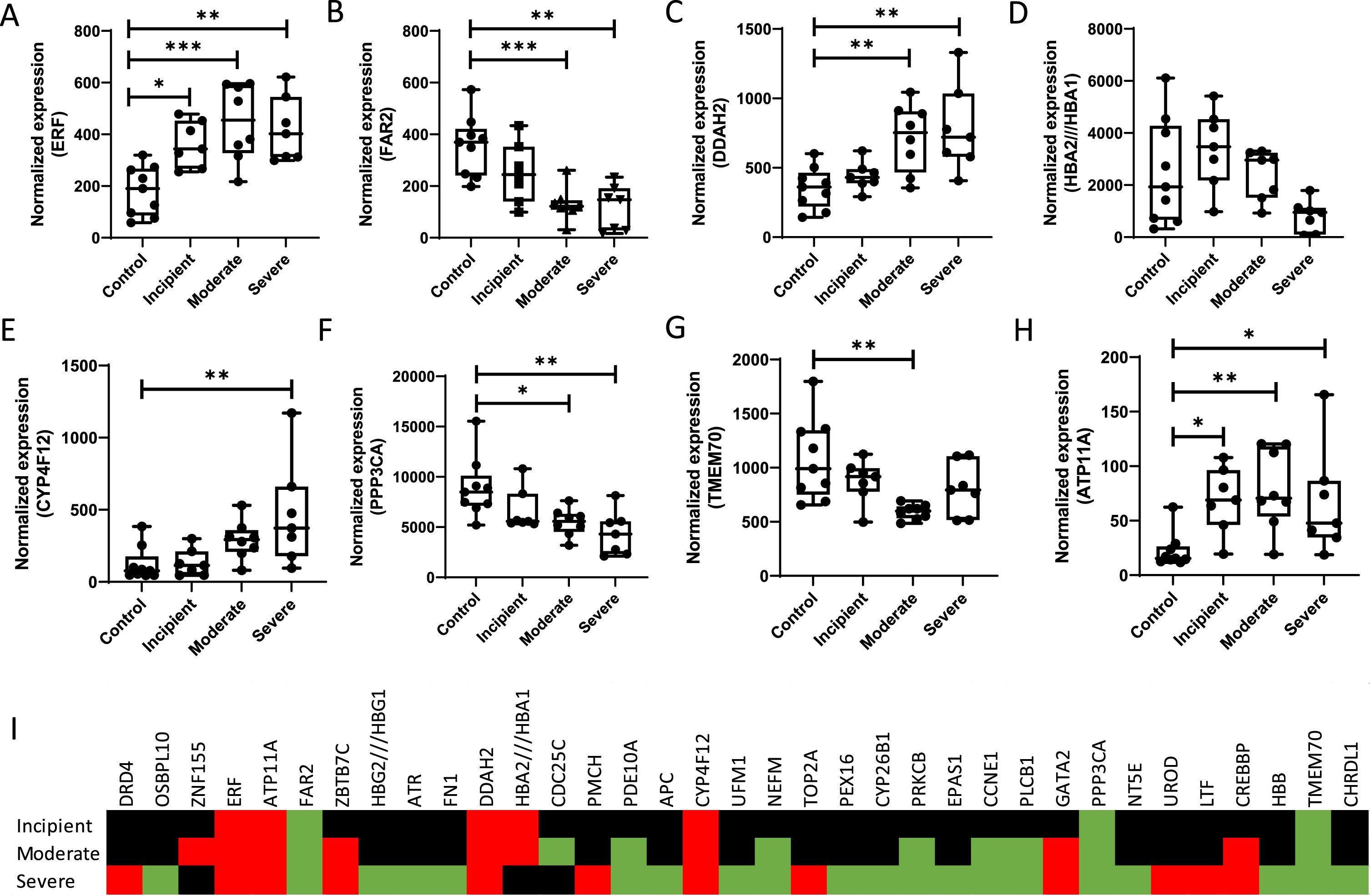
mRNA expression levels of some genes are affected as a function of AD progress. (A) ERF, (B) FAR2, (C) DDAH2, (D) HBA2/HBA1, (E) CYP4F12, (F) PPP3CA, (G) TMEM70, (H) ATP11A, (I) Heatmap of significant genes being up (red), down (green), or unaltered (black) regulated in various stages of AD. mRNA expression levels were assessed in the publicly available transcriptome datasets of brain from patients with various stages of AD (GSE1297) as obtained from the National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo). Patients were categorized into four groups: healthy individuals (n = 9), incipient AD patient (n = 7), moderate AD patients (n = 8) and severe AD patients (n = 7), and data was analyzed using online database GEO2R.
