
Abstract
Background:
The link between cholesterol and Alzheimer’s disease (AD) has received much attention, as evidence suggests high levels of cholesterol might be an AD risk factor. The carriage of cholesterol and lipids through the body is mediated via lipoproteins, some of which, particularly apolipoprotein E (ApoE), are intimately linked with AD. In humans, high density lipoprotein (HDL) is regarded as a “good” lipid complex due to its ability to enable clearance of excess cholesterol via ‘cholesterol reverse transport’, although its activities in the pathogenesis of AD are poorly understood. There are several subclasses of HDL; these range from the newly formed small HDL, to much larger HDL.
Objective:
We examined the major subclasses of HDL in healthy controls, mild cognitively impaired, and AD patients who were not taking statins to determine whether there were HDL profile differences between the groups, and whether HDL subclass levels correlated with plasma amyloid-β (Aβ) levels or brain Aβ deposition.
Methods:
Samples from AIBL cohort were used in this study. HDL subclass levels were assessed by Lipoprint while Aβ1–42 levels were assessed by ELISA. Brain Aβ deposition was assessed by PET scan. Statistical analysis was performed using parametric and non-parametric tests.
Results:
We found that small HDL subclass is reduced in AD patients and it correlates with cognitive performance while plasma Aβ concentrations do not correlate with lipid profile or HDL subfraction levels.
Conclusion:
Our data indicate that AD patients exhibit altered plasma HDL profile and that HDL subclasses correlate with cognitive performances.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, pathologically characterized by the extracellular deposition of amyloid-β (Aβ) in the brain and the accumulation of hyperphosphorylated tau filaments in neurons. While a small portion of AD cases can be attributed to a genetic predisposition, sporadic or late-onset AD (LOAD) accounts for the majority of cases.
In the periphery, cholesterol is transported to the cells where it accumulates through the action of very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL), which are referred to as ‘bad cholesterol’ as their action increases the cholesterol levels and the risk for several diseases associated with it. The cholesterol balance is maintained by the action of high-density lipoproteins (HDL), which are referred to as ‘good cholesterol’, as their duty is to remove excess cholesterol by transporting it to the liver for its elimination. However, the lipid environment in the brain is very different compared to the lipid environment in the periphery, and these differences are maintained by the protective features of the blood-brain barrier (BBB), which isolates the brain from the periphery and forces the brain to generate cholesterol in situ. In the brain, cholesterol is carried by HDL-like particles, in which Apolipoprotein E (ApoE), produced by astrocytes and microglia [1–4], is the main apolipoprotein (whereas in plasma it is ApoA-I) and cholesterol-carrier in the brain, followed by ApoA-I, which is not produced in the brain but it can be transported across in the choroid plexus [5]. These differences are at the basis for four different CSF lipoprotein classes, CSF-Lp ApoA-I, CSF-Lp ApoE, CSF-Lp ApoE/ApoA-I, and CSF-Lp (without ApoE and ApoA-I) [6]. Additionally, other lipoproteins that are common in the peripheral circulation, such as LDL and VLDL, are not present in the brain environment, leaving the whole load of cholesterol transport duties to CSF-Lp particles. Finally, the elimination of excess of cholesterol requires the conversion of cholesterol to a more soluble 24S-hydroxycholesterol, which allows it to cross the BBB into the peripheral circulation, where it is picked up by plasma lipoprotein and redirected to the liver for its elimination [7].
