
Abstract
Alzheimer’s disease is the most common neurodegenerative disorder that can cause dementia in elderly over 60 years of age. One of the disease hallmarks is oxidative stress which interconnects with other processes such as amyloid-β deposition, tau hyperphosphorylation, and tangle formation. This review discusses current thoughts on molecular mechanisms that may relate oxidative stress to Alzheimer’s disease and identifies genetic factors observed from in vitro, in vivo, and clinical studies that may be associated with Alzheimer’s disease-related oxidative stress.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder with memory deficits and executive dysfunction as characteristic clinical features [1]. Investigations show that hallmarks of oxidative stress are observed early in the progress of AD [2–7]. Related to this, pre-symptomatic AD has been associated with mitochondrial deficiency resulting in disturbed bioenergetics [8]. Apart from reducing the generation of ATP, mitochondrial deficiency results in excessive production of reactive oxygen species (ROS). These ROS, in turn, have been related to membrane damage, cytoskeletal alterations, and cell death [9]. Other than features of oxidative stress, progress of AD is characterized by extracellular accumulation of aggregated amyloid-β (Aβ), and intracellular neurofibrillary tangles containing hyperphosphorylated tau. The precise nature of the association between oxidative stress and other hallmarks of AD pathology is unknown although some molecular mechanisms have been suggested which will be discussed in this review. Further, the excessive generation of ROS as well as the neutralization of their damaging effects in a neurodegenerative condition such as AD will be covered.
OXIDATIVE STRESS
Oxidative stress is a state in which either increased levels of cellular ROS are generated and/or cellular mechanisms to reduce the potentially damaging impact of ROS are of insufficient capacity [10, 11]. This definition inherently dictates that as long as the antioxidant defense system is sufficiently capable of scavenging the generated reactive species, increased ROS or reactive nitrogen species (RNS) production generally does not provide sufficient leverage to cause pathology. However, in some cases aging-related increased production of free radical species coincides with a decreased capacity of the endogenous antioxidant defense system [12, 13]. Even though normal aging coincides with increased levels of oxidative stress, a very diverse range of diseases demonstrates a more pronounced level of oxidative stress including attention deficit hyperactivity disorder (ADHD) [14], cancer (e.g., [15]), Parkinson’s disease [16], atherosclerosis [17], myocardial infarction [18], sickle cell disease [19], Down’s syndrome [20], depression [21], and diabetes mellitus [22].
OXIDATIVE DAMAGE TO BIOMACROMOLECULES IN ALZHEIMER’S DISEASE
Even though low levels of ROS are crucial for normal physiological functioning, increased ROS levels are associated with oxidative damage of various cellular compartments and molecules. For example, structural and functional impairments of membrane-associated macromolecules such as lipids and proteins in several regions of the brain have been observed in response to ROS associated damage [23–25]. Analysis of lipid rafts in AD brain tissue samples showed that increased levels of membrane-associated oxidative stress correlated with accumulation of cholesterol and ceramides into clustered microdomains which could be prevented by the antioxidant vitamin E and ceramide inhibitors [26]. The mechanisms by which such microdomains assemble have been elaborately studied but perhaps one of the key observations was that extracellular Aβ aggregation in close proximity of the cell membrane induces membrane-associated oxidative stress. Membrane-associated oxidative stress involves lipid peroxidation and generation of aldehyde 4-hydroxynonenal (HNE), a neurotoxic aldehyde that can be detected at early stages of disease progress in the AD brain [27]. Interestingly, HNE levels were observed to be proportional to the extent of neuronal lesions [28, 29]. Oxidative stress can also lead to activation of pathways involved in AD pathogenesis. For example, one member of the mitogen-activated protein kinases (MAPKs) family, namely p38, is activated during Aβ-mediated oxidative stress. Among the different roles of p38, it was observed to induce tau phosphorylation in a primary neuronal model, which could be prevented by pretreatment with an inhibitor of p38 or vitamin E [30]. These findings were confirmed in vivo using a transgenic APP/PS1 mouse model for AD [31]. AD-related oxidative stress is also reflected by extensive oxidative damage to nucleic acids leading to alterations in DNA structure [32, 33]. Apart from in AD, oxidation of mitochondrial DNA and RNA are observed in a number of other pathologies [34]. One feature of DNA/RNA oxidation is the oxidation of the base guanosine to produce 8-hydroxyguanosine (8-oxoG) [35]. High levels of 8-oxoG were observed in neurons within the hippocampus, subiculum, entorhinal cortex, and frontal, temporal, and occipital neocortex in autoptic brain tissues of patients affected by AD [36]. Moreover, RNA oxidation was found to be significantly increased in the hippocampus, cortical neurons, white matter and in the frontoparietal cortex of aged rats [37]. These findings imply a role of oxidative-stress induced damage of DNA and RNA in neurodegenerative disease and aging.
Also, Aβ and tau have been reported to undergo a number of modifications as a function of oxidative stress. Tau plays a role in microtubule organization by dynamically interacting with the formed microtubules [38]. Intracellular dynamics of microtubule organization were observed to be disrupted in AD patients [39]. Various cell lines, including ventricular myocytes, neuro-2A cells, rat pheochromocytoma PC12, and pancreatic epithelial cell line AR42J, when exposed to H2O2 or HNE, show a decreased growth of the microtubular network as a result of increased microtubular catastrophe rate [40–45] largely mediated by Michael addition reactions [45]. This paragraph discusses the types of modification that tau and Aβ are subject to under conditions of oxidative stress.
Copper-induced dityrosine cross-linking of Aβ
A specific type of Aβ assembly involves dityrosine cross-linking which has been associated with clinical markers of oxidative stress in AD but also other neurodegenerative diseases [46]. Increased levels of oxidative stress in the brain are reflected by increased brain content of copper (Cu) and zinc (Zn), specifically in the neuropil and in AD plaques [47, 48]. Copper was shown to catalyze hydroxyl radical, peroxynitrite, nitrosoperoxycarbonate, and lipid hydroperoxide-mediated dityrosine cross-linking [49, 50] in monomeric and, at a lower rate, fibrillar Aβ1-40 [51] in a concentration-dependent manner [51]. The precise mechanism of crosslinking has been subject of study [52], but it was shown that the picomolar affinity of Aβ for copper [53] drives the generation of H2O2, which, in turn, promotes the formation of SDS-resistant dityrosine cross-linked Aβ1-28, Aβ1-40, and Aβ1-42 [54, 55]. It has also been shown that Aβ1-42, the 42-residue more amyloidogenic version of Aβ, has higher affinity to bind Cu2+ than Aβ1-40, the 40-residue version of Aβ [55]. One of the hypotheses by which binding of Aβ to Cu2+ can induce the formation of H2O2 required for Aβ crosslinking is by its ability to undergo Fenton redox cycling [56]. Consistent with this thought, histidines 6, 13, and 14 in Aβ that were identified to be involved in the redox cycling of bound Cu2+ [43] are located in close proximity to tyrosine 10. Density functional theory calculations and tyrosine-to-alanine mutational studies experimentally demonstrated that indeed tyrosine residue 10 in Aβ critically determines the generation of H2O2 mediated by Aβ-Cu2+ interaction [57]. The resulting crosslinked species were shown to accumulate in the AD brain, and to exert high levels of toxicity to neuronal cells [54, 59]. Using tandem mass spectrometry, it was observed that dityrosine cross-linked forms of Aβ can also be generated in vitro under conditions of oxidative stress induced by enzymatic peroxidation [60]. A recent paper showed that exposure of in vitro generated Aβ1-40 fibrils to Cu2+ significantly reduced fibril length as a result of fibril fragmentation [51]. Even though exposure of Aβ1-40 to Cu2+ was shown to induce thioflavin T (ThT) positive fibril assembly [51, 62], the addition of H2O2 inhibited the further assembly process [51] possibly stabilizing potent neurotoxic Aβ species.
