
Abstract
Background:
Glucocerebrosidase (GBA) gene mutations and APOE polymorphisms are common in dementia with Lewy bodies (DLB), however their clinical impact is only partially elucidated.
Objective:
To explore the clinical impact of mutations in the GBA gene and APOE polymorphisms separately and in combination, in a cohort of Ashkenazi Jewish (AJ) patients with DLB.
Methods:
One hundred consecutively recruited AJ patients with clinically diagnosed DLB underwent genotyping for GBA mutations and APOE polymorphisms, and performed cognitive and motor clinical assessments.
Results:
Thirty-two (32%) patients with DLB were carriers of GBA mutations and 33 (33%) carried an APOE ɛ4 allele. GBA mutation carriers had a younger age of onset (mean [SD] age, 67.2 years [8.9] versus 71.97 [5.91]; p = 0.03), poorer cognition as assessed by the Mini-Mental State Examination (21.41 [6.9] versus 23.97 [5.18]; p < 0.005), and more severe parkinsonism as assessed with the Unified Parkinson’s Disease Rating Scale motor part III (34.41 [13.49] versus 28.38 [11.21]; p = 0.01) compared to non-carriers. There were statistically significant interactions between the two genetic factors, so that patients who carried both a mild GBA mutation and the APOE ɛ4 allele (n = 9) had more severe cognitive (p = 0.048) and motor dysfunction (p = 0.037).
Conclusion:
We found a high frequency of both GBA mutations and the APOE ɛ4 allele among AJ patients with DLB, both of which have distinct effects on the clinical disease phenotype, separately and in combination.
INTRODUCTION
Dementia with Lewy bodies (DLB) is the second most common neurodegenerative dementia after Alzheimer’s disease (AD) [1]. Pathologically, it is characterized by the accumulation of Lewy bodies and Lewy neurites, though concomitant AD pathology is commonly present. This mixed pathology leads to a heterogeneous clinical presentation, resulting in low diagnostic accuracy [2]. For example, increasing AD pathology, specifically neuritic plaque pathology, is associated with a lower likelihood of occurrence of the DLB clinical syndrome [3], and therefore a lower chance of correct diagnosis.
Mutations in the GBA gene are frequent among patients with DLB and Parkinson’s disease (PD) [4–6], whereas the APOE ɛ4 allele is a significant risk factor for development of AD [7]. Several studies have demonstrated that patients with PD or DLB who carry mutations in the GBA gene have a younger age of onset and a more severe disease phenotype [8–10]. Penetrance estimations indicate that up to 30%of GBA mutation carriers will go on to develop PD by age 80 [11]. While the data regarding the penetrance of GBA mutations in DLB is less clear, it remains likely that the majority of GBA mutation carriers will not go on to develop Lewy body disease. As to whether APOE polymorphisms have any effect on the clinical features of PD, the data is conflicting, with several studies describing a detrimental effect on the clinical phenotype of the disease, with others finding no effect [12–17].
In contrast to PD, there may be a more convincing association with the APOE ɛ4 allele for DLB. Studies have reported a higher APOE ɛ4 frequency in DLB than that found either in the healthy population or in patients with PD or Parkinson’s disease dementia (PDD) [18–23]. A previous study examining the clinical impact of APOE polymorphisms in DLB found no association of the APOE ɛ4 allele with age of onset or disease duration [24]; however, the presence of the APOE ɛ4 allele has been associated with shorter survival [25]. Another study examining the effect of APOE ɛ4 across the whole AD/DLB spectrum found smaller hippocampal volumes in APOE ɛ4 carriers relative to non-carriers, as well as worse long-delay free word recall in the APOE ɛ4 carriers. In this study the authors did not look at the DLB group separately, so this effect may have been driven by differences in the AD group [26]. Apart from these studies, few clear-cut correlations have been found between the presence of the APOE ɛ4 allele and the clinical syndrome in DLB.
Therefore, the clinical effect of APOE ɛ4 polymorphisms on DLB remains less clear, and any possible clinical interaction with GBA mutations has not been fully addressed in the literature to date. In this study we aimed to examine the clinical effects of GBA mutations and APOE ɛ4 polymorphisms on Ashkenazi Jewish (AJ) patients with clinically diagnosed DLB, and to explore the potential clinical effect of these genetic factors in combination.