Overall, lipid metabolism has been linked to AD pathogenesis in several ways. Several reports suggest that high levels of intracellular cholesterol cause an increase in Aβ deposition in the brain [8–10], whereas low cholesterol levels have been shown to increase processing of the amyloid-β protein precursor (AβPP) via the non-amyloidogenic pathway [11–14]. In addition, longitudinal studies have shown that mid-life obesity and cardiovascular disease are also risk factors for AD [15, 16] and peripheral biomarker studies have found preclinical AD-related biomarkers are associated with lipid metabolism [17–19]. To date, studies of cholesterol levels in AD have produced conflicting results. Some studies have found increased levels of cholesterol in AD [20–22], while other reports have not [23–25]. It has also been reported that individuals who subsequently develop AD exhibit decreased cholesterol levels before clinical manifestation of the disease [26]. Despite such reports, hypercholesterolemia is still considered an early risk factor for developing AD [27] and has been associated with impaired memory recall in the elderly [28]. In support of these studies, mice fed with high-fat diets exhibit altered brain mass and amyloid levels in the brain [9, 30], while elevated plasma triglyceride (TG) levels have been shown to precede amyloid deposition [31]. Consistent with the notion that high cholesterol levels increase disease risk, statins have shown the potential to reduce the risk for AD, though their effectiveness appears to require administration well before clinical manifestation of symptoms [32–35]. More recently, statins have also been shown to be capable of raising HDL levels, most likely through a mechanism independent of lowering LDL levels [36, 37]. There are studies which have reported altered HDL levels in AD compared to controls [21, 22], and some that have reported no such association [23, 38]. It is, however, important to note that low plasma HDL levels have recently been associated with higher cerebral Aβ deposition [39] and that ApoA-I, the major constituent of plasma HDL, has been reported to be lower in AD compared to controls [40–43]. ApoA-I binds to the Aβ peptide as well as its precursor protein AβPP, and has been shown to reduce Aβ-induced toxicity and Aβ aggregation into β-pleated sheets [44, 45]. In AD mouse models, ApoA-I overexpression reduces cognitive deterioration and neuroinflammation, whereas its deficiency increases cognitive defects and cerebral amyloid angiopathy [46, 47].
It is most likely the first clear link between AD and lipid metabolism was reported when APOE polymorphisms were shown to influence AD [48]. The APOE gene is located on chromosome 19, and there are three allelic variations of APOE (ɛ2, ɛ3, ɛ4), with the possession of the ɛ4 allele (APOE ɛ4) to be the predominant genetic risk factor for the late-onset form of the disease (carried by ∼50% of all sporadic AD patients, yet is only found at a frequency of around 14% in the general population), whereas possession of the APOE ɛ2 allele appears to be protective [49, 50]. It is still not clear how possession of the APOE ɛ4 allele increases AD risk, but ApoE appears to be required for Aβ aggregation, and ApoE ɛ4 has been shown to be the most efficient at promoting such oligomerization; ApoE also helps remove Aβ from the brain to the periphery, yet again, ApoE ɛ4 is the least efficient at this task [51, 52]. The ɛ4 allele is also the least efficient in promoting cholesterol efflux from neuronal cells [53], and cholesterol bound to ApoE ɛ4 displays a lower rate of cellular uptake [54].
Clearly, there is still a need for further studies to increase our understanding of changes to plasma cholesterol levels in AD. Therefore, we evaluated plasma cholesterol and lipoproteins further, by investigating plasma lipoprotein subgroups, to provide more insight into the early pathogenic events involved in the development of AD. An aspect overlooked by most previous studies is that cholesterol is transported by at least four different subclasses of lipoproteins in the blood stream: chylomicrons, VLDL, LDL, and HDL. HDL also exists in different forms, from the newly generated HDL small (which have been reported to display anti-oxidant features), to the more mature HDL large. Changes in the levels of these subclasses may be missed if not measured individually. In this study, we evaluated the plasma levels of HDL subclasses, as well as levels of cholesterol, LDL, total HDL, and TG, to determine whether more detailed lipid profiles would reveal differences between healthy controls, MCI, and AD patients. In addition, we evaluated the cerebral Aβ deposition in this cohort, measured as neocortical standardized uptake value ratio (SUVR) score from 11C-PiB positron emission tomography (PET) analysis and investigated correlations with regards to HDL levels to determine whether HDL subclasses levels could reflect brain amyloid deposition.