ERF and target genes
ERF is significantly upregulated at early-stage AD and levels peak at moderate AD compared to healthy control subjects. ERF encodes for the Ets-2 repressor factor protein that is ubiquitously expressed in various organs and tissues. Ets2 is a member of the Ets family of transcription factors that is characterized by an evolutionary conserved characteristic 85-amino acid Ets domain that aids the binding to DNA sequences rich in purine with a GGAA/T consensus core with flanking nucleotides [145]. While co-activator cyclic adenosine monophosphate (cAMP)-response element binding (CREB)-binding protein/p300 has been described to activate Ets2-mediated transcription [146, 147], Ets-2 repressor factor was observed to bind to the c-ets-2 promoter and is involved in the regulation of cell proliferation [148]. The consensus core GGAA/T recognized by Ets is also recognized by heat-shock factors [149] raising the possibility that cellular stress response may be associated with differential regulation of Ets expression levels. While extensive research covered the response of Ets-1 to various types of stress including oxidative stress showing for example, that Ets-1 is directly responding to hydrogen peroxide, H2O2 [150], the availability of literature on Ets-2 in such conditions is limited. In a 3T3 fibroblast cell model, exposure to H2O2 was found to induce mRNA expression of Ets-2 and that the resulting induced Ets-2 expression (Fig. 3), in turn, sensitizes cells to apoptosis mediated by H2O2 [151]. Another study demonstrating a relation between Ets-2 and oxidative stress similarly showed induction of Ets-2 by oxidative stress related to an ALS model [152]. Target genes of Ets-2 include those containing the Tie1 promoter [153], tumor-suppressor p53 [154], GATA-binding factor 1 [155], transcription factor JunB [156], vascular endothelial growth factor receptor 1 (VEGF-R1) [157], Intercellular Adhesion Molecule 1 (ICAM-1) [158], von Willebrand factor [159], and cyclin D1 [160]. Even though studies on the direct involvement of Ets-2 in oxidative stress response are sparse, a number of reports show the involvement of gene products that are the result of Ets-2-mediated transcriptional activity. For example, mouse embryonic fibroblasts exposed to H2O2 show p53 transcriptional response [161] while primary cells derived from p53-/- mice are resistant to H2O2 exposure. P53, in turn, is reported to play a key role in regulation of cellular levels of ROS, e.g., by partial inhibition of glycolysis and increased production of nicotinamide adenine dinucleotide phosphate (NADPH) [162] (Fig. 3). Specifically in relation to AD, p53 levels were observed to be elevated in AD brains [163] and p53 was shown to interact with AD-hallmark proteins tau and Aβ in the AD brain. GATA-binding factor 1, a master erythroid transcription factor, has previously been associated with the expression of the SNCA gene [164] which codes for the protein α-synuclein, implicated in Parkinson’s disease. Using Snca knock-out mice, α-synuclein, and specifically aggregates of α-synuclein, were observed to be associated with generation of oxidative stress [164, 165].
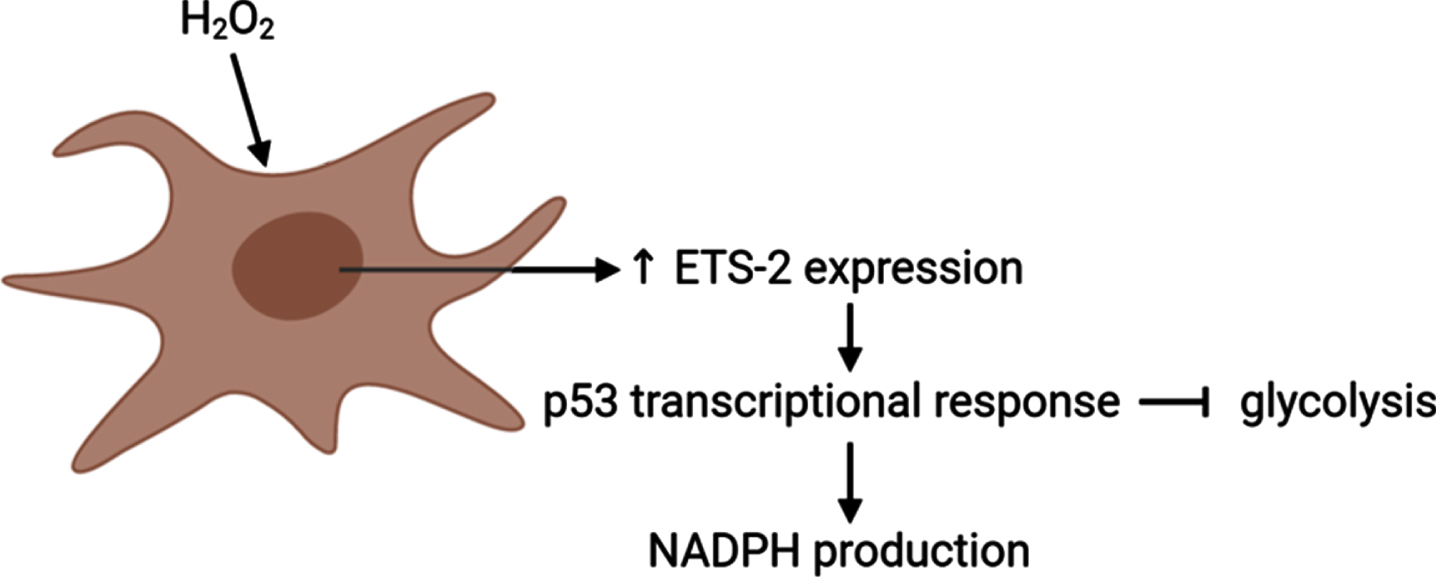
ETS-2 is suggested to regulate levels of reactive oxygen species in a p53 transcriptional response-mediated manner. Exposure of cells to H2O2 induces Ets-2 expression, which, in turn, induces p53 transcriptional response. P53, in turn, plays a key role in regulation of cellular levels of ROS, by partial inhibition of glycolysis and increased production of NADPH. Image created using BioRender.