METHODS
Standard protocols approvals, registrations, and patient consent
The Institutional and National Helsinki Committees for Genetic Studies approved the study protocols. All participants provided signed informed consent prior to participating in the study, and patients with DLB only participated in the current study if they were able to give valid consent.
The cohort included 100 consecutively recruited patients of AJ descent with DLB, all unrelated for three generations, who attended the Cognitive Neurology Unit at the Neurological Institute, Tel Aviv Medical Center, between July 2013 and January 2020. Thirty-four of these 100 patients have been reported in our previous analysis in which GBA carrier rate was explored [8].
Neurological evaluation
All participants underwent full neurological examinations. An experienced neurologist obtained a detailed medical history, including symptoms and medications. Age at onset was defined as the onset of either cognitive decline or parkinsonism. All patients fulfilled the 2017 McKeith clinical criteria for possible or probable DLB [27] (with criteria retrospectively applied to those recruited before 2017), and all had onset of motor symptoms less than one year prior to onset of cognitive decline. Motor symptoms were assessed using the Unified Parkinson’s Disease Rating Scale (UPDRS motor part III) [21]. Global cognitive function was assessed by the Mini-Mental State Examination (MMSE) [29] and by the Montreal Cognitive Assessment (MoCA) [23]. The presence of REM sleep behavior disorder (RBD) was assessed by the RBD Questionnaire (RBDQ) [24], and the existence of hallucinations, current or past, was assessed by direct questions to the patients and the primary care giver.
Genotyping
Genotyping of founder GBA mutations was performed as previously described [32, 33]. Briefly, patients were tested for the 84GG, IVS2 + IG>A, p.N370S, p.L444P, p.V394L, p.R496H, and 370Rec GBA mutations, using the “Gaucher kit” (Catalog number 800672, Savyon Diagnostics, Israel, https://www.savyondiagnostics.com/product/nanochip-gaucher). In addition, the E326K and T369M GBA mut-ations were genotyped using gene-specific TaqMan assay, followed by Sanger sequencing of gene specific PCR products, to confirm all carriers. These nine mutations cover over 96%of the GBA mutations among individuals of full Ashkenazi origin [34] [Goldstein and Orr-Urtreger, unpublished data]. In view of the high carrier rate of LRRK2 mutations among AJ patients with PD, existence of the G2019S mutation was also examined. Genotyping of the APOE polymorphisms was performed by PCR amplification of a specific fragment, using the following primers: forward 5’ GGCACGGCTGTCCAAGGAGCT 3’ and reverse 5’ GCCCCGGCCTGGTACACTGC 3’ and Sanger sequencing. All samples were also genotyped for rs429358 and rs7412, using TaqMan assays (C___3084793_20 and C____904973_10, respectively, Applied Biosystems) to establish their APOE haplotypes.
Statistical analysis
Normality and homogeneity of variance were explored using Q-Q plot and Levene’s homogeneity test. Demographic and clinical characteristics of the patients were analyzed using a two-tailed Student’s t-test or chi-squared test. Motor and cognitive characteristics of all patients were analyzed with an analysis of covariance (ANCOVA), in which the presence of GBA mutations and APOE polymorphisms served as the two between-subject factors, and cognitive tests (MMSE, MoCA) and motor tests (UPDRS) served as the dependent variables. In the analysis of the cognitive tests, age, time since diagnosis, and treatment with acetylcholinesterase inhibitors (AChEI) served as covariates. In the analysis of the motor test, age and time since diagnosis served as covariates.
For the longitudinal follow up of MMSE scores, we used an ANCOVA with GBA and APOE as between-subject factors and the change in MMSE as the dependent variable. Two follow up MMSE points were examined, at one year (range 8–16 months) and at two years (range 20–28 months) since first testing. Covariates in this analysis included age and time since diagnosis, but not medication as this was generally changed during the follow up time.
Effect size was calculated using eta squared. All analyses were conducted with SPSS software (version 25; SPSS, Inc.), and the alpha level was set at 0.05.