Methionine-35 oxidation of Aβ
A second commonly detected Cu2+-induced modification of Aβ in plaques is the reversible modification of oxidation-sensitive methionine 35 to its sulfoxide [48, 63] or its further irreversible oxidation product methionine sulfone. APP23 transgenic mice show methionine oxidized forms of Aβ1-40 [64] and methionine oxidized Aβ is also abundantly detected in AD patient brains [38, 64]. The sulfoxide intermediate can be reduced by the action of peptide–methionine sulfoxide reductase [65], although levels of this enzyme in the AD brain were reportedly reduced [66]. In line with this observation, upon knock-out of methionine sulfoxide reductase A in a human amyloid-β protein precursor (AβPP) mouse model, levels of soluble methionine sulfoxide Aβ were increased and associated with defects in mitochondrial respiration and cytochrome c oxidase activity [67]. In turn, exposure of rat neuroblastoma cell line IMR-32 to methionine-oxidized Aβ1-42 induced an increase in levels of mRNA expression and activity of methionine sulfoxide reductase type A [68], suggesting that levels of methionine sulfoxide reductase A and methionine-oxidized Aβ1-42 may affect each other in a bidirectional manner. Somewhat conflicting results have been published on the effect of methionine-35 oxidation on Aβ aggregation. For example, it was shown that H2O2/Cu2+-induced methionine-35 oxidation slows down ThT-positive Aβ fibril formation of commercially derived Aβ1-40 and Aβ1-42 compared to wild type Aβ without affecting morphological features of the formed Aβ fibrils as observed by transmission electron microscopy (TEM) [69]. Marked differences in response to H2O2 induced methionine oxidation of Aβ1-40 and Aβ1-42 were observed in a different study showing that oxidation of Aβ1-40 increases fibril formation kinetics while slowing down fibril formation of Aβ1-42 [70] suggesting an isoform differential effect. Imaging of the resulting fibers using TEM showed that fibers generated by oxidized Aβ1-40 and Aβ1-42 were both highly fragmented compared to unoxidized peptide [70]. In another study, methionine 35 of synthesized Aβ1-42 was oxidized by exposure to H2O2 and oxidation was validated using mass spectrometry. Subsequent atomic force microscopy (AFM) and circular dichroism spectroscopy showed that methionine oxidation in this way hindered the typical random coil to β-sheet conversion and filamentous morphology characteristic for Aβ fibril formation [71]. Early aggregate formation of methionine oxidized Aβ1-40 was studied using electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry showing that trimer formation was inhibited without affecting dimer assembly [72]. One of the mechanisms suggested to affect the decreased aggregation propensity of Aβ upon oxidation of methionine 35 was that oxidation results in a reduced hydrophobicity of Aβ [71, 73], while hydrophobicity is one of the main driving forces for Aβ self-assembly. An oxidation-induced change in hydrophobicity was experimentally illustrated for apolipoprotein A-I, which, upon oxidation, affected the ability of this protein to interact with lipids [73]. It is difficult to delineate the precise origin of the diversity in results that have been obtained in aggregation studies of Aβ in response to methionine oxidation, but it is likely that variations in sample preparation, origin and incubation conditions may contribute as aggregation properties are sensitively affected by these parameters.
4-hydroxynonenal modification of Aβ
A third type of oxidative stress related feature in the AD brain is the accumulation of HNE [29, 75]. HNE generation has been detected both in vitro and in vivo as a result of lipid peroxidation [76, 77]. A 1990 hypothesis paper proposed several multi-step iron-catalyzed chemical routes for the generation of HNE through the oxidation of n-6 polyunsaturated fatty acids, particularly linoleic, γ-linoleate, and arachidonic acid [78]. Further, the presence of Aβ was shown to induce Cu2+-mediated production of HNE from lipids [79], and that, in turn, the released HNE can conjugate with Aβ and induce assembly of Aβ into high molecular weight species and increase the generation of Aβ by modulating β-secretase (BACE) activity [80–82]. Collectively, these data suggest that a number of in-brain factors interrelate to generate a downward spiral that is possibly associated with the observed pathogenic progress of AD. Metals were shown to regulate HNE modification of Aβ. For example, it was observed that HNE modification of Aβ in vitro can be achieved by means of coincubation of Aβ with HNE upon overnight incubation only in PBS that is free from magnesium and calcium [83] consistent with the finding that physiological levels of calcium effectively inhibit HNE modification of Aβ [82]. Also HNE conjugation to a truncated form of Aβ, Aβ1-16, was shown by means of mass spectrometry to be prevented by calcium and copper [83]. Of interest then was the observation that HNE conjugation of Aβ is a ROS-induced modification often encountered in amyloid plaques [2, 85], while in plaque levels of calcium and copper are reportedly high. A study mimicking in vivo in plaque conditions, involving physiological levels of calcium and high levels of copper, demonstrated indeed that HNE-adducts and Aβ were both recognized, though not colocalized, in cerebral vessels [83]. These data perhaps demonstrate that the raised levels of metals in plaques locally inhibit HNE conjugation to Aβ in these plaques [82]. Lysine and histidine residues in Aβ seem to be the most reactive residues toward HNE adduct formation, and it was suggested that the microenvironment of a specific residue determines the actual reactivity to HNE [86]. Two chemical reactions were identified that dictate the HNE-Aβ adduct formation: via formation of a Schiff’s base or by Michael addition [86]. Consistent with this thought, the conjugation reaction can be quenched by azide, primary amines, ammonia, Tris, DTT [83] or trifluoroacetic acid [79]. Also the addition of antioxidants hydralazine [83] or 3,5-di-tert-butylhydroxytoluene (BHT), or copper gelator diethylenetriaminepentaacetic acid (DTPA) [79], were reported to inhibit HNE-modification of Aβ.