Silencing of transcription factor JunB expression in human lymphoma cells was shown to increase the level and rate of loss of mitochondrial membrane potential upon exposure to H2O2 [166]. While no studies to date report on a direct association between ERF, oxidative stress, and AD specifically, a few interactions between gene products of ERF and regulation of oxidative stress levels and response to oxidative stress have been reported but require further investigation.
FAR2
Our transcriptomic analysis shows that mRNA expression levels of FAR2 are progressively downregulated throughout the progress of AD and this gene has also been identified in a gene-based genome-wide association study into resilience of executive functioning [167]. Furthermore, FAR2 was found to be downregulated in patients with human immunodeficiency virus (HIV)-associated dementia, AD, and Parkinson’s disease [168], and the gene was identified to affect the hippocampus in AD in a bioinformatic analysis [169]. Despite repeated identification of this gene in various studies, the molecular mechanical pathway by which this gene may be involved in AD pathogenic processes remains enigmatic. FAR2 encodes the peroxisomal protein fatty acyl coenzyme A (CoA) reductase 2, an enzyme that reduces saturated fatty acids of 16 or 18 carbons, and, with lower affinity, shorter saturated fatty acids, into fatty alcohols [170]. While the enzyme is most highly expressed in eyelid, skin, and small intestine, the brain also expresses relatively high levels of FAR2 mRNA [170]. Thus far, FAR2 has only been investigated in some detail in relation to renal diseases [171] and renal aging [172]. In renal aging, FAR2 expression levels appeared to associate with mesengial matrix expansion characterized by aberrant accumulation of extracellular matrix proteins in mesangial cells [172]. FAR2-mediated activity is suggested to result in the generation of platelet activating factor precursors, which are inflammatory mediators, and transforming growth factor (TGF)-β, a cytokine, which collectively drive deposition of mesangial matrix [172]. Receptors for platelet activating factors have been identified in the rat brain [173] and mediate the induction of hippocampal long-term potentiation processes [174, 175]. A number of studies reported an association between platelet-activating factor and AD or other types of dementia [176]. FAR2 plays a role in the stepwise biosynthesis of plasmalogens, a glycerophospholipid class containing sn-1-associated vinyl-ether and sn-2-associated ester on a glycerol backbone [177, 178]. An antioxidant role has been ascribed to plasmalogens by virtue of the oxidation state of the vinyl ether bond [179, 180]. In turn, circulating and brain plasmalogen levels were observed to be decreased in 40 AD patients with cognitive decline [181 –183]. Overall, a number of observations indicate that FAR2 may be associated with AD and maybe with AD-related oxidative stress albeit potentially by an indirect mechanism.