MATERIALS AND METHODS
The AIBL study was approved by the ethics committees of St. Vincent’s Health and Austin Health in Melbourne, Hollywood Private Hospital and Edith Cowan University in Perth (Australia). All volunteers gave written and informed consent before participating in our study. A total of 486 participants, divided into healthy controls (HC), mild cognitive impaired (MCI), and Alzheimer’s patients (AD) from the AIBL cohort were used, after ensuring that this subset was not undergoing statin treatment, and that conversion to a different status did not occur in the following 18 months. A full description of the recruitment process has already been published. Exclusion criteria included a history of non-AD dementia, schizophrenia, bipolar disorder, current depression (GDS score above 5/15), Parkinson’s disease, uncontrolled hypertension (systolic BP > 170 or diastolic BP > 100), cancer (other than basal cell skin carcinoma) within the last two years, symptomatic stroke, uncontrolled diabetes, or current regular alcohol use exceeding two standard drinks per day for women or four per day for men [55]. The AIBL Study clinical panel meets on a monthly basis to discuss baseline classification for each set of patients recently tested and ensures diagnoses were made in accordance with the NINCDS-ARDA criteria [56, 57]. Various body parameters were evaluated at the examinations, such as weight, height, blood pressure, and pulse rate. Blood was drawn from overnight fasting participants and collected to obtain plasma or serum for our analysis. APOE status was determined by genotyping cells from whole blood as previously described [17]. Total cholesterol, LDL, HDL, and TG levels were assessed in plasma.
The Lipoprint system (Quantimetrix, Redondo Beach, CA, USA) was used to examine the serum HDL profiles of study participants following the kit instructions. Briefly, 25μl samples of serum were combined with 200μL of the supplied loading buffer in glass gel tubes and allowed to polymerize for 30 min. Samples were then separated in an electrophoretic chamber for 1–1.5 h at 3 mA per tube. Finally, gels were scanned and the band intensity of each subfraction was obtained using the supplied software. The HDL analysis provided values for 10 subclasses of HDL which we divided into three major groups named HDL Large (subclasses 1 through 3), HDL Intermediate (subclasses 4–6), and HDL Small (subclasses 7–10). Initial analysis indicated a good correlation between HDL Large and HDL-2 (1.063–1.125 g/ml) and between HDL Intermediate + Small and HDL-3 (1.125–1.21 g/ml) (personal communication). Upon assessing the subclass percentage in serum, absolute levels (mg/dl) in each subclass (L, I, and S) were determined using the plasma HDL concentration measured in the initial blood drawn analysis which was routinely performed in all participants. Although plasma and serum are different biological fluids, it has been reported that HDL concentration is almost identical [58, 59].
Plasma Aβ1–42 concentrations were measured using a commercially available ELISA kit (INNO-BIA, Innogenetics, Gent, Belgium) following manufacturer’s instructions.
PET scans consisting of 30 min acquisitions were performed 40 min after injection of 370 MBq 11C-PiB. PET images were processed using a semi-automatic region-of-interested method as previously described [60]. Standardized uptake values (SUV) for 11C-PiB were calculated for all brain regions examined. The SUV ratio (SUVR) was calculated dividing all regional SUV by the cerebellar cortex SUV. However, the centiloid scale was recently proposed to provide a standard quantification of Aβ-PET images. In the centiloid scale, the Aβ burden can be expressed with values ranging from 0 (the typical Aβ burden in young controls) to 100 (the typical Aβ burden in mild AD patients) [61]. Centiloid values were generated using CapAIBL as described elsewhere [62].
Statistical comparison of means in different groups was based on ANCOVA (Analysis of Covariance) where adjustment was made for covariates such as age, gender, site, and APOE status. For non-normal distributions, a non-parametric ANOVA (Kruskal-Wallis) and non-parametric U Test (Mann-Whitney) were used. Associations between continuous variates were assessed using Spearman’s correlation. Due to the non-normal distribution of our variables, most of our analysis were performed using non-parametric tests, which limits our power to adjust for several covariates. We acknowledge this is a limitation of our study. A p-value less than 0.05 was regarded as significant. Analyses were carried out using TIBCO Spotfire S+ version 8.2 (Boston, MA) and SPSS version 25 (Chicago, IL, USA).