RESULTS
Participants
One-hundred patients (72 [72%] men) were included in the study. Average age at disease onset was 69.74 (range 44–85, SD 7.18) years. Average age at enrollment was 73.10 (range 57–89, SD 6.94) years.
Thirty-two (32%) DLB patients carried mutations in the GBA gene, 27 carried mutations considered to be mild, 4 carried mutations considered to be severe [35], and 1 was heterozygote for the T369M mutation of unknown significance (Table 1A). Two of the patients had a clinical diagnosis of Gaucher disease and 1 patient who was homozygote for the N370S mutation had not been diagnosed with Gaucher disease prior to the DLB diagnosis. GBA mutation carriers were younger at symptom onset than were non-carriers (t = 2.21, p = 0.03) (Table 2).
GBA mutation type distribution in the cohort
Patient demographic and clinical results, by GBA mutation status
All p-values reflect two-tailed t-test results unless marked with an asterisk; *p-values obtained by chi-squared test; **p-values obtained by ANCOVA with age, disease duration, and ACEI use as covariates; +p-values obtained by ANCOVA with age and disease duration as covariates. MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; UPDRS, Unified Parkinson Disease Rating Scale; RBDQ, REM sleep behavior disorder questionnaire; AChEI, acetylcholine esterase inhibitors.
Thirty-three (33%) DLB patients carried an APOE ɛ4 allele (Table 1B and Table 3). Nine of them were also carriers of mild GBA mutations (N370S, R496H, T369M) one of whom was homozygote for the N370S mutation but had no formal diagnosis of Gaucher disease.
APOE polymorphism distribution in the cohort
Patient demographic and clinical results, by APOE ɛ4 allele carrier status
All p-values reflect two-tailed t-test results unless marked with an asterisk; *p-values obtained by chi-squared test; **p-values obtained by ANCOVA with age, disease duration, and ACEI use as covariates; +p-values obtained by ANCOVA with age and disease duration as covariates. MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; UPDRS, Unified Parkinson Disease Rating Scale; RBDQ, REM sleep behavior disorder questionnaire; AChEI, acetylcholine esterase inhibitors.
Firstly, we examined MMSE scores, showing that after controlling for age, disease duration and AChEI use, GBA carriers performed worse than non-carriers, F(1,84) = 8.124, p = 0.005,ŋ2 = 0.088. There was no main effect of the APOE ɛ4 allele, F(1,84) = 0.129, p = 0.72, ŋ2 = 0.002. Nevertheless, the interaction between the two mutations was significant, F(1,84) = 4.030 p = 0.048, ŋ2 = 0.046. An analysis examining MoCA scores did not reach significance on any of the comparisons: main effect of GBA, F(1,77) = 2.695, p = 0.105, ŋ2 = 0.034, main effect of APOE F(1,77) = 0.118, p = 0.733, ŋ2 = 0.002, interaction, F(1,77) = 1.595, p = 0.210, ŋ2 = 0.02.
Secondly, we examined motor UPDRS scores, sho-wing that after controlling for age and disease duration, GBA mutation carriers performed worse than non-carriers, F(1,87) = 6.560, p = 0.012, ŋ2 = 0.07. There was no main effect of the APOE ɛ4 allele, F(1,87) = 0.052, p = 0.820, ŋ2 = 0.001, but once again we found that the interaction between the two mutations was significant, F(1,87) = 4.48, p = 0.037, ŋ2 = 0.049 (Fig. 1).
