
Abstract
Background:
Right temporal variant frontotemporal dementia (rtvFTD) has been generally considered as a right sided variant of semantic variant primary progressive aphasia (svPPA), which is a genetically sporadic disorder. Recently, we have shown that rtvFTD has a unique clinical syndrome compared to svPPA and behavioral variant frontotemporal dementia.
Objective:
We challenge the assumption that rtvFTD is a sporadic, non-familial variant of FTD by identifying potential autosomal dominant inheritance and related genes in rtvFTD.
Methods:
We collected all subjects with a diagnosis of FTD or primary progressive aphasia who had undergone genetic screening (n = 284) and subsequently who had a genetic variant (n = 48) with a diagnosis of rtvFTD (n = 6) in 2 specialized memory clinics.
Results:
Genetic variants in FTD related genes were found in 33% of genetically screened rtvFTD cases; including MAPT (n = 4), GRN (n = 1), and TARDBP (n = 1) genes, whereas only one svPPA case had a genetic variant in our combined cohorts. Additionally, 4 out of 6 rtvFTD subjects had a strong family history for dementia.
Conclusion:
Our results demonstrate that rtvFTD, unlike svPPA, is not a pure sporadic, but a heterogeneous potential genetic variant of FTD, and screening for genetic causes for FTD should be performed in patients with rtvFTD.
Keywords
INTRODUCTION
Frontotemporal dementia (FTD) is a syndrome caused by degeneration of the frontal and/or temporal lobes [1]. Patients with predominant behavioral disturbances and frontotemporal atrophy on neuro-imaging are classified as behavioral variant FTD (bvFTD) [2], whereas the language predominant subtypes of FTD are classified under the umbrella of primary progressive aphasia (PPA) and have been associated with left hemisphere atrophy [3].
Over the years, the genetics of FTD have been broadly explored. The autosomal dominant inherita-nce pattern has been found higher in bvFTD, whereas semantic variant PPA (svPPA) is typically a non-familial sporadic disease [4 –7]. Pathogenic variants are most common in the microtubule associated protein tau gene (MAPT), the progranulin gene (GRN), and a hexanucleotide repeat expansion in the chromosome 9 open reading frame 72 gene (C9orf72), whereas a variety of rare pathogenic variants has been described as well [5].
Currently, diagnostic criteria for a variant of FTD presenting with behavioral changes, memory deficit, and prosopagnosia in the presence of right temporal atrophy (rtvFTD) are lacking [8]. Because of the atrophy pattern, theoretically, rtvFTD is considered a right variant of svPPA [3 , 10] and the general assumption is that it is also a sporadic disease.
Only one study focusing on the underlying genetic and pathological features in rtvFTD, showed a positive family history in 45% of the patients with pos-tmortem diagnostic confirmation [11]. Thus, we set out to investigate whether rtvFTD could be potentially a genetic disorder.
METHODS
In this report, out of 636 patients from the Amsterdam dementia cohort (ADC) with a clinical diagnosis of bvFTD (n = 450), non-fluent variant PPA (n = 32), logopenic variant PPA (n = 18), svPPA (n = 65), and rtvFTD (n = 71) (January 2000–November 2019) [12], we included 148 cases who had undergone gen-etic screening. Additionally, 136 FTD/PPA patients with genetic screening from the Istanbul University dementia cohort (IUDC) (November 1999-January 2020) [13] were included (total genetically screened patients, n = 284). Genetic screening was offered in case of a positive family history or when this was requested by the patient/caregiver. All included pat-ients were screened for a variant in the GRN and MAPT genes. Additionally, a subset of patients was screened for the hexanucleotide repeat expansion in the C9orf72 gene (n = 189) and/or the variants in other dementia genes with whole-exome sequencing (WES) (n = 77) (Supplementary Material 1). In 48 patients, pathogenic variants or variants of unknown significance (VUS) [14] in the FTD related genes were identified and six out of them met the clinical and the radiological characteristics of rtvFTD [8] (Supplementary Figure 1). Of note, in all subjects, the atrophy scores of the right temporal lobe [15 –17] were higher (at least 1 grade) than the left temporal lobe and the frontal lobes that were assessed by a well experienced neuroradiologist who was blind to the clinical diagnosis (FB). Additionally, in our sample, the frontal atrophy scores were less than grade-1 [16] and none of the subjects met the diagnostic criteria of svPPA [3], while all of the fulfilled at least two symptoms out of prosopagnosia, episodic memory impairment, and behavioral change [8], even if they had an accompanying left temporal atrophy on the initial MRI. All subjects gave their written informed consent for the use of their clinical and genetic data for research purposes. Details of the genetic and pathological assessment are reported in Supplementary Material 1.