RESULTS
The basic demographics of the study participants are summarized in Table 1. In total, 347 HC, 55 MCI, and 84 AD patients were studied (all participants were 65-years old and older). All samples were evaluated for total cholesterol, LDL, HDL, and TG levels. Using Kruskal-Wallis analysis, we compared the levels of cholesterol, LDL, HDL, and triglycerides among HC, MCI, and AD patients and we did not observe any significant difference (Table 1). Additionally, the effect of APOE genotype was assessed in each clinical group by Mann-Whitney U Test. The levels of cholesterol and LDL were significantly higher APOE ɛ4-carrier in HC only, but not in MCI or AD. Conversely, HDL and TG levels were not affected by the presence of APOE ɛ4 allele in any clinical group (Supplementary Table 1). To determine whether the number of APOE ɛ4 alleles affected the lipid profile, we performed the Kruskal-Wallis analysis in each clinical group. Again, the cholesterol and LDL levels were significantly affected by APOE genotype in HC only with APOE ɛ4 homozygous individuals displaying highest levels of cholesterol and LDL. HDL and TG levels were not affected by the number of APOE ɛ4 alleles in any clinical groups (data not shown).
Comparison of demographic characteristics and cholesterol, LDL, HDL, and triglyceride levels among HC, MCI, and AD participants
Values are presented as mean±S.D or as frequency. Non-parametric ANOVA (Kruskal-Wallis analysis) was performed.
The levels of HDL subgroups, expressed as a percentage of the total HDL or in terms of concentration, are listed in Table 2 (top). These analyses were performed using ANCOVA for normal distributions (HDL L% and HDL I%) or Kruskal-Wallis analysis for non-normal distributions (HDL S%, HDL L mg/dl, HDL I mg/dl, and HDL S mg/dl). Using the data reported in percentage (Table 2, bottom), analyses found that differences between the HC, MCI, and AD patients are associated with the HDL Small group only (p < 0.001). A pairwise comparison with Mann-Whitney U Test within the HDL Small group shows that the AD participants exhibit significantly less HDL Small particles when compared to the HC (p < 0.001) (–26%) and MCI (p = 0.004) (–25%) participants. In contrast, comparison of HC with MCI participants showed no differences (p = 0.77). Conversely, there were no significant differences associated with HDL Large (p = 0.10) or HDL Intermediate (p = 0.21). Analysis of the absolute values (Table 2, bottom) resulted in the same conclusion; significant differences associated with the HDL Small values (p = <0.001), while no differences are associated with HDL Large (p = 0.12) or HDL Intermediate (p = 0.56). A further post hoc analysis of the HDL Small group demonstrated the same decrease in HDL Small particles in AD versus HC (p < 0.001) (–24%) and in AD versus MCI (p = 0.006) (–21%), while HC versus MCI displayed no significant difference (p = 0.58). The significance of our results was not affected by performing the same analysis in non-smokers only, which removed an important factor known to modulate HDL [63] (HDL S% : Kruskal-Wallis p < 0.001; Mann-Whitney U Test, HC versus AD p < 0.001, MCI versus AD p = 0.009; HDL S mg/dl: Kruskal-Wallis p < 0.001; Mann-Whitney U Test, HC versus AD p < 0.001, MCI versus AD p = 0.026). We have also performed the same analysis in APOE ɛ4-carriers and APOE ɛ4-non-carriers but we did not find any difference between those groups (Supplementary Table 2). A more detailed analysis assessing the number of APOE ɛ4 alleles (ANCOVA for HDL L% and I% and Kruskal-Wallis for HDL S% and HDL L, I, S mg/dl) did not alter our previous findings in HC and AD, suggesting that HDL subclasses distributions are not affected by APOE genotype in these clinical groups. In MCI, the distribution of HDL L or I (expressed as % or mg/dl) was not affected by the number of APOE ɛ4 alleles. However, Kruskal-Wallis analysis unexpectedly indicated that in MCI, HDL S% subclasses are affected by APOE genotype (p = 0.03) (with APOE ɛ4 homozygous displaying the highest levels), while HDL S mg/dl distribution are not (p = 0.107) (data not shown). However, the low number of APOE ɛ4 homozygous (n = 6) in the MCI group may have affected the results and further analysis is needed.