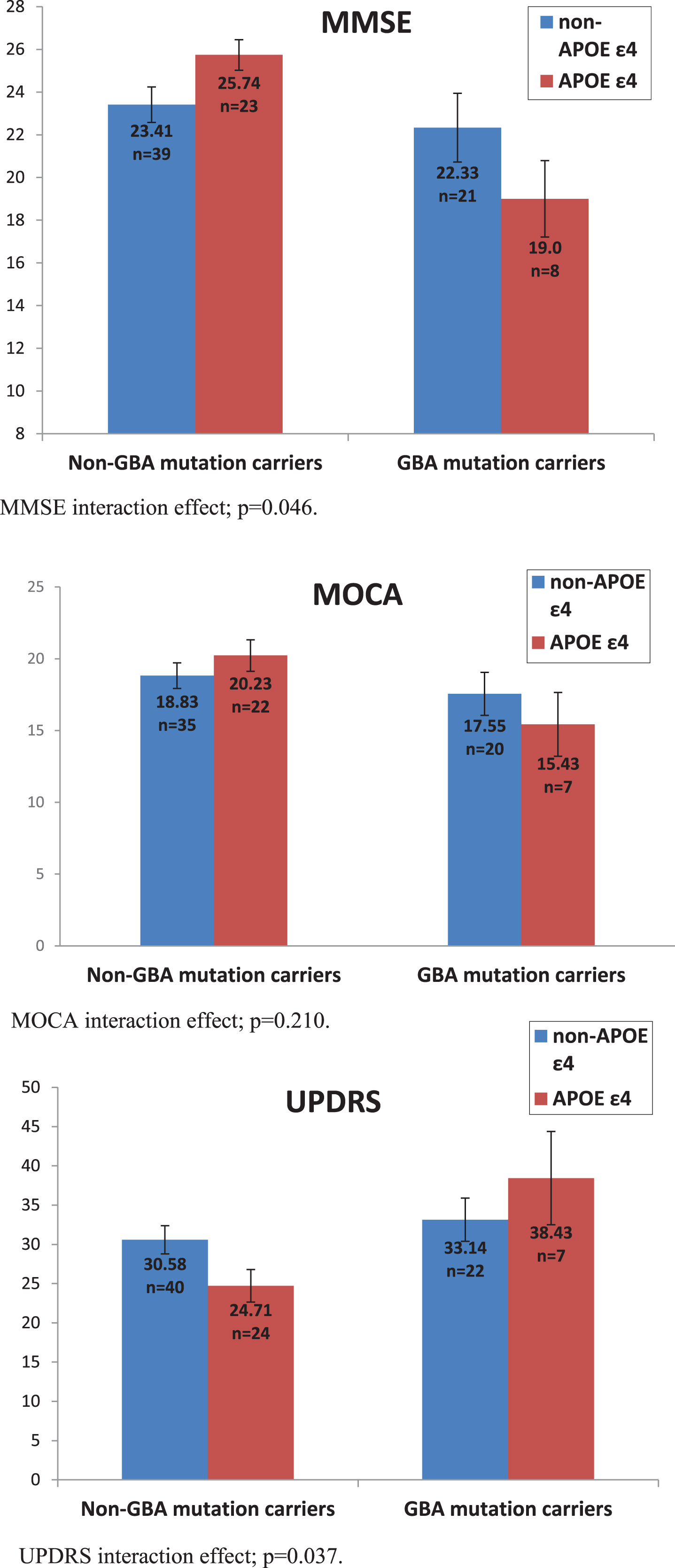
Effect of GBA mutations and APOE ɛ4 polymorphisms on MMSE, UPDRS, and MOCA scores. MMSE interaction effect; p = 0.046. MOCA interaction effect; p = 0.210. UPDRS interaction effect; p = 0.037.
The majority of patients with DLB continued to be followed up in the clinic at regular intervals, however we have limited data on serial MMSE scores due to various reasons, including inconsistent visit times, rapid deterioration precluding test administration, and death. We examined the change in MMSE scores from baseline to the one-year follow up (34 patients) and to the two-year follow up (23 patients), while controlling for age at testing and disease duration. We found no significant differences in MMSE change at one year between GBA carriers and non-carriers, F(1,33) = 2.305, p = 0.140, ŋ2 = 0.076, or between APOE ɛ4 carriers and non-carriers, F(1,33) = 0.060, p = 0.809, ŋ2 = 0.002), and no significant interaction between the two genetic profiles, F(1,33) = 2.757, p = 0.108, ŋ2 = 0.02. The same analysis was carried out for the MMSE score at the two year follow up. We again found no significant differences between GBA carriers and non-carriers, F(1,17) = 0.192, p = 0.667, ŋ2 = 0.011, or between APOE ɛ4 carriers and non-carriers, F(1,17) = 0.165, p = 0.689, ŋ2 = 0.01, and no significant interaction, F(1,17) = 0.654, p = 0.43, ŋ2 = 0.037.