Heme-complex formation of Aβ
Heme-complexed Aβ adducts have been postulated to affect cytochrome c oxidase (COX) activity, a mitochondrial electron transport chain enzyme which is significantly decreased in the AD brain [87]. COX requires heme-a, of which regulatory heme is a precursor, to assemble and perform its function [88, 89]. In turn, heme-a levels were observed to be significantly decreased in the temporal lobes of AD patient brains compared to age-matched controls [90]. A potential Aβ-mediated role in the availability of heme in the AD brain came to light when it was shown that the presence of heme dose-dependently inhibits oligomer formation of both Aβ1-40 and Aβ1-42 in an immunoassay and prevented loss of cellular viability upon addition of the complex to human neuroblastoma cell line IMR32 [91]. It was thought that, by competitive binding to heme, Aβ could deplete the availability of regulatory heme leading to deprived COX functionality and energy deficiency in AD. Similarly, the presence of heme was found to inhibit activation of Aβ-induced inflammatory response in primary mouse astrocytes [92]. At the same time, Aβ and heme, in the presence of H2O2, were reported to increase tyrosyl radical formation in Aβ1-16 and mediate its dimerization through 3,3’-dityrosine cross-linking [93–95], a reaction that was observed to be competitively inhibited by NaNO2 [94]. A direct and rapid interaction between heme and Aβ was shown upon addition of heme-a or heme-b to Aβ1-40 or a mixture of Aβ1-40 and Aβ1-42 which resulted in an immediate spectral shift of heme [90]. Aβ histidine residues were speculated as potential binding site via involvement of the π-electrons of the histidine imidazole rings, as addition of copper and zinc ions competitively inhibited the interaction of Aβ with heme, but only when heme was added to the reaction mixture after copper and zinc [90, 91]. In a subsequent site-directed mutagenesis study, using voltammetry, histidines 13 and 14 were specifically identified as heme binding sites in Aβ [96]. In addition to this, an NMR-based spectroscopic study showed that heme-b binds to Aβ1-16 with higher affinity compared to free histidine or other histidine-containing peptides indicating that other parts of the Aβ peptide contribute to the interaction with heme-b [95]. Heme-Aβ conjugates have also been found in AD plaques and conjugation to heme was shown to inhibit Aβ aggregate formation in a cell-free system and to dissociate existing aggregates [97]. Spectroscopic studies shed more light on the structural implications for Aβ1-16 upon interaction with heme-b [95]. This study showed that two complexes can be formed that exist in equilibrium, a low spin six-coordinated 1:2 heme/Aβ1-16 stoichiometry and a high-spin heme-(Aβ1-16) species. Aβ-heme adducts were found to exercise peroxidase activity [98] although in vivo relevance of this catalytic activity was questioned as a result of the reported very low k cat value of the complex of 0.016 s-1 at 278 K compared with a reference value of 45.5 s-1 for horse radish peroxidase [95]. However, a substantial effort has since gone into understanding the structural basis of this peroxide activity. One study showed that mutation of either histidine 13 or 14, but not both, does not affect peroxidase activity of the Aβ1-16-heme complex [94]. Free histidine, similar to the unmutated Aβ1-16-heme complex, induced peroxidase activity as observed using an 2,2’-azinobis(3-ethylbenzothiazoline-6-sulphonic acid) diammonium salt (ABTS) oxidation assay [94]. Apart from a regulating role by histidines, peroxidase activity of the Aβ-heme complex was shown to involve arginine 5 as proton donating residue cleaving the O-O bond of the peroxide [94, 98]. At the same time, the addition of free arginine to heme failed to induce peroxidase activity demonstrating that the structural incorporation of arginine 5 within a protein environment is somehow relevant for its action [94, 95]. Peroxidase activity of the complex was reported to depend on the heme-Aβ ratio and temperature, with increasing Aβ1-16 to heme and temperature inducing more potent peroxidase activity [95, 99].
Oxidative damage colocalizes with tau neurofibrillary tangles
Oxidative damage was found to colocalize with tau enriched neurofibrillary tangles [100]. In this study, hippocampal tissue from AD patients was subjected to postmortem analysis investigating the localization of the enzyme dimethylarginase. This enzyme regulates the activity of nitric oxide synthase [101]. In AD hippocampal tissue, neurons that contain neurofibrillary tangles also stain positive for dimethylargininase providing a first indication that nitric oxide is generated in close proximity to the tau that makes up the neurofibrillary tangles. In line with these observations, an antibody that recognizes an HNE-lysine adduct was found to colocalize with endogenously obtained paired helical tau filaments from AD brains [29]. Also, acrolein, which is an aldehyde product of lipid peroxidation, was observed to colocalize with neurofibrillary tangles in AD patient brains [102]. Further, the antibody Alz50 [103], which recognizes a conformational change in tau [104], coincides with heme oxygenase-1 (HO-1), which is an antioxidant enzyme [29], levels of which are strongly increased in the AD and mild cognitive impaired (MCI) brain [105]. Whether HO-1 activity is beneficial in terms of alleviating oxidative stress or can induce neurotoxicity in the MCI and AD brain has been subject of debate as increased HO-1 levels were also correlated with increased phosphorylation of tau serine residues [105].
Oxidation of tau affects filament assembly
Ascorbate/Fe(III)/O2-induced oxidation of bovine tau was shown to induce the assembly of tau into filaments in vitro [106]. The oxidation of one of the cysteine residues was found to be involved in the induction of the assembly of a recombinant fetal isoform of tau into such assemblies [107]. These data suggest that the generation of tau filaments is a disulfide bond mediated process while oxidation modulates the ability to self-assemble. Consistent with this thought it was recently shown that Zn2+ interacts with the cysteine residue of a truncated version of tau containing only the third repeat unit of the microtubule-binding domain accelerating its aggregation rate and toxicity in a Neuro-2A cell line [108]. A more direct role for oxidative stress in tau assembly was demonstrated upon administration of the anti-oxidants 2,4-disulfonyl α-phenyl tertiary butyl nitrone and N-acetylcysteine which reduced immunoreactivity against tau oligomers [109]. Of interest was the observation that peroxynitrite treatment of tau induced nitration, S-nitrosylation and oxidation of methionine, as observed by HPLC-electrospray ionization tandem mass spectrometry while markedly reducing aggregation, as analyzed by light scattering and electron microscopy [110]. Collectively, these observations suggest that oxidation of tau may modulate aggregation by either inducing or inhibiting the self-assembly process and that the specific outcome may depend on the type of oxidant and the specific amino acid residue involved. Phosphorylation was shown to importantly regulate HNE-induced assembly of tau as exposure of phosphorylated tau, as opposed to unmodified tau, induced misfolding of tau which was recognized by antibody Alz50, and the formation of tau aggregates [29]. In line with this, the self-assembly of a tau fragment including the first and third tubulin-binding domains showed that the presence of HNE mediated polymerization of phosphorylated tau [111]. At a molecular level, a link between phosphorylation and oxidative stress was revealed when a study showed that the activity of alkaline phosphatase was inhibited in the presence of HNE. Exposure of tau to HNE hence resulted in the generation of a tau species resistant against dephosphorylation [112].