Comparison of HDL sub-distribution in the HC, MCI, and AD groups, expressed as % or mg/dl
ANCOVA (analyses were adjusted for sex, age, site, and APOE ɛ4-carrier status) or non-parametric ANOVA (Kruskal-Wallis analysis) were used and regarded as significant when p < 0.05 (bold). When Kruskal-Wallis analysis was significant, individual comparison was performed using Mann-Whitney U Test and considered significant when p < 0.05 (bold). For HDL S (% and mg/dl), HDL L (mg/dl) and HDL I (mg/dl) non-parametric tests were used. Values are presented as mean±S.D.
Based on the evidence that HDL Small levels were affected by clinical classifications, with levels lower in AD versus HC, we also evaluated if HDL subclass levels were affected by brain amyloid deposition in HC (HC Aβ–versus HC Aβ+). These analyses were performed using ANCOVA for normal distributions (HDL L% and HDL I%) or Mann-Whitney U Test analysis for non-normal distributions (HDL S%, HDL L mg/dl, HDL I mg/dl, and HDL S mg/dl). As shown in Table 3 (top), there was no different distribution of any HDL subclass with regards to brain amyloid deposition. ANCOVA analysis (performed adjusted for age, gender, site, and APOE status) or Mann-Whitney U Test analysis did not reveal any significant difference (Table 3, bottom).
Comparison of HDL sub-distribution in HC with low (Aβ–) and high (Aβ+) Aβ deposition, expressed as % or mg/dl
ANCOVA (analyses were adjusted for sex, age, site, and APOE ɛ4-carrier status) or non-parametric Mann-Whitney U Test were used and regarded as significant when p < 0.05. For HDL S (% and mg/dl), HDL L (mg/dl) and HDL I (mg/dl) non-parametric tests were used. Values are presented as mean±S.D.
Using Spearman’s correlation, SUVR-Centiloid values were correlated with HDL or HDL subfractions (HDL Large, Intermediate, and Small) to determine whether alteration in the HDL profile was associated with a different SUVR score in HC and in combined MCI/AD groups. No significant association with HDL, HDL Intermediate, or HDL Small in any of the groups, nor with HDL Large in HC was observed (Table 4, top). However, there was a significant negative association between SUVR and HDL Large in the MCI/AD group only (p = 0.037). We also evaluated if plasma levels of Aβ1–42 correlated with HDL or HDL subclasses in HC or MCI/AD. Again, we did not find any significant correlation between plasma levels of Aβ1–42 and HDL (or any HDL subclasses) in any clinical classification (Table 4, bottom).
Correlations between brain Aβ deposition, plasma Aβ1–42 levels, and HDL subclasses
Spearman’s correlation evaluating brain amyloidosis or plasma Aβ1–42 levels with HDL (or HDL subgroups) was performed in HC, MCI, and AD and considered significant when p < 0.05 (bold).
Cognitive performance scores (MMSE) were correlated with the HDL or HDL subfractions in HC and MCI/AD groups to determine whether cognitive performances were associated with HDL profile. HDL Small levels significantly positively correlated with MMSE in the MCI/AD group (ρ= 0.171, p = 0.044), but not in the HC group (ρ= –0.052, p = 0.334). However, we did not find any significant association between HDL, HDL Large and HDL Intermediate with MMSE in any clinical group (in HC: ρ= –0.001, p = 0.989; ρ= –0.004, p = 0.934; and ρ= 0.001, p = 0.986 for HDL, HDL L, and HDL I, respectively; in MCI/AD: ρ= 0.070, p = 0.416; ρ= –0.038, p = 0.653; and ρ= 0.051, p = 0.551 for HDL, HDL L, and HDL I, respectively (Fig. 1).