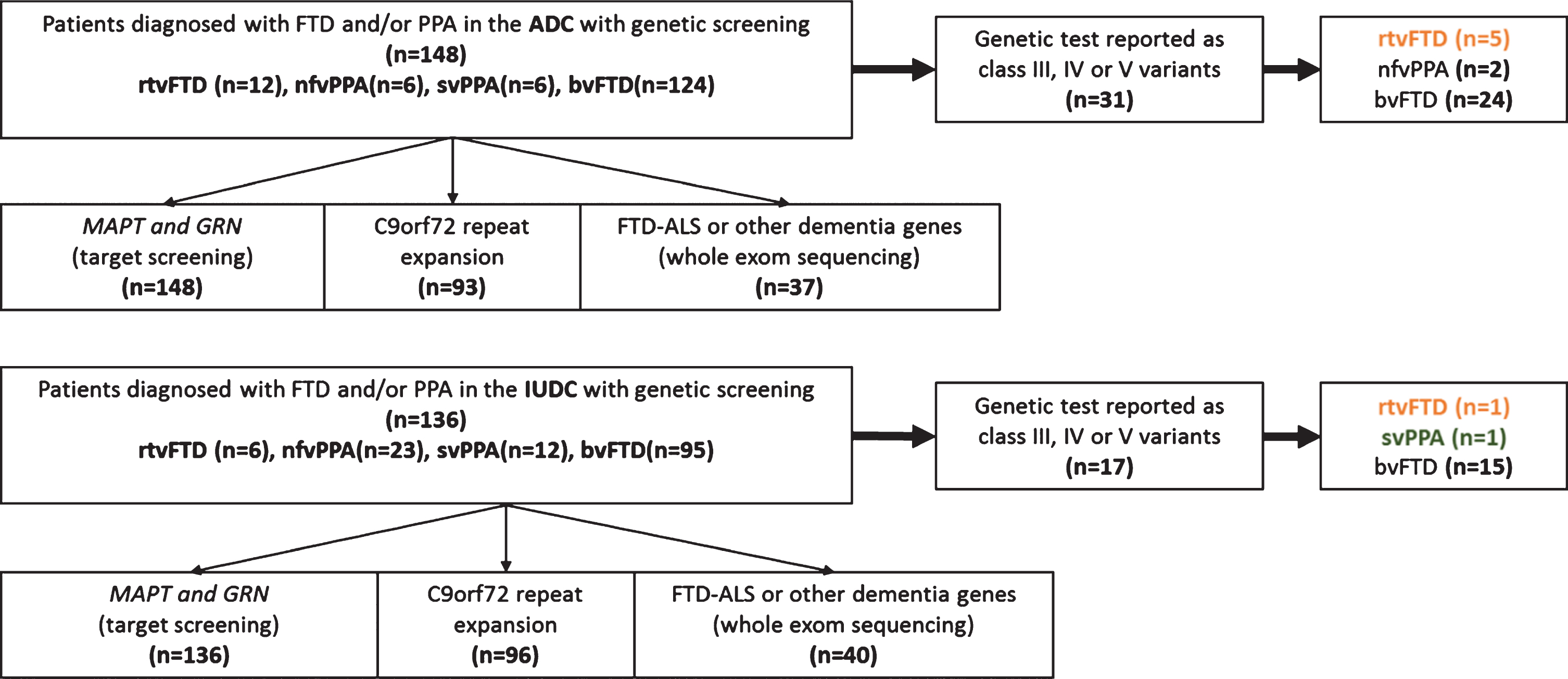
Patient selection.
RESULTS
Demographic, clinical features are displayed in Table 1, and detailed case histories are reported in Supplementary Material 2.
Demographic and clinical data
ADC, Amsterdam Dementia Cohort; IUDC, Istanbul University Dementia Cohort; MTA, mesial temporal atrophy; PET, positron emission tomography; APOE, Apolipoprotein E; CSF, cerebrospinal fluid; Aβ42, amyloid-β 42; P-tau, phospho tau; MMSE: Mini-Mental State Examination; FAB, Frontal assessment battery; TMT, Trail making test; VAT, Visual association test; RAVLT, Dutch version of the Rey Auditory Verbal Learning Test; VOSP, Visual objective and space perception; FL, Fragmented letters; L, Low; VL, Very low; HA, High average; LA, Low average; A, Average. *Cutoff value for CSF Aβ42 indicating Alzheimer’s disease pathology is <550 pg/mL, Tau >375 pg/mL, P-tau>52 pg/mL.
In our combined cohorts, genetic variants in FTD related genes were found in 33% of genetically sc-reened rtvFTD subjects (6 out of 18 genetically scr-eened rtvFTD), whereas only one svPPA (1 out of 18 genetically screened svPPA) subject had a genetic variant.