AGE-conjugated tau is associated with oxidative stress markers
Advanced glycation end products (AGEs) are the oxidation product of sugars that interact with proteins and their accumulation has been related to amyloid deposition in AD [113]. Tau assembled into paired helical filaments has been shown to be immunoreactive against Nɛ-(carboxymethyl)lysine, one of the major AGEs [114]. Interaction of recombinantly produced tau with ribose-derived AGE products was shown to result in the generation of reactive oxygen intermediates, which, in turn, activate NFκb to induce amyloidogenic processing of AβPP to generate Aβ [115]. Uptake of AGE-glycated tau into SH-SY5Y neuroblastoma cells was associated with malondialdehyde and HO-1 detection which was prevented by the exposure of these cells to N-acetylcysteine and probucol, two antioxidant compounds [116]. Diffuse cytosolic immunoreactivity against AGE was shown in many neurons of post-mortem AD brains that also contain hyperphosphorylated tau [117]. Astrocytes residing in the temporal cortex of medium to severely affected AD subjects were found to be immunoreactive for inducible nitric oxide synthase (iNOS) as well as AGEs [118]. Thus far it is unclear whether AGE-glycation of tau has implications for the physiological role of this tubulin binding protein in the cytoskeletal organization or what the hierarchical correlation is between AGE formation and tau assembly into filaments.
Collectively, a clinical link between mitochondrial dysfunction and AD has been firmly established, with a central role for AD hallmark proteins Aβ and tau. While various types of ROS-mediated modifications of Aβ and tau have been investigated and play a potential role the precise implications of these species on disease progress have not been investigated.
EFFECT OF OXIDATIVE STRESS ON MITOCHONDRIA IN ALZHEIMER’S DISEASE
AD brain originating neurons containing defective mitochondria show loss of dendritic spines and abbreviation of dendritic arborization [119]. Differences in CA1 hippocampal mitochondria structure have been detected using 3-dimensional electron microscopy. Instead of the uniformly elongated mitochondrial morphology observed in wild type mice, human AD brain and hippocampal mitochondria in mice carrying mutations for presenilin-1 (psen1), AβPP, and tau, have an ovoid or teardrop profile [115]. Further, AD mouse models and AD patients show the presence of multiple small mitochondria and exaggerated mitochondrial division [120] suggesting that the mitochondrial fission process is altered in AD. The mitochondrial fission process relies on dynamin related protein 1 (Drp1) and mitochondrial fission protein 1 (Fis1) [121, 122]. Recent research observed the presence of elongated interconnected organelles where multiple teardrop shaped mitochondria were connected by thin double membranes. This structure, referred to as “Mitochondria-on-a-string (MOAS)”, has been identified in an AD mouse model together with increased Drp1 phosphorylation, causing incomplete fission. Even though altered mitochondrial fission processes in neurodegenerative diseases have been viewed primarily as a pathological feature, in cardiomyocytes Drp1 induced mitochondrial fission was shown to exert a protective effect against cellular apoptosis by enabling the cells to meet altered energetic demands [123]. An alternative role of Drp1 was suggested with the observation that reduced association of Drp1 with the mitochondrial membrane induced a lack of mitochondrial fusion, which, in turn, induces high levels of mitochondrial oxidative stress [124]. The fusion process should be in balance with mitochondrial fission to maintain mitochondrial homeostasis. Mitochondrial fusion is mediated by inner membrane fusion factor optic atrophy-1 (OPA1). Addition of H2O2 to an osteosarcoma and a cardiomyoblast cell line lead to inhibited mitochondrial fusion as a result of loss of OPA1 activity through cleavage mediated by metalloendopeptidase OMA1 [125, 126].
ENDOGENOUS ANTIOXIDANT ACTIVITY IS COMPROMISED IN ALZHEIMER’S DISEASE
The endogenous antioxidant capacity is a multi-component system targeted at neutralizing ROS and RNS to prevent damage of cellular compartments. Many of the factors involved in endogenous antioxidant capacity are affected in AD, and experimental evidence for this will be discussed in this section.
Glutathione
Glutathione (GSH) is one of the prime endogenous antioxidants in the brain. GSH is a tripeptide thiol-containing antioxidant that is synthesized by the conjugation of the amino acids glutamate, cysteine, and glycine mediated by the enzymes γ-glutamyl cysteine synthetase and glutathione synthetase [127]. GSH acts by scavenging ROS, and, in the process, becomes reversibly oxidized to form glutathione disulfate (GSSG) [127, 128]. Oxidative stress induces the expression of the NADPH-dependent enzyme glutathione reductase, which reverts oxidized GSSG to its reduced form GSH [129]. A study involving 74 human subjects demonstrated that GSH levels of autopsied brains did not significantly decrease with aging [128]. At the same time, whole-brain GSH levels were shown to be profoundly reduced in individuals suffering from AD compared to age-matched controls [130] although another study reports that GSH levels in AD brains are not significantly different from those found in age-matched control brains [131]. Region-specific differences were identified showing increased GSH levels in the hippocampus and midbrain of AD patients without significant difference in GSSG levels [132]. Moreover, a correlation between peripheral and brain levels of GSH exists as it was demonstrated that levels of erythrocytic GSH in elderly patients with MCI and AD were substantially decreased compared to a control group [130]. A study investigating human AD patient lymphocytes showed that decreased GSH levels correlated with increased GSSG levels [133]. Moreover, basal blood levels of GSSG/GSH ratios in control, mild, moderate or severe dementia patients showed a significant correlation with progression of disease [134]. Aging related reduction of brain GSH was shown to go hand in hand with decreased gene expression of γ-glutamyl cysteine synthetase in the brain [135]. While whole-brain levels of GSH transferase in AD brains were not significantly different from age-matched control brains [131], mRNA expression levels of γ-glutamyl cysteine synthetase vary per region in the brain with high expression levels in cortex, cerebellum and hippocampus and low expression in the neostriatum of mice [136, 137], and it was suggested that these regional differences in de novo GSH generation can explain regional differences in susceptibility to oxidative stress [137].
Melatonin
Melatonin, or N-acetyl-5-methoxytryptamine, is involved in various homeostatic functions to aid cellular protection. It is an electroreactive neurohormone with antioxidant activity that is synthesized and secreted in the brain from mitochondria of pinaelocytes, cells of the pineal gland [138–140]. Also the metabolites of melatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), demonstrate antioxidant activity, either directly by scavenging a variety of free radicals including hydroxyl, peroxyl, superoxide, peroxide and peroxynitrite (ONOO-) [141, 142], or indirectly by inducing antioxidant enzymes including superoxide dismutase (SOD), glutathione peroxidase (GPx), and GSH reductase [143], increasing GSH synthesis [144], and inhibiting prooxidant enzymes RNS, xanthine oxidase, and myeloperoxidase [145]. Even though aging is related to a decrease in CSF melatonin levels, presenile and senile AD patients demonstrated an even stronger reduction in melatonin levels that was shown to be dependent on apolipoprotein genotype [146], one of the strongest identified genetic correlates with AD. How these factors and processes are associated is currently unclear.