DISCUSSION
The aim of this study was to further explore the clinical impact of GBA mutations in a cohort of AJ patients with DLB known to have a high frequency of GBA mutations, and to evaluate any potential clinical effect of the presence of the APOE ɛ4 allele in this cohort. We also aimed to explore whether the presence of both of these common genetic factors would have an impact on the clinical phenotype in DLB.
In this work we confirm the findings of our previous report [8], and find a very high frequency (33%) of GBA mutations in a larger cohort of AJ patients with DLB (n = 100). This is compared to the reported carrier rate of 7.8%in the unaffected AJ population [26]. We have established in a much larger cohort that patients with DLB and GBA mutations have a younger age of symptom onset as well as a more severe cognitive and motor course than non-carriers.
Pathologically, GBA mutations in patients with DLB are associated with more extensive cortical Lewy bodies on postmortem [6]. Furthermore, patients who carry GBA mutations are less likely to meet pathological criteria for AD, and these mutations are generally associated with lower levels of amyloid pathology on postmortem [28, 29]. These findings would suggest that dementia in DLB patients with GBA mutations is primarily mediated by diffuse alpha synuclein pathology and not by AD-type pathology.
We found that 33%of AJ patients with DLB carried an APOE ɛ4 allele, leading to an APOE ɛ4 allele frequency of 17.9%in our current cohort, which is higher than the rate reported for the unaffected AJ population [38], but lower than that described in other cohorts of patients with DLB [19, 31]. The frequency of the APOE ɛ4 allele among the healthy population has been shown to vary widely among different cohorts [7]. Our finding of a lower rate of the APOE ɛ4 allele in this patient cohort, relative to that described in the literature, may reflect the relatively lower APOE ɛ4 allele frequency described in the unaffected AJ population, which stands at 9.03%[30]. Other studies in Jewish cohorts in Israel have also found a relatively low APOE ɛ4 allele frequency of 6%[40], which is different from that found in other typical Caucasian populations in which the allele frequency is quoted as around 15%[41]
In AD, the presence of the APOE ɛ4 allele increases the risk of developing cognitive impairment and dementia, and affects the age of onset of the disease, which is 5–15 years earlier in carriers versus non-carriers [42]. Reports have been conflicting regarding the effect of the APOE ɛ4 allele on the clinical profile in AD. Studies have shown an effect on the rate of conversion from mild cognitive impairment (MCI) to dementia [43], and an effect on the progression of cognitive decline in AD [44], yet other studies have not replicated these findings [45, 46].
There is inconsistent evidence as to whether APOE polymorphisms have an effect on the clinical syndrome in PD and DLB. In familial PD, the presence of the APOE ɛ4 allele has been shown to increase the risk of dementia and has been associated with an earlier age of onset [13]. A study in sporadic PD, however, found no association between APOE ɛ4 and MCI, dementia, or any other specific neuropsychological deficits [14]. Furthermore, in PD, the APOE ɛ4 allele frequency has been shown to be similar to that of the general population [12]. There have also been conflicting reports regarding the association between the APOE ɛ4 allele and the presence of hallucinations in PD, with some studies describing increased frequency in APOE ɛ4 carriers [15], while others not describing such effects [16, 17].
Of interest, we did not see a difference in the longitudinal data between GBA and non-GBA mutation carriers, so that both groups demonstrated similar change in MMSE scores over time. There are several possible explanations for this finding. One possibility is that the mutation status does not affect the rate of deterioration in DLB, although GBA mutations have been associated with more rapid progression in PD [47]. Another possibility is that the groups for whom we had longitudinal data were too small or that a two-year follow up was not a long enough period in order to observe significant differences.