Summary of the cases
Case 1: A 59-year-old male presented with be-havioral problems, memory deficit, depression, topo-graphagnosia, and developed swallowing problems and mutism. The modified Goldman score [4] for fa-mily history was 2. We identified a heterozygous pat-hogenic variant in the GRN gene (NM 002087.3) c.388_391del, p.(Gln130Serfs*125).
Case 2: A 64-year-old female presented with prosopagnosia, behavioral changes, memory deficit, depression, and developed topographagnosia and motor restless. The modified Goldman score [4] for family history was 1. We identified a heterozygous likely pathogenic variant in the MAPT gene (NM 005910.5) c.914G>C, p.(Ser305Thr).
Case 3: A 58-year-old male presented with behavioral changes, depression, memory deficits, and dev-eloped prosopagnosia and atypical parkinsonism. The modified Goldman score [4] for family history was 4. We identified a heterozygous VUS in the MAPT gene (NM 005910.5) c.1055C>T, p.(Ser352Leu). In addition, extensive 3R and 4R tauopa-thy was reported in his autopsy which is suggestive for a pathogenic mutation in the MAPT gene [18] (Fig. 2).
Case 4: A 53-year-old female presented with memory deficits, depression, apathy, and developed anomia and several behavioral problems. The modified Goldman score [4] for family history was 1. We identified a heterozygous pathogenic variant in the MAPT gene, (NM 005910.5) c.1216C>T, p.(Arg406Trp).
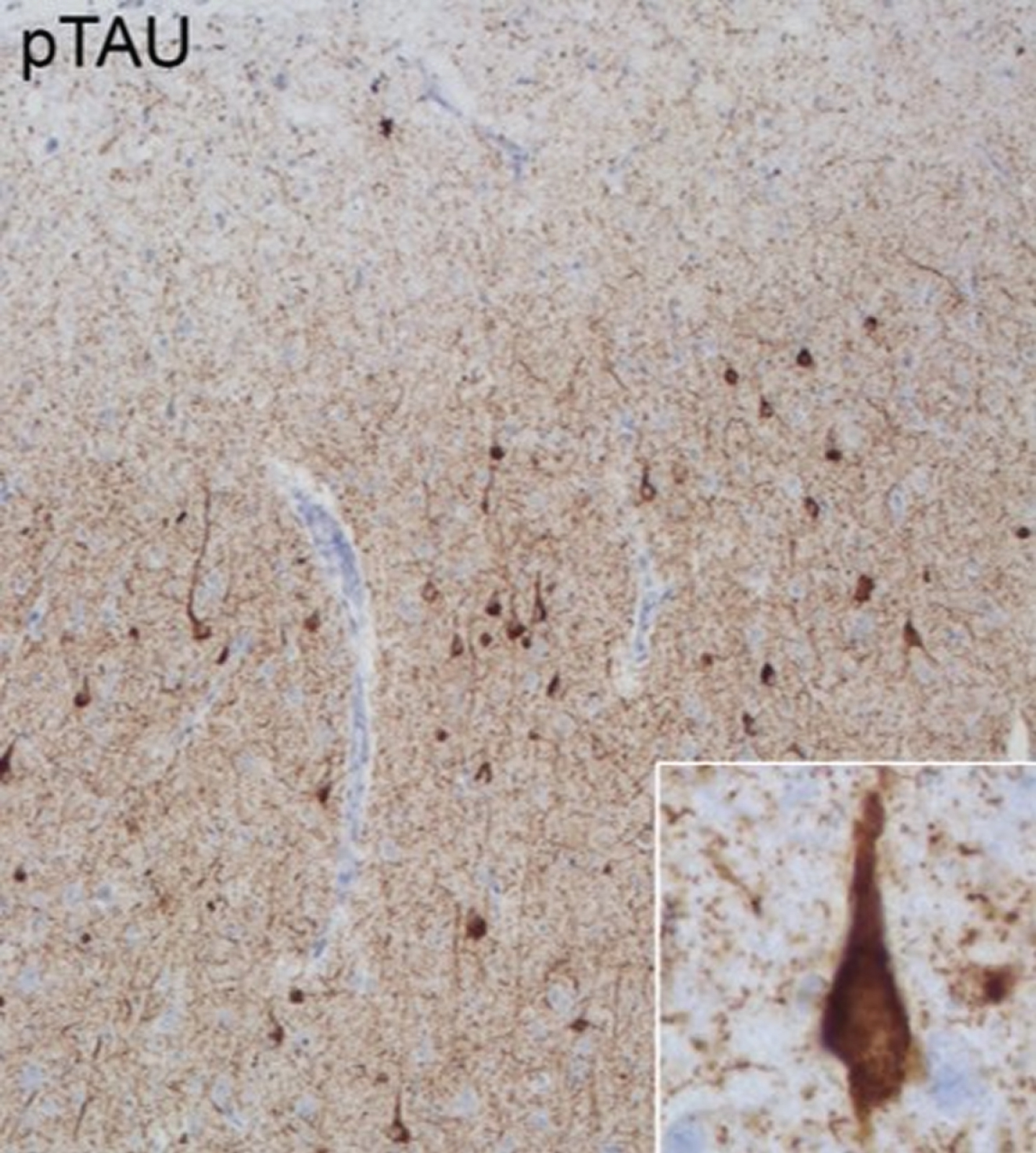
Pathological features of Case 3. Anterior cingulate cortex stained with phospho-tau (p-tau) monoclonal antibody (AT8: Pierce Biotechnology, Rockford, IL, USA). Extensive 3R and 4R tauopathy which is characteristic for MAPT related frontotemporal lobar degeneration is observed in neurons across all layers.