Transcriptional control of the endogenous antioxidant system by Nrf2
The neuron-glial unit, the main interaction site between neurons and cells of glial origin such as astrocytes, regulates oxidative stress levels through an intimately linked intercellular mechanism for maintaining redox homeostasis [147, 148]. Brain oxidative stress levels are maintained within strict limits as a result of the astrocytic nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2) homeostatic pathway [149]. Upon translocation to the nucleus, Nrf2 binds to antioxidant response element (ARE), a promotor element present on antioxidant genes [150]. Nrf2 degradation is controlled by ubiquitin-mediated degradation, which, in turn, is regulated by cytoskeleton associated Kelch-like protein, Keap1 [151–154]. In the absence of oxidative stress, Nrf2 is transcriptionally inactive as its activity is repressed by Keap1 [154]. Under conditions of oxidative stress Keap1 is oxidized inhibiting the degradation of Nrf2. Transcriptional activity of Nrf2 was shown to decline upon aging [155, 156]. One study showed that AD progression was linked with haplotype allele variation in the NFE2L2 gene promotor which encodes for NRF2 [157] while therapeutic administration of a lentiviral vector encoding for human Nrf2 was shown to improve cognitive dysfunction in APP/PS1 [158], and APP/PS1DeltaE9 mice [159]. Furthermore, a recent transcriptomics study demonstrated that NRF2 knockout leads to early onset cognitive dysfunction, plaque deposition and tau tangle formation [160]. Other recent experimental evidence linking AD to the Nrf2 pathway showed that methysticin, a kavalactone activating the Nrf2 signaling pathway, reduced neuroinflammation, loss of memory and damage as a result of oxidative stress in the hippocampus of APP/Psen1 mice [161]. Even though the Nrf2 signaling pathway is highly active in astrocytic cells, this pathway is virtually absent in cells of neuronal origin [162, 163] while the capacity of neurons to degrade Nrf2 is high as a result of abundant neuronal expression of the protein cullin 3 which leads to destabilization of neuronal Nrf2 [162]. These observations argue for a high level of functional integration of astrocytes and neurons in the brain to regulate oxidative stress levels.
ALZHEIMER’S DISEASE RELATED OXIDATIVE STRESS
Disturbed metal ion homeostasis in Alzheimer’s disease
Metal ions such as Cu2+ and Zn2+ play an important role in regulating synaptic functioning by inhibiting the rat excitatory NMDA receptor [164], and rat GABA receptor [164, 165]. Iron ion (Fe2+) has been documented to regulate synaptic plasticity and synaptogenesis as well as myelination [166] as illustrated by the neuronal expression of iron transporter DMT1 [167–169]. The levels of these metal ions are normally strictly regulated to prevent oxidative stress resulting from interaction of Fe2+ or Cu2+ with oxygen to generate radicals such as superoxide ions or hydroxyl radicals. Disruption of metal ion homeostasis has been observed in various neurodegenerative disorders including AD [170]. A patient study using instrumental neutron activation analysis demonstrated that levels of Cu2+ were decreased while Zn2+ and Fe2+ levels were elevated in the hippocampus and amygdala of AD patients which correlated with observed histopathological changes in these regions [171]. On the other hand, serum levels of Cu2+ were shown to be increased in AD patients compared to control subjects [172]. Also in preclinical stages and MCI Fe2+ levels were increased in the cortex and cerebellum and correlated with generation of radicals [173]. Compared with the neuropil of the amygdala of AD patients, senile plaques were observed to contain increased levels of Zn2+, Fe2+, and selectively in the rim of the plaques, Cu2+ [47]. As Zn2+, Fe2+, and Cu2+ have been shown to interact with Aβ in vitro [174], metal ion dyshomeostasis has been postulated as potential mechanism by which AD pathology may be modulated.
Spatial link between amyloid plaques and cells exhibiting oxidative damage
A multiphoton microscopy-based study using the genetically encoded calcium indicator Yellow Cameleon 3.6 packaged into an adeno-associated virus (AAV2) and expressed in the brains of adult transgenic APP/PS1 mice showed that calcium overloaded neurites in living animals were more likely to be located in close proximity (<25μm) of a plaque [175]. This observation suggests a direct or spatial link between pathological alterations in neurons and the formation of senile plaques. A second marker that indicates that there is a spatial link between AD-related deposits in the brain and neuronal functioning was the receptor for advanced glycation end products (RAGE). Neuronal cells adjacent to senile plaques display increased RAGE expression while little change in expression was demonstrated in brain regions remote from plaques [176]. Two other markers that have been used to topologically differentiate subpopulations of cells affected by oxidative stress include p50, which is a DNA binding subunit of transcription factor NFκB [177, 178], and HO-1. Cellular structures containing accumulations of Aβ displayed increased levels of oxidative stress as demonstrated by elevated levels of HO-1, and p50 [176]. Inactive NFκB resides in the cytosol and is bound to inhibitory protein IκB which prevents nuclear translocation of NFκB. Phosphorylation, ubiquitination, and degradation of IκB drives the activation of NFκB [179]. The redox state regulates activation and nuclear translocation of NFκB [180], and, as such, ROS was found to induce phosphorylation of IκB via activation of responsible kinases [181, 182]. Using p50 and HO-1, it was observed that the spatial link found between Aβ deposits and induction of cellular ROS is not limited to CSF residing neurons, but this observation extends to endothelial and smooth muscle cells in cerebral blood vessels. The expression of HO-1 was found to be elevated in AD injured neuronal cells, a feature that was more pronounced in regions close to neurofibrillary tangles and Aβ plaque deposits [183].
Oxidative stress is an early stage pathological feature
A redox proteomics study of the brain of Down syndrome (DS) patients prior to onset of AD provided insight into the role of oxidative damage in the development of DS related early onset AD [184]. Male and female DS and control brains were analyzed postmortem for carbonylation levels of proteins as hallmark of oxidative stress. DS brains showed increased carbonylation of six proteins including cathepsin D, glial fibrillary acidic protein and succinyl-CoA:3-ketoacid-coenzyme A transferase 1 mitochondrial protein. Carbonylation affected protein functionality, while at the same time, proteasome activity and autophagy activity were decreased [184] potentially leading to loss of functional protein. Even though this study was conducted on a small number of subjects, it did provide important insight into the potential role of oxidative stress in early stages of disease. A larger scale study using human peripheral blood mononuclear cells (PBMCs) derived from 104 MCI subjects similarly showed increased oxidative stress markers as detected by the fluorescent probe DCFH2-DA [185]. Also, in MCI and mild AD patient PBMCs homeostasis of ER stress-mediated Ca2+ was disturbed with decreased SOD1 levels [185]. Analysis of lymphocytes obtained from MCI subjects and AD patients similarly showed increased ROS levels, detected by 8OHdG, compared to lymphocytes derived from an age-matched control population [186]. The validity of using 8OHdG brain levels as a biomarker to detect oxidative stress-related damage to DNA in AD patients has been questioned [187]. However, the detection of increased levels in the frontal cortex of other modified macromolecules such as F2-isoprostanes as well as 3-NT and oxidized glutathione detected in patients with probable AD further corroborates the thought that oxidative stress is an early stage pathological feature of AD [188]. The work by Ansari and Scheff also compared oxidative stress levels in age-matched groups with progressive forms of cognitive disorder, from non-cognitively impaired to AD, and showed that oxidative stress progressively worsened with cognitive decline. In addition to this, activities of SOD and catalase in post mitochondrial supernatant and in mitochondrial and synaptosomal fractions of the frontal cortex were significantly declined already in MCI subjects [188]. Consistent with this, an earlier longitudinal study on autopsied control and patient brains demonstrated that levels of isoprostane (F2) and F4-neuroprostane were increased in both amnestic MCI and late stage AD patients in various regions of the brain [189].