In our study, we found an interaction between mild GBA mutations and the APOE ɛ4 allele, such that patients who carry both had the most severe cognitive and motor impairment. Of note, we only found a difference in the MMSE and not in the MoCA. Although the GBA mutation carriers had lower scores on the MoCA than non-carriers, similar to the direction of results for the MMSE scores, this difference did not reach significance, possibly due to the smaller sample size of those who completed the MoCA versus the MMSE. Another possibility is that the increased weighting of the visuospatial tasks in the MoCA compared to the MMSE makes it a less effective test at discriminating between patient groups because in this patient population visuospatial functions are commonly impaired [27]. We did not find a main effect of the APOE ɛ4 allele on the clinical characteristics of the patients, so that carriers and non-carriers did not differ when the GBA mutation was not taken into account. A possible explanation for these findings lies in the effect of the APOE polymorphisms on the neuropathology of PDD and DLB. Neuropathological studies have shown that over the Lewy body spectrum, the presence of the APOE ɛ4 allele is associated with increased levels of AD neuropathology [36]. However, even in “pure” DLB and PDD (i.e. in those cases in which the overall brain neuritic plaque burden is low), there is an increased frequency of the APOE ɛ4 allele [19]. Furthermore, the presence of the APOE ɛ4 allele has been found to be associated with an increased risk of diffuse Lewy body pathology even in patients with low AD pathology [39]. These findings suggest that APOE ɛ4 may function as a modifier of the spread of Lewy body pathology, and that APOE ɛ4 may contribute to neurodegeneration through mechanisms that are unrelated to amyloid processing [19, 39].
As summarized above, APOE ɛ4 appears to have an effect on levels of AD pathology in DLB, as well as through mechanisms unrelated to amyloid processing. An important open question is which pathology likely dominates in patients who carry both genetic factors. One possibility is that the clinical effect is seen due to the diffuse Lewy body pathology associated with GBA mutations (the so called ‘pure’ synucleinopathy), with APOE ɛ4 acting as a modifier that affects the spread of Lewy body pathology. Alternatively, it may be that the more severe disease is mediated by increased amyloid pathology found in association with the presence of the APOE ɛ4 allele, as has been previously found in the pathological studies looking at the entire Lewy body spectrum [36]. We note that in the non-GBA mutation carriers in our sample, the APOE ɛ4 allele appears to slightly improve performance. In AD, the absence of an APOE ɛ4 allele has been shown to affect the spatial distribution of AD pathology by increasing the frequency of hippocampal sparing AD [48]. It is therefore possible that in DLB the presence of APOE ɛ4 allele without the presence of a GBA mutation leads to a different distribution of pathology, however further research is required in order to examine this possibility.
As far as we know, there are no studies examining the clinical effects of the presence of both GBA mutations and the APOE ɛ4 allele on patients with DLB. We acknowledge that there are limitations to this study. The main limitation in our study is the low number of patients who carry both a GBA mutation and the APOE ɛ4 allele. Nevertheless, we believe that even this small number of patients reveals a significant and important finding, which has particular implications to our understanding of the phenotypic variability in DLB. A further limitation is that we do not have confirmatory post-mortem pathology on the subjects, and the diagnosis was based on clinical criteria and supporting features. This is likely to have led to under-diagnosis of cases, as has been described previously [49]. We ascertained GBA mutation status by screening for the common nine AJ mutations and not through whole gene sequencing. This method has been validated previously in the AJ population; however, we acknowledge that it is possible that additional genetic factors could have contributed to the findings. Finally, we had follow-up data on a small subset of the sample, which unfortunately did not allow for a definitive conclusion on the longitudinal outcomes in the GBA mutation and APOE ɛ4 polymorphism carriers. We continue to follow this cohort and hope to have more longitudinal data in the future.
CONCLUSION
In this study, we aimed to examine the clinical effect of the two most common genetic factors associated with DLB, mutations in the GBA gene and the presence of the APOE ɛ4 allele, as well as the interaction between them. We confirmed the detrimental impact of GBA mutations on the clinical phenotype of DLB and found that these mutations act in a complex interaction with APOE ɛ4 allele, so that patients who carry both genetic risk factors had a more severe cognitive and motor involvement. These findings contribute to our understanding of the effects of these common genetic factors but leave many open questions regarding the specific role of each one of the factors, as well as the functional interplay between them and its impact on the clinical and pathological syndrome in DLB. These findings may also have implications for suitability of patients for specific therapeutic interventions in the future.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1295r3).