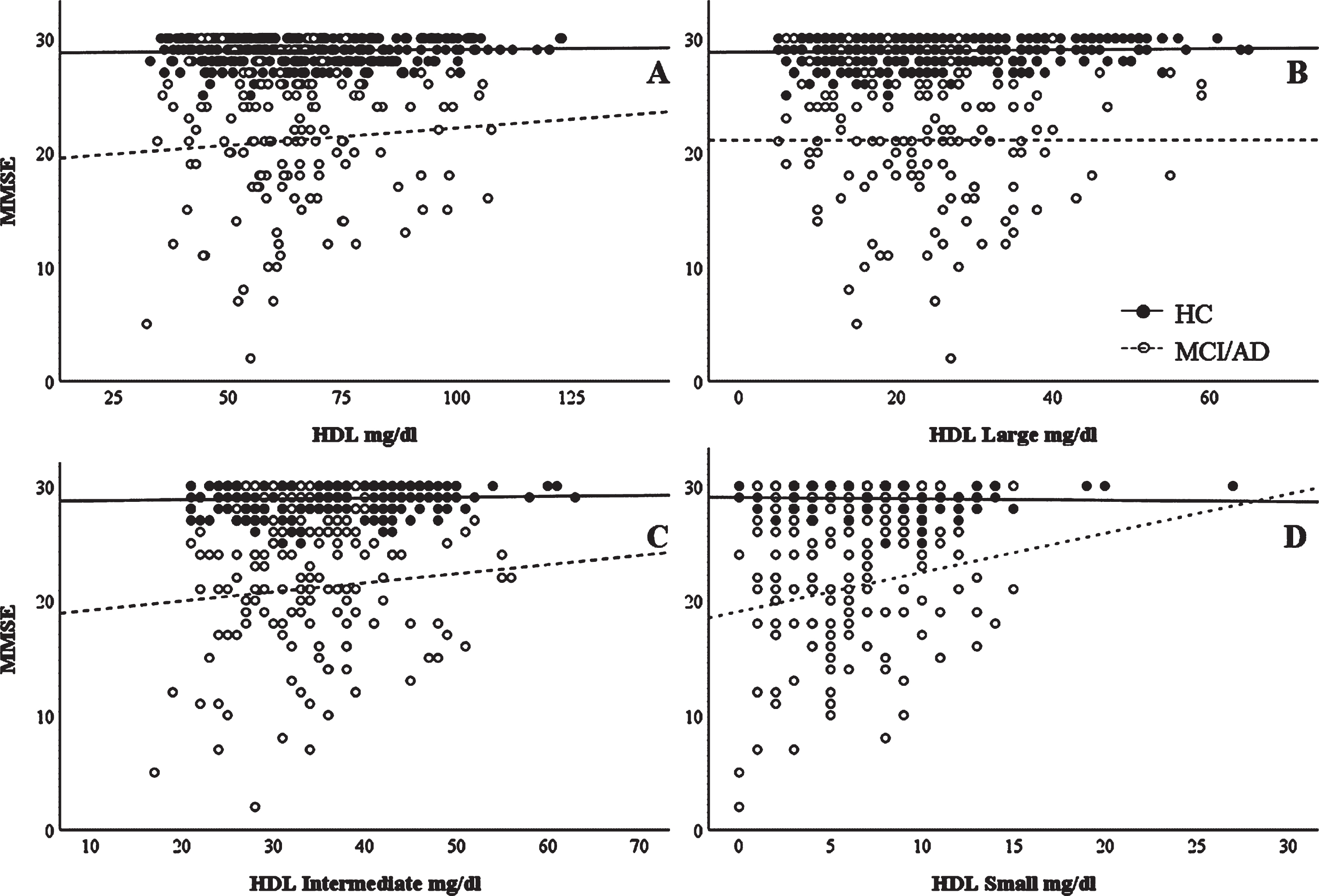
Correlations between MMSE scores and HDL subclasses. Unadjusted correlation between MMSE scores and (A) HDL, (B) HDL Large, (C) HDL Intermediate, and (D) HDL Small subclasses. Spearman’s correlation data are reported in the text.
DISCUSSION
The finding over two decades ago that APOE polymorphisms can influence the risk of AD strongly implied a link between lipid metabolism, particularly cholesterol metabolism, and AD. The later findings that brain Aβ plaque deposits can be found commonly in coronary artery disease (CAD) as well as in hypertension patients strengthened this concept, suggesting a neuropathologic link between CAD, hypertension, and AD. It is now known that many related conditions, including CAD, hypertension, mid-life obesity, type 2 diabetes, and insulin resistance all increase AD risk to some extent [64].