DDAH2
DDAH2 is a gene coding for the cytosolic protein dimethyl arginine dimethyl aminohydrolase 2. This enzyme functions in the metabolism of nitric oxide (NO) [184]. NO has been shown to affect learning and cognitive function [185] and DDAH2 may therefore potentially be correlated with the increased levels of oxidative stress observed in the AD brain. In particular, DDAH2 is responsible for the hydrolysis of the intracellular asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase (NOS) [186]. Arlt and co-workers assessed the levels of ADMA in plasma and CSF in 80 AD patients and compared the results to an age-matched healthy control group [187]. They found that while ADMA plasma levels were increased, AMDA CSF levels were reduced in AD subjects [187]. Similar results were reported in another study in a sample of 14 AD patients [188]. Decreased levels of ADMA may lead to a higher central nervous system (CNS) NOS activity and thus higher CNS levels of NO, which, by reacting with superoxide, result in the formation of ROS, causing the oxidative damage to biomolecules observed in brain tissue of AD patients [189]. Therefore, reduced levels of CNS ADMA may lead to increased oxidative stress and AD pathology. Furthermore, a recent study investigated protein co-expression and new biomarkers in AD. A tandem mass tag mass spectrometry-based quantitation (TMT-MS) approach allowed the identification of 532 proteins in the CSF of 297 subjects consisting of controls and AD patients. Results of this proteomic analysis revealed DDAH2 among the most significantly increased proteins playing a role in glucose and carbohydrate metabolism [190]. Our transcriptomic analysis showed that DDAH2 gene expression is increasingly upregulated throughout the entire progress of AD. Analysis of DDAH2 levels in postmortem AD brain samples showed regional differences, where the occipital lobe showed reduced and the frontal lobe increased levels of DDAH2. Inhibition of DDAH2 activity was reported in cardiovascular and the renal system as a result of its high sensitivity to oxidative stress [191]. It is plausible that inhibition of activity of DDAH2 is compensated for by upregulation of DDAH2 gene expression. These suggestions were substantiated by the observation that neuroblastoma SH-SY5Y cells treated with interleukin (IL)18 showed a robust increase of DDAH2 production [192]. Another study, exposing HUVEC to 4-HNE, showed that expression of intracellular ADMA was increased concurrent with a decrease in expression of DDAH2, both at mRNA and protein level [193]. DDAH2 was previously observed to metabolize ADMA into citrulline and dimethylamine [186]. Nevertheless, the notion that AD is correlated with an increased generation of DDAH2 seems to be contradictory to the observation that exposure of young rats to ROS with bile-duct-ligation showed downregulation of DDAH2. This same study showed that in vitro expression and activity of DDAH were inhibited by exposure to superoxide and H2O2 [194]. Precisely how levels of DDAH2 expression and activity are regulated in the brain is perhaps difficult to predict based on studies that focus on peripheral systems.
HBA2/HBA1
The genes HBA1 and HBA2 encode for two subunits of the heme scavenging protein hemoglobin. Free hemoglobin was reported to induce damage to cells because the heme moiety is able to catalyze the peroxidation of lipids [195]. Various reports describe that hemoglobin levels are affected as a function of AD. In line with this, a rat-based study observed that expression levels of hemoglobin and cognitive impairment were positively correlated [196], and a prospective cohort analysis showed that both higher and lower levels of hemoglobin were associated with the risk to develop AD [197] suggesting that maintaining hemoglobin levels within strict threshold values is critical. This notion was further underlined by the finding that both anemic participants as well as those with high hemoglobin levels in this study showed an increased risk for developing AD which also correlated with the rate of cognitive decline [197]. An association between anemia and AD was also observed in a cross-sectional cohort study including 211 participants with AD [198]. These data were partially reproduced in a study among an Indian 55 + year old population showing that low levels of hemoglobin could be linked with risk for AD [199]. The correlation between serum hemoglobin and risk of developing AD was further confirmed by a large study involving 12,305 participants. The outcome of this research similarly revealed that both low and high hemoglobin levels were associated with increased dementia risk. In particular, subjects with high levels of hemoglobin were 20%more likely to develop dementia than those with hemoglobin levels within the physiological range, while anemia was associated with 41%risk for AD onset [200]. Hemoglobin arises from the combination of two distinct globin chains, named α and β. A study using immunofluorescence and confocal microscopy imaging of neuronal cells, showed that decreased expression levels of both α and β hemoglobin chain coincided with hyperphosphorylated tau deposits in the frontal cortex and hippocampus in AD. On the other hand, hemoglobin was unaltered in neurons with no intracytoplasmic inclusions [201]. CSF [202] and sensorimotor cortex [203] consistently showed an increased level of hemoglobin β chain levels in AD patients. Further, mRNA levels of hemoglobin were found to be increased in the inferior temporal gyri of AD patients compared with non-affected subjects [204] while hemoglobin protein concentrations were highest in the hippocampal formation and the parietal gray matter in AD patients [204]. Interestingly, it was found that the in vitro deposition of synthetic Aβ1–40 is promoted by high hemoglobin levels [204 –206], and that hemoglobin is co-localized with Aβ-containing senile plaques as well as cerebral amyloid angiopathy in human brain material [204]. Consistent with this, it was shown that β hemoglobin levels were higher in the plasma of AD patients compared to individuals in a control group and that α hemoglobin was associated with amyloid-depositions [205]. How hemoglobin crosses the blood-brain barrier is unclear, but researchers speculate that hemoglobin infiltration into the brain could be the result of the report loss of blood-brain barrier integrity which is observed at early stage AD [207].