A vascular component
The microcerebrovascular structure showed age-dependent changes [190] which are more pronounced in cognitive disorders such as dementia [191, 192]. For example, the basement membranes of cortical capillaries of patients suffering from cognitive disorders were significantly thicker than those of age-matched controls [192]. Smooth muscle atrophy and general disorganization of these cells was consistently observed in AD subjects although these features seemed unrelated to the deposition of Aβ [193]. These structural changes translate into a decreased capillary flow in aged (16 months old) compared to young (2 months old) mice [194] as well as aggravated loss of blood flow rate in an aged APPswe/PS1ΔE9 transgenic mouse model [195]. The observed structural and functional changes in the microvascular organization thus lead to hypoperfusion and a general inability of the cerebral vasculature to meet the metabolic needs of the brain while this was partly compensated for by an increased ability to extract oxygen from the remaining blood flow [196]. However, the remaining metabolic deficiency is of sufficient magnitude to result in neural hypoxia [196]. Various conditions have been associated with increased brain oxidative stress and neuronal apoptosis in response to hypoxia, including sleep apnea [197, 198], exposure to carbon monoxide [199], and ischemia [200]. Sleep apnea co-occurs frequently with AD [201] while prevalence of sleep apnea positively correlates with aging [202, 203], and treatment of sleep apnea slows down the rate of cognitive decline in patients diagnosed with mild-to-moderate AD [201]. Further, hypoxia induced oxidative stress in the brain has been shown to induce cognitive deficits in rats [204]. The mechanisms by which the brain adapts to hypoperfusion-induced hypoxia have been explored and most proposed mechanisms are centered around the thought that activation of hypoxia-inducible factor 1α (HIF-1α) plays an important role. HIF-1α is a component of a heterodimeric complex with the aryl hydrocarbon nuclear translocator (ARNT or HIFβ) [205]. Under normoxic conditions, HIFα is dissociated from this complex and unstable as a result of its hydroxylation which targets it for ubiquitination and proteasomal degradation [206–208]. Hypoxia prevents hydroxylation of HIFα by inhibition of the two hydroxylating enzymes, factor inhibiting HIF-1 and prolyl hydroxylase enzymes [209, 210]. This stabilization induces its nuclear localization and heterodimeric complexation with ARNT. Subsequent co-recruitment of this complex with transcription coactivators p300 and CREB binding protein (CBP) initiates gene transcription. Hypoxic conditions are considered to raise cytosolic ROS levels and, in this way, induce the activation of HIF (reviewed in [211]) probably in a mitochondrial complex III-dependent manner [212]. The HIF-dependent hypoxia-inducible genes are generally involved in processes aimed at promotion cellular survival under hypoxic conditions. A study investigating mRNA expression in adult rat brains upon occlusion of the middle cerebral artery demonstrated that glucose transporter-1 (GLUT-1) and glycolytic enzymes (phosphofructokinase, aldolase, and pyruvate kinase) were upregulated to increase transport of glucose and glycolysis [213].
GENETIC AND OTHER FACTORS THAT CORRELATE OXIDATIVE STRESS TO ALZHEIMER’S DISEASE
This paragraph will first review the established experimental evidence that has demonstrated a connection between oxidative stress and AD, such as clusterin, apolipoprotein E (ApoE), and genes related to AβPP processing machinery. Second, this paragraph will also highlight some potential interactions that have yet to be experimentally established but for which observations have shown to connect to both AD and oxidative stress. These factors include Klotho, and circadian clock genes and we envisage that future investigation into these factors and their relation to AD and oxidative stress levels may highlight alternative or additional mechanisms for the interaction of these clinical features. Figure 1 summarizes the genetic factors associating AD and oxidative stress to date.
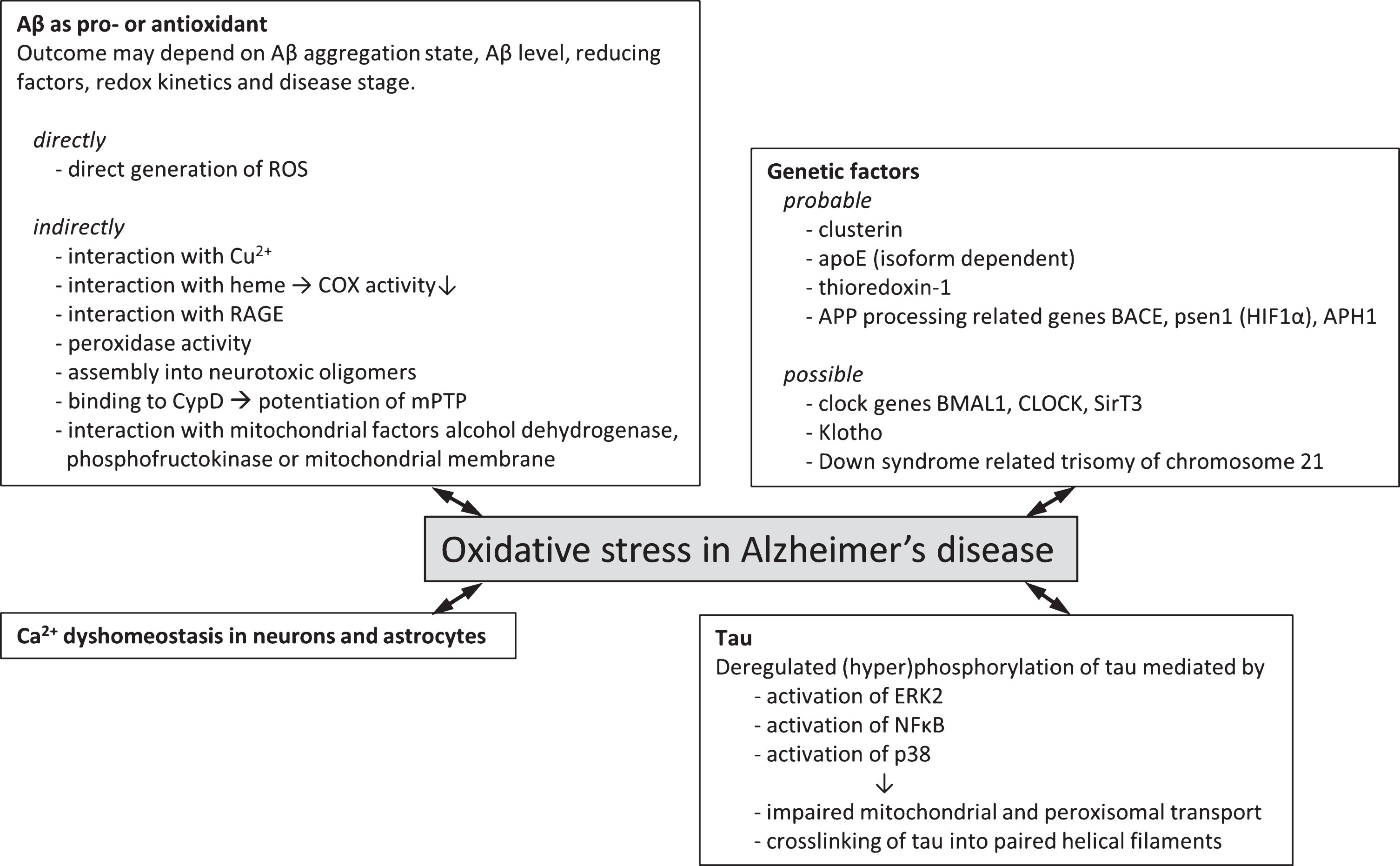
Genetic factors and molecular mechanisms of oxidative stress in Alzheimer’s disease. Overview of probable (experimental evidence available in literature) and possible (no direct experimental evidence available) genetic factors that associate oxidative stress with Alzheimer’s disease. Various molecular mechanisms by which oxidative stress and Alzheimer’s disease may be associated have been described. These often involve the two hallmark proteins Aβ and tau and effects may be directly involving the generation of ROS or indirectly via interaction with various cellular factors giving rise to increased ROS generation or lowered endogenous antioxidant capacity.
Clusterin