However, cholesterol metabolism is substantially different in the periphery and in the brain. While in the periphery, chylomicrons, VLDL, LDL, and HDL are all responsible for cholesterol transport, in the brain the whole burden is carried by HDL-like particles. Additionally, these HDL-like particles have ApoE generated in the brain as the major apolipoprotein, while ApoA-I relies upon transport across the choroid plexus to enter the brain as it is not generated in situ. Furthermore, cholesterol in the brain can be eliminated only after conversion to 24S-hydroxycholesterol, which allows it to cross the BBB and reach the periphery where it is directed to the liver for its excretion [7]. These differences altogether indicate that with regards to cholesterol and its metabolism, the periphery and the brain may display different behaviors that can modulate the risk for AD in different ways.
The notion of a linkage between cholesterol and AD is reported in studies indicating that high-fat diets in animal models induce the deposition of Aβ [9, 66]. Other studies looking for a mechanistic link found that cholesterol can modulate AβPP processing toward the amyloidogenic pathway [27, 67–69], and the conclusions from all these studies were that high levels of cholesterol in the circulation are most likely a risk factor for AD.
Many studies have tested the effects of lowering cholesterol levels on AD or AD risk through the use of statins, either in clinical trials or in vitro studies; however, results have been inconsistent [11–13, 70–74]. A possible reason for the conflicting data may be due to the way in which statins function. In clinical studies, many reports linked the use of statins to a decreased risk for AD, whereas other reports did not observe changes in symptoms, or any altered AβPP processing in AD patients. Overall, the results suggest that statins may be protective before the onset of the disease but become less efficient or ineffective once the disease has started [32–35]. Interestingly, while statins may have beneficial effects in preventing AD due to cholesterol-lowering properties, this may have little to do with cholesterol effects on Aβ production in the brain, as positive results were seen in some studies despite the fact that the statins being tested were not able to cross the BBB, hence effects must have been due to peripheral changes. Alternatively, statins may be able to reduce AD risk due to influences on other biochemical pathways, for example, statins also alter the isoprenylation of small GTPases, another pathway that has been linked to AD [75–78]. In order to eliminate this confounding factor, our sample population excluded all participants who were undergoing statin treatment. In our cohort, we did not observe any marked alteration in total cholesterol, total LDL, total HDL, or TG levels (Table 1) between the different groups. Other studies have reported mostly unchanged levels of lipoproteins, with one or two exceptions. For example, one study found that the charge-based major LDL subfraction, as characterized by capillary isotachophoresis, was associated with both MCI and AD, yet did not find differences in other lipoprotein classes [22]. Another study detected mild hypercholesterolemia in AD patients, with no changes in triglycerides [20–22]. One study demonstrated that low levels of HDL are associated with lower grey matter volume in cognitively healthy adults [79].
It is important to note that comparisons of apparently similar studies are complicated to an extent by the presence of other conditions, and also by the type of lipoprotein measure used, such as HDL-cholesterol levels versus HDL particle levels. For example, in one study of a cohort enriched for cerebrovascular disease and elevated vascular risk, statistical models that controlled for age and APOE ɛ4 alleles revealed independent associations among the levels of LDL-cholesterol, HDL-cholesterol, and the level of Aβ deposition as measured by PiB-PET: in this study, higher LDL-cholesterol and lower HDL-cholesterol levels were both associated with a higher Aβ deposition [39].