CYP4F12
CYP4F12 is a member of the p450 cytochrome family [208]. The p450 superfamily is a series of proteins involved in oxidative metabolism of substrates of various origins. The CYP4F family is specifically involved in ω-hydroxylation of eicosanoids [209]. CYP4F12 was observed to catalyze, upon consumption of NADPH, albeit with low activity compared to other CYP4 family members, the ω-hydroxylation of leukotriene B4, an inflammatory regulator, and arachidonic acid [210], a 20-carbon long polyunsaturated fatty acid, when expressed in Saccharomyces cerevisiae and activity is inhibited by ligands that overlap inhibiting ligands for other p450-family enzymes [211, 212]. Other than its endogenous substrates CYP4F12 metabolizes ebastine, an antihistamine drug [213] and antiallergen astemizole [212]. Rigidity of the active site of CYP4F12 compared to other p450 enzymes was considered to explain the limited catalytic activity of CYP4F12 over other CYP4 enzymes [212]. Expression of CYP4F12 is induced by activation of the p53 pathway [214, 215]. P53 signaling pathways have been implicated in oxidative stress on various occasions. For example, p53 was found to be involved, together with p66Shc, a protein that induces apoptosis based on oxidative signals, to genetically regulate steady state levels of intracellular oxidants [216]. P450 activity has been demonstrated in the brain, both in neuronal and glia cells [217, 218]. CYP4F12 was originally identified in the human liver [208, 210], colon, kidney, heart, and small intestine with negligible expression levels in the brain [210, 219]. P450 family member CYP2C19 was shown to affect brain Aβ homeostasis [220], and polymorphism of CYP46A1 increased risk to develop AD mediated by disturbance of brain cholesterol homeostasis [221, 222]. Using an AD mouse model, it was shown that overexpression of CYP46A1 improved cognitive functioning and ameliorated Aβ and tau pathologies. However, specific involvement of CYP4F12 in AD has not been reported yet while we found that mRNA levels of this protein are upregulated throughout AD progression. The precise role of CYP4F12 in AD-related oxidative stress warrants further exploration.
ATP11A
Eukaryotic membranes are characterized by an asymmetrical distribution of phospholipids in the plasma membrane which is essential for various cellular processes. Maintenance and generation of asymmetric lipid distribution in mammalian cells requires ATP-powered enzymes with translocase activity [223, 224]. Concentration of the lipids phosphatidylethanolamine and phosphatidylserine in the cytoplasmic leaflet of the membrane bilayer is aided by P4-ATPases which actively translocate lipids from the membrane leaflet exposed to the extracellular space to the cytoplasmic leaflet [225]. Our transcriptomic analysis indicates that AD is characterized by upregulation of ATP11A throughout disease progression. No publications to date are known that report on the relevance of ATP11A expression levels to AD progression but various studies do show that an indirect link between ATP11A expression, Ca2 + signaling, oxidative stress, and apoptosis may exist. The gene ATP11A encodes for the protein ATPase phospholipid transporting 11A, which is a class 6 P4-ATPase plasma membrane-localized phosphatidylserine and phosphatidylethanolamine translocase [226]. To be able to actively perform inward translocation of phosphatidylserine, ATP11A requires co-chaperoning by the colocalized plasma membrane glycoprotein Cell Cycle Control Protein (CDC)50A (internalization receptor transmembrane protein (TMEM) 30A) [226, 227], to ensure endoplasmic reticulum (ER) export and localization at the Golgi and plasma membrane. A molecular description of the direct effect of oxidative stress on ATP11A gene expression has not been reported but oxidative stress-mediated dysregulation of intracellular calcium homeostasis may play a role. Calcium- and ROS-mediated signaling pathways show bidirectional crosstalk which makes physiological sense as intracellular Ca2 + homeostasis is regulated by a redox-dependent mechanism. Increased intracellular Ca2 + levels are related to increased intracellular generation of ROS by stimulation of respiratory chain activity. An increased intracellular Ca2 + concentration was reported to inhibit ATP11A activity in a dose-dependent manner, presumably by direct binding of Ca2 + to ATP11A [226]. Increased Ca2 + and ROS levels simultaneously activate the mitochondrial permeability transition pore (mPTP) releasing pro-apoptotic factors such as procaspases and cytochrome c (Fig. 4). The sequence of ATP11A contains two evolutionary conserved caspase cleavage sites [228]. When mouse cells were exposed to leucine-zippered human Fas ligand, which induces caspase 3 activation, ATP11A was found to be cleaved and inactivated and lost the ability to translocate phosphatidylserine [226] (Fig. 4) demonstrating that ATP11A plays a role in efficient clearance of dead cells in apoptotic processes. Lethal defects are observed in mouse embryos upon deletion of Atp11a [229] as a result of the essential role of ATP11A-CDC50 complex in the regulation of a mechanosensitive Ca2 + channel involved in myotube formation [230]. At the same time, some mutations in ATP11A, when co-expressed in COS cells with CDC50A, were observed to be retained at the ER, or to display reduced phospholipid flippase activity [231]. Transgenic mice carrying loss of function mutations of P4-ATPase show neurodegenerative disorders such as axonal degeneration in the central and peripheral nervous system [232]. Also in humans missense mutations in P4-ATPases have been reported to affect neuronal functioning and to lead to atropic processes [233, 234]. However, all these studies have been limited to ATP8A2, another member of the P4-ATPase family and while ATP11A functioning resembles that of ATP8A2 fairly well, it remains to be shown that mutations in ATP11A similarly lead to neuronal dysfunction. Tissue-specific mRNA expression patterns show that ATP11A mRNA is expressed in various regions of the brain [235] indicating that a brain-related function may be apparent. Until now, evidence to show a role of ATP11A in oxidative stress seems rather circumstantial than direct and indicates a response of ATP11A gene expression levels to be a downstream consequence of oxidative stress.