Apolipoprotein J is a ubiquitously expressed secreted glycoprotein which is also known as clusterin (CLU). Aging induces elevated levels of CLU gene expression [214, 215] and plasma CLU [216]. In a genome-wide association study CLU has been identified as a genetic determinant of AD [217] and plasma CLU levels were associated with atrophy of the entorhinal cortex and clinical progression of the disease [218] as well as with longitudinal brain atrophy in MCI patients [219]. Apart from aging, expression of the CLU gene was demonstrated to be sensitive to heat-shock induced changes in the organism or the direct environment of the organism as a result of the presence of activating protein-1 (AP-1) and CLU-specific element regulatory elements in its promotor [220]. Consistent with the idea that CLU plays a role in stress-associated coping of cellular response a study in H9c2 cardiomyocytes revealed that the Akt/GSK-3β pathway may be involved in the anti-oxidant and anti-apoptotic effect of CLU in a megalin-dependent manner [221]. Multi-ligand receptor megalin has been identified to also act as receptor of clusterin [222]. Various cellular stress stimuli have been shown to regulate transcriptional activity of AP-1 [223]. Differential CLU expression was similarly observed in other oxidative stress-related pathologies including asthma [224], atopic dermatitis [225], diabetes type 2 [226], coronary heart disease [226], and cancer [227]. Oxidative stress increases CLU expression. This was demonstrated in a study in which human diploid fibroblasts were treated with H2O2 which resulted in increased mRNA levels of CLU [228]. In human neuroblastoma cell lines SH-SY5Y and IMR-32 both mRNA and protein levels of CLU were found to be upregulated in response to pro-oxidant pair iron-ascorbate [229]. In line with these observations, CLU was originally identified to function as a chaperone protein where its activity was reported to depend on cellular redox state [230]. CLU was shown to protect against oxidative stress in various cellular systems including fibroblasts and prostate cancer cells [221, 231] but also in vivo in a Drosophila melanogaster model [232]. In neuroblastoma N2a and SH-SY5Y cells knockdown of CLU by short hairpin RNA interference was found to down-regulate antioxidant capacity [233]. The precise anti-oxidant mechanism of CLU is not known although blockage of the sulfhydryl groups contained in the sequence of the protein resulted in abolishment of its oxidative stress preventive activity [232]. A review covering the involvement of CLU in oxidative stress detection and action has been published before [234]. Apart from an antioxidative effect of CLU, an indirect role of CLU actually promoting oxidative stress has been described showing that the presence of CLU induces the formation of slowly sedimenting complexes composed of SDS-resistant synthetic Aβ assemblies that, in turn, induced oxidative stress in PC12 cells [235].
Apolipoprotein E and Thioredoxin-1
Apolipoprotein E4 (ApoE4) was identified as one of the major genetic risk factors for AD [236–239]. Apolipoprotein E exists in three isoforms, ɛ2, ɛ3, and ɛ4, which vary in their amino acid composition. Carriers of the ɛ4-allele have an increased risk of developing AD [236] as well as a decreased age of AD onset [237] compared to non-ɛ4 carriers. The pathogenic origins of ApoE4 have been studied to great length and indicate that ApoE4 is involved in processes such as aggregation and clearance of Aβ [240, 241], mitochondrial dysfunction, and impairment of calcium [242, 243] or cellular iron homeostasis [244], and ApoE4 affects synaptic architecture and functioning [245, 246]. A potential connection between the ApoE allele, AD, and oxidative stress was first deduced from the observation that the extent of oxidative stress and anti-oxidant defense is related to ApoE genotype in mice and in patients [13, 247–249]. ApoE was demonstrated to act, directly or indirectly, as an antioxidant against hydrogen peroxide-induced cytotoxicity in a B12 ApoE expressing cell line [250]. Elevated levels of peroxidized plasma low-density lipoproteins were observed in ApoE-deficient mice [251]. Levels of lipid oxidation were significantly increased in the frontal cortex of AD patients that were homozygous or heterozygous for the ɛ4-allele of ApoE compared to homozygous ɛ3 carriers and controls [13]. Upregulation of catalase activity was exclusively observed in frontal cortex tissue of homozygous ApoE4 carriers while SOD activity and concentrations of glutathione were not different from that of controls [13]. Additionally, levels of HNE were increased in ɛ4-carriers [84]. Further, mouse brain synaptosomes expressing human ApoE4 were more susceptible to Aβ42-associated oxidative stress than synaptosomes from mice expressing human ApoE2 or ApoE3 [252]. Various experimental findings shed light on the potential molecular mechanism underlying these observations. They show involvement of thioredoxin-1 (Trx1), an endogenous antioxidant with a downregulating role in apoptosis signal-regulating kinase-1 (Ask-1) [253]. Thioredoxin reductases are reducers of Trx1 [254]. Levels of Trx1 were reduced in AD brains [255, 256] depending on ApoE genotype, but also in ApoE4 expressing mouse hippocampi, and human primary cortical neurons and neuroblastoma cells to which ApoE4 was supplemented with the culture medium, compared to ApoE3 [257]. At the same time, Trx1 mRNA levels in ApoE4 TR mouse hippocampi were elevated consistent with findings reporting increased Trx1 expression in conditions of oxidative stress [258]. Persson and colleagues suggested that increased mRNA levels of Trx1 potentially act as a compensatory mechanism for the increased cathepsin D-induced Trx1 turnover as observed in SH-SY5Y neuroblastoma cells [257]. Moreover, Aβ1-42 was demonstrated to cause transient oxidation of Trx1 [255] as well as ApoE4-induced downregulation of this protein which resulted in activation of an apoptotic pathway involving the translocation of Death-Domain Associated Protein-6 [255, 259] without affecting catalase and GSH activities [257].