Intrigued by the apparent lack of change in total HDL levels, we then compared levels of the respective HDL subclasses between AD, MCI, and controls. As shown in Table 2, HDL exhibited a different profile in the plasma of AD participants. Examination of the HDL profiles revealed lower levels of the smaller HDL particles (HDL Small) in AD participants compared with the MCI or healthy controls. Other studies of plasma HDL have reported, for example: lower overall levels of HDL in vascular dementia, but not AD [21, 80], lower levels of HDL in AD with cardiovascular comorbidities but not in AD alone [81], or lower levels of HDL-cholesterol and ApoA-I in AD [82], showing that results have not been conclusive. These studies did not investigate HDL subclasses, however, and further lipoprotein subclass studies such as ours may reveal more information concerning plasma lipoprotein changes in various conditions. It is interesting to note that these small, dense HDL particles have been shown to be beneficial against atherosclerosis and vascular related oxidative stress [83], which fits with the hypothesis that AD is intimately linked to metabolic syndrome [84]. As metabolic syndrome (as well as type 2 diabetes) linked oxidative stress and vascular damage (leading to vascular insufficiency) are believed to be risk factors for AD, more detailed studies might reveal other changes in small HDL particles which occur at earlier stages of AD pathogenesis. In accordance with the notion that small HDL display protective features, our data indicated that higher levels of HDL small particles were significantly associated with higher MMSE scores in MCI/AD, suggesting a beneficial role of these small dense HDL in the disease.
In other studies, aging has been shown to affect the composition and function of HDL [85], and HDL levels in general have been shown to decrease with aging [86]. In people with exceptional longevity (age > 95), lower HDL levels do appear to be associated with lower cognitive function [85–89]. Furthermore, phospholipid transfer protein (PLTP) is responsible for HDL remodeling and HDL enlargement, and it is also possible that lower levels of small HDL in AD are a consequence of higher PLTP activity, which has already been reported in AD [90]. Additionally, cholesterol efflux has been shown to be abnormal in aging and has been associated with decreased levels of HDL in aged individuals [86].
It has been reported that increased cerebral Aβ deposition is associated with high levels of LDL and low levels of HDL [39]; however, our analysis did not find any association between Aβ deposition levels and the levels of cholesterol, LDL, HDL, or TG in any clinical group (data not shown). We then evaluated whether levels of the HDL subgroups correlated with levels of Aβ deposition in the brain. From our results, cerebral Aβ deposition level appears to be independent of levels of HDL sub-fractions in almost every clinical group, with the sole exception of a significant negative association between SUVR and HDL Large in the MCI/AD group (p = 0.037). Additionally, we did not observe any correlation between Aβ1–42 plasma levels and HDL or HDL subclasses. We have also considered HDL subclasses as potential biomarkers for HC with high amyloidosis for whom the conversion to AD is more likely. However, we did not observe any different HDL subclasses distribution with regards to brain amyloid deposition. These data indicated that, at least in our cohort, in spite of an altered HDL metabolism, there is no correlation between HDL subclasses levels and brain amyloid deposition or plasma levels of Aβ1–42. However, we cannot exclude that a more detailed HDL analysis that includes additional factor would unveil a link between HDL and amyloid deposition. The fact that HDL subclasses are altered in AD but not in HC Aβ+ suggests that an altered HDL metabolism may be a consequence of the disease progression.
Taken together, our data support previous studies which have shown that patients with AD exhibit alterations in their plasma lipoprotein profile, and we suggest that HDL changes can be attributed to lower levels of small HDL particles. Further studies will be necessary to determine at what stage of AD pathogenesis these alterations to small HDL occur, and how they are involved in the progression of the disease.
Footnotes
ACKNOWLEDGMENTS
We thank all the participants who took part in this study and the clinicians who referred participants. The AIBL study (http://www.AIBL.csiro.au) is a collaboration between CSIRO, Edith Cowan University (ECU), The Florey Institute of Neuroscience and Mental Health (FINMH), National Ageing Research Institute (NARI) and Austin Health. The authors acknowledge the financial support of the Cooperative Research Centre for Mental Health (CRCMH), the CRCMH program is an Australian Government Initiative, McCusker Alzheimer’s Research Foundation, Alzheimer’s Australia Research Foundation (AARF), the Science and Industry Endowment Fund, CSIRO, Brightfocus, USA and the WA Dept. of Health. FINMH acknowledges the funding support from the Victorian Government’s Operational Infrastructure Support program. Pfizer International has contributed financial support to assist with analysis of blood samples and to further the AIBL research program. Authors’ disclosures available online (![]() ).
).