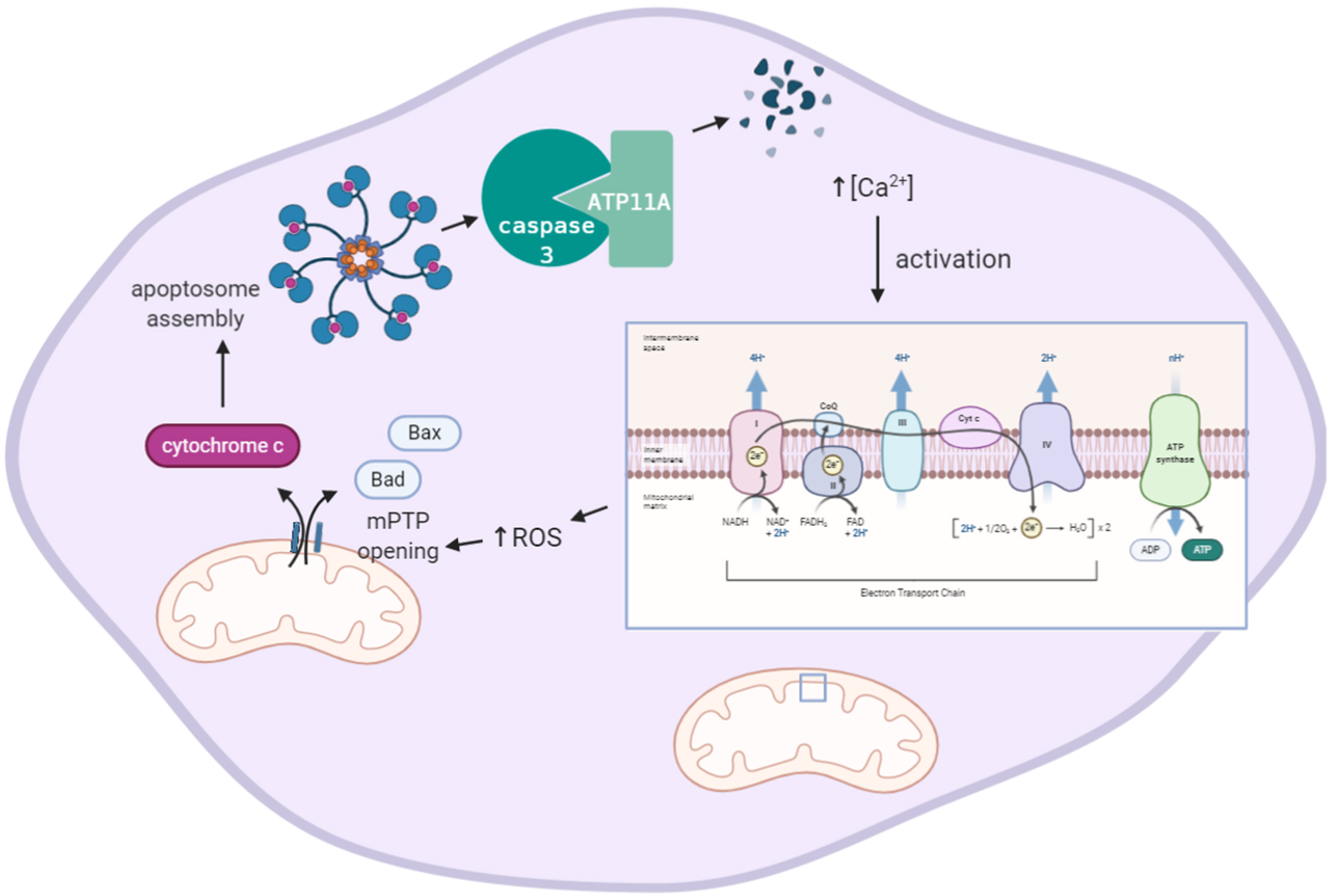
Increased intracellular Ca2 + levels involve formation of ROS and induce caspase 3-mediated degradation of ATP11A activity. Increased intracellular Ca2 + levels increase electron transport chain activity mediating the generation of mitochondrial ROS. ROS induces opening of the mitochondrial Pore Transition Protein (mPTP), which, in turn, results in release of apoptotic mediators Bax, Bad, and cytochrome c. Cytochrome c induces assembly of a large protein complex termed apoptosome which mediates cleavage of pro-caspases into active caspases (not shown). Activated caspase 3 degrades ATPase phospholipid transporting 11A (ATP11A). Image created using BioRender.