Case 5: A 63-year-old male presented with beh-avioral changes, prosopagnosia, anomia, and single word comprehension deficit, and developed topogra-phagnosia. The modified Goldman score [4] for family history was 1. We identified a heterozygous pathogenic variant in the MAPT gene (NM 005910.5) c.902C>T p.(Pro301Leu).
Case 6: A 58-year-old female presented with somatic and behavioral problems, memory deficit, and motor restless. The modified Goldman score [4] for family history was 3. We identified a heterozygous VUS in the TARDBP gene, (NM007375.3) c.1147A>G, p.(Ile383Val).
DISCUSSION
RtvFTD and svPPA are generally considered sporadic, non familial variants of FTD. In our combined cohorts, we can confirm that in svPPA rarely (∼5%) class III-V genetic variants in FTD related genes are found. However, 33% of rtvFTD patients that were screened for genetic mutations in FTD genes had a genetic variant. Moreover, these variants were in three different genes (MAPT, GRN, and TARDBP). This demonstrates that rtvFTD patients, unlike svPPA, are a heterogenous group that should be screened for genetic mutations.
The genetic diagnosis of four out of six rtvFTD cases was FTLD-MAPT. Previous clinico-rad-iological studies have shown that FTLD-MAPT links to bilateral anterior temporal atrophy [19], which might include rtvFTD. Moreover, the relationship between rtvFTD and MAPT mutations has been previously reported [11].
Besides the MAPT gene, the association between rtvFTD with variants in the GRN gene has been confirmed in separate case reports [20 –23]. In many cases with a variant in the GRN gene, the asymmetric atrophy extends to the parietal lobe, which was not the case in our patient. Our finding underscores the observation that a pathogenic variant status in the GRN gene may be associated with an asymmetric atrophy pattern [24, 25], which can also involve uniquely the temporal lobe.
Although TARDBP gene mutations have been described in sporadic and familial amyotrophic lateral sclerosis (ALS) in early studies [26, 27], it has subsequently been associated with FTD without ALS [28 –33]. Additionally, the heterozygous variant of Case 6 has been reported in subjects with temporal variant FTD without ALS [28 –30].
In our study, four out of six patients had a strong family history for dementia. In the literature, a positive family history was reported in 37.5% (15 out of 40) of patients with rtvFTD [combined Chan et al. [34] and Josephs et al. [11]]. This percentage is quite high compared to svPPA in which a suggestive family history is identified in less than 5% of patients [6, 35]
Nonetheless, it is still unknown whether rtvFTD and svPPA share the same pathophysiology. A recent GWAS metadata analysis [36] has revealed that the svPPA gene network is uniquely associated with TAR DNA binding protein 43 metabolism. From this perspective, accompanying tauopathy in rtv-FTD resembles the heterogeneous pathophysiology of bvFTD, rather than svPPA. On the other hand, although C9orf72 is the most common worldwide cause of genetic FTD [5], it should be noted that this variant was not found either in our study or other rtvFTD cohorts [11, 34]. Therefore, further research into the pathophysiological background of rtvFTD and how this relates to the other FTD subtypes is warranted.
In conclusion, currently, there is no consensus on whether rtvFTD is a mirror variant of svPPA or should be lumped with svPPA. Although reminiscent of svPPA, our findings show that rtvFTD, unlike svPPA, often has a genetic basis and the genetic variants are found in multiple genes. Therefore, genetic screening is essential in patients with rtvFTD.
Footnotes
ACKNOWLEDGMENTS
We are very grateful for the generous contribution of the patients and their relatives. Research of the Alzheimer Center Amsterdam is part of the neu-rodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. WF holds the Pasman chair. Dr. HUE has received research support from the Turkish Neurological Society. FB is supported by the NIHR biomedical research center at UCLH.