PPP3CA
Our transcriptomic analysis indicated that mRNA levels of the gene PPP3CA, encoding the protein calcineurin A α, are downregulated in incipient, moderate, and severe cases of AD in a progressive manner. Calcineurin 3 A α is one out of three different isoforms of calcineurin A, which interacts to form a heterodimer with calcineurin B forming a calmodulin-regulated protein phosphatase [236]. Within this complex, calcineurin B forms the Ca2 +-binding regulatory subunit, while calcineurin A comprises the catalytic site. In T lymphocytes, calcineurin was shown to act, when Ca2 + levels rise, by dephosphorylating the DNA-binding protein nuclear factor of activated T cells (NF-AT) inducing its translocation from the cytosol to the nucleus [237]. Truncated forms of calcineurin are found in the brains of AD patients [238]. Various functions of this protein have been ascribed to its abundant presence in the brain [239] and various tissues containing neuroendocrine cells [240]. N-methyl-D-aspartate receptor-mediated increase of intracellular Ca2 + levels was suggested to activate calcineurin [241]. Calcineurin-related signaling pathways have been implicated in AD on various occasions. For example, neuronal damage was observed to irreversibly activate calcineurin [242] and that soluble oligomeric forms of Aβ are directly responsible for this process. As demonstrated in AD hippocampal brain tissues and primary astrocyte/hippocampal cell cultures, exposure to soluble Aβ resulted in nuclear accumulation of NF-AT3 that positively correlated with calcineurin A α levels [243, 244]. As Ca2 + signaling events, apoptosis, and oxidative stress are interlinked, as was demonstrated in a cardiomyocyte system showing that apoptosis is induced by an increase in intracellular Ca2 + level [245], the role of calcineurin in AD-related oxidative stress becomes palpable. A study using SH-SY5Y cells showed that pharmacological inhibition of calcineurin activity decreased the expression of APP, while activation of calcineurin-NF-AT signaling reduced the expression of tau and neprilysin [246]. Levels of NF-AT, the target of calcineurin, show differential expression and nuclear localization in MCI and AD hippocampi in an NF-AT isoform-dependent manner [247]. Further, a role of calcineurin in the regulation of NF-AT levels was detected by the presence of fragments of the isoform calcineurin A α indicating the activation of calcineurin-NF-AT pathways [247, 248]. While increased NF-AT expression was also associated with astrocyte activation in an inflammatory brain damage mouse model, the cytoplasmic NF-AT3 transcripts were found to be up-regulated [249] and nuclear translocation of NF-ATs is induced by ATP that activates of purinergic type 2 (P2)X7 receptor which stimulates the generation of oxygen radicals [250, 251]. The overproduction of ROS and the consequent oxidative damage resulting in HNE products led to derailed glutamate uptake due to impairment of GLT1/EAAT2 [252]. Hippocampal EAAT2 expression was observed to be downregulated in AD coinciding with increased calcineurin-NF-AT signaling [243]. The interaction between EAAT2 expression levels and calcineurin-NF-AT signaling may be explained by the observation that EAAT2 reduction is linked to astrocyte activation and EAAT2 is transcriptionally regulated by NF-AT as evidenced by the detection of an NF-AT binding site in the human EAAT2 promotor [253]. The process of calcineurin-NF-AT signaling is further regulated by calcineurin 1 (RCAN1) protein, which, when expressed at high levels, can also act as a mediator of oxidative stress as observed in primary neuronal cultures [254].
TMEM70
Our transcriptomic analysis suggests that TMEM70 mRNA levels are downregulated in all stages of AD, but most significantly during moderate stages of AD. This nuclear gene on chromosome 8q21.11 contains 3 exons encoding for a 260 amino acid (AA) mitochondrial localized 21 kDa transmembrane protein 70. TMEM70 is initially synthesized as a 29 kDa precursor protein [255 –257], which is thought to be involved in the biogenesis of mitochondrial ATP synthase [258], in particular F1 subunit assembly or F1-F0-subunit interaction. TMEM70 deficiency, as a result of mutations in the TMEM70 gene, was first identified to result in deficiency of ATP synthase activity [258]. Patients suffering from such mutations are characterized by abnormally shaped mitochondria [259] and nucleoid disorganization [260]. A genetic defect of the TMEM70 gene in neonatal patients showed an almost complete ATP synthase deficiency in mitochondria [259, 260]. Moreover, the role of the TMEM70 gene in the assembly of ATP synthase, or complex V, subunits was reported in extracted mitochondria from Hela cell cultures expressing the human TMEM70 gene. Patients carrying homozygous and heterozygous mutations of the gene presented with a decreased expression of ATP synthase α and β subunits [260] of F1 culminating in ATP synthase deficiency [261]. The oxidative phosphorylation process for producing ATP comprises a crucial function by the F1-F0-subunits of ATP synthase which is the result of oxidative phosphorylation. While in oxidative stress the disruption of electron transport chain is explored widely in neurological pathologies, it has been observed in mitochondria of postmortem cortical brain tissue with bipolar disorder that there was not only inflected oxidative damage to proteins [262] but also decreased expression of mRNA levels coding subunits of mitochondrial complexes. Impaired expression was found to be the result of damage to their genes including that of complex V, the ATP synthase membrane subunit c locus 3 (ATP5G3), that encodes the catalytic F1 subunit [263]. Collectively, these observations suggest that reduced TMEM70 mRNA expression levels to transcript the assembly precursor of F1-F0 subunits can be regarded as a possible biomarker of oxidative stress. Even though, no earlier publications reported an association between AD and TMEM70. However, the generation of ROS has been observed as a result of upregulated mitochondrial membrane potential in response to ATP synthase activity modulation [264]. In line with the suggested role of TMEM70 in ATP synthase biogenesis, Tmem70-/- mouse embryos showed upregulation of SOD2 without significantly affecting SOD1 levels [265].
CONCLUSIONS AND FUTURE PERSPECTIVES
Excessive generation of mitochondrial ROS, as observed in AD, can result in detrimental effects on lipids and proteins in the brain. These structures are affected in early-stage AD as the biomarkers of their peroxidation and oxidation are readily detectable at incipient and MCI stages. Many of the currently known biomarkers that can be used to assess oxidative stress status of the brain lack specificity toward the AD brain as also other forms of neurodegeneration are known to show differential levels compared to healthy individuals. This observation raises questions as to the isolated use of oxidative stress biomarkers to specifically and unequivocally diagnose AD. However, together with known AD biomarkers for Aβ1–42 and hyperphosphorylated tau, oxidative stress-related biomarkers may be useful to assess the health status of an AD patient and progression of disease and support determination of individual therapeutic needs.
Most data on oxidative stress levels in AD have been based on postmortem measurement of biomarker levels in the brain. Obtaining patient material from the brain to allow insight into disease state is rather invasive and peripheral measurement of biomarker levels, such as in plasma or urine, are thus preferrable to obtain insight into oxidative stress in living patients. A protocol for the measurement of malondialdehyde in body fluids has been reported [266]. However, information on peripheral biomarker levels in relation to disease status and brain oxidative stress levels is limited and the observed lack of correlation of peripheral biomarker levels with oxidative stress status of the brain further hampers implementation of this route.
Brain microvascular pathologies are often found to co-occur in AD patients and such microvascular lesions are correlated with raised levels of oxidative stress [267]. As particularly aged AD patients often suffer from a range of co-morbidities that, in turn, have also been related to oxidative stress including vascular disease [268], it would be of interest to study oxidative stress levels in young AD patients to unravel the contribution of the AD pathogenic process in isolation.
Molecular mechanisms of oxidative stress level regulation and deregulation in AD have been explored but remain enigmatic. Elucidation of additional biomarkers may provide a more detailed insight into molecular details and generate potential additional read-outs for oxidative stress more specific to AD at incipient stages. Using a transcriptomic approach, genes that could act as potential new biomarkers were identified that show either increase (e.g., ERF, ATP11A, CYP4F12) or decrease (e.g., FAR2, TMEM70) in mRNA levels depending on disease stage, compared to healthy subjects. While a direct correlation between some of the products of the identified genes and oxidative stress level regulation have already been reported, other genes are relatively underexplored. For many of these genes it also remains to be revealed how mRNA expression levels correlate with protein expression. Further research is warranted to establish the potential of these novel biomarkers for determination of oxidative stress and disease status of AD patients.