Down syndrome, a trisomy of AβPP encoding chromosome 21
Individuals with DS are prone to develop early-age AD with pronounced oxidative stress. DS is characterized by trisomy of chromosome 21 (HSA21), which encodes AβPP as well as some proteins of relevance to redox homeostasis providing an interesting group of patients to study early stage aspects of oxidative stress in AD pathogenesis in response to a defined genetic condition. Comparable to observations in AD patients, mouse models of DS demonstrated deficits in hippocampal learning and memory as well as neurodegeneration of cholinergic basal forebrain neurons [260, 261]. DS patients display features of cellular energy impairment [4]. Recent transcriptomic profiling of the skeletal muscle of a DS mouse model showed that among the identified differentially expressed protein-coding genes in this tissue, two, Sod1 and Runx1, were implicated in oxidative stress [262]. Chromosome HSA21 also codes for SOD explaining why expression levels of SOD are increased in DS [263]. Transgenic mice overexpressing SOD1 demonstrate excessive levels of oxidative stress [264] because concentrations of CAT and GPx, two enzymes that act to neutralize hydrogen peroxide, the product of SOD1 activity, do not rise accordingly. Besides SOD1 fifteen other genes on HSA21 were predicted to play a role in mitochondrial energy generation and the metabolism of ROS [265]. Levels of various ROS, RNS and aldehyde products of lipid peroxidation were found to be increased in brain [20, 266] and urine [267] of DS humans and animals [266] indicating that oxidative stress may play a role in the pathogenesis of DS associated AD. Levels of oxidative stress, i.e., high ratio between SOD1 and GPx, correlated with cognitive phenotype [268]. However, a recent study showed that administration of melatonin at the pre- and post-natal stages partially alleviated oxidative stress but did not improve cognitive function in a mouse model [269]. The process of programmed cell death was found to coincide with increased levels of oxidative stress, compared to control cells. Programmed cell death could be rescued by administration of free radical scavengers including vitamin E, and N-tert-butyl-2-sulphophenylnitrone [263] but dietary parameters did not alleviate oxidative stress biomarkers in young adult DS patients [20]. Similar to AD, the DS brain shows features of oxidative stress at very early stage. For example, the DS fetal brain cortex was observed to show increased levels of thiobarbituric acid reactive substances (TBARs), HNE, and protein carbonyl groups compared to controls [270]. Also end-products of non-enzymatic glycation, pyrraline and pentosidine, were increased in DS fetal tissue [270] and in amniotic fluid of DS pregnancies [271].
AβPP processing machinery
Cells of both neuronal and non-neuronal origin exposed to H2O2 or HNE generate increased levels of intracellular and secreted Aβ [272–275]. The role of oxidative stress in Aβ generation was further demonstrated in Tg19959 mice, which overexpress a double mutated form of AβPP. Upon crossing this mouse line with a mouse in which one allele of MnSOD was knocked out, brain Aβ levels and Aβ plaque load were significantly increased [276]. Similarly, hypoxia treated transgenic APP23 mice that were subjected to hypoxia conditions demonstrated increased memory deficits and deposition of Aβ into plaques [277]. Repeated exposure to hypoxia facilitated progress of AD-like pathology in aged APPSwe/PS1A246E transgenic mice [278]. Vascular deposition of Aβ on the surface of cerebral endothelial cells results in vascular degeneration which has been observed in AD and leads to a condition termed cerebral amyloid angiopathy [279]. Exposure of primary cerebral endothelial cells derived from 2-month-old Tg2576 mouse brains to H2O2 resulted in upregulation of AβPP expression and altered AβPP processing to favor the amyloidogenic pathway [280]. Also in humans it was found that oxidative stress induced by hypoxia due to cardiac arrest increased serum Aβ levels [281] suggesting that the machinery that generates this peptide is upregulated under pro-oxidative stress conditions in a wide range of disease models affecting various regions of the brain.
Oxidative stress has an Aβ species specific effect
Aβ is generated as a heterogeneous pool of peptides which vary in the number of C-terminal amino acids. The two most prevalent types of Aβ are the 40-amino acid (Aβ1-40) and the 42-amino acid (Aβ1-42) isoforms. It has been demonstrated that the longer Aβ1-42 peptide is inherently more amyloidogenic than Aβ1-40. Analysis of the specific species of Aβ that were generated under HNE-induced oxidative stress conditions using a TN2 cell culture revealed a 70-80% and 60-140% increase of intracellular Aβx-40 and Aβx-42, respectively. Secreted levels of Aβx-40 were not affected by oxidative stress while secreted Aβx-42 was increased by approximately 50% compared to control cells [274]. These findings are potentially pathologically relevant as it was reported previously by our group and others that a marginally increased Aβ1-42:Aβ1-40 ratio has severe implications for synaptotoxic response [282–284]. Aβ is generated by sequential cleavage of AβPP by two enzymes, γ-secretase and BACE, by a process termed amyloidogenic pathway. Alternatively, AβPP can be cleaved into an N-terminally truncated fragment of the Aβ peptide, called the p3 peptide, by γ- and α-secretase-mediated processing. Details of AβPP processing have been extensively covered in a number of reviews [81, 285].
Both presenilin and anterior pharynx-defective-1, components of γ-secretase, are upregulated in conditions of oxidative stress
Psen1 constitutes the catalytic site of the AβPP cleaving enzyme γ-secretase. In concerted action with BACE, psen1 is responsible for the generation of Aβ (reviewed in [286]). Clinical mutations in psen1 cause familial forms of early onset AD (reviewed in [287]) and can affect γ-secretase mediated processing of AβPP in various ways [288]. Generally, mutations in psen1 comprise the composition of the heterogeneous Aβ mixture by shifting the ratio between the various Aβ peptides generated [289–291]. In human SK-N-BE neuroblastoma cells, which were exposed to HNE- or H2O2-mediated oxidative stress, an increase in Aβ level was found that could be attributed indirectly to γ-secretase in a c-jun N-terminal kinase (JNK)/c-jun pathway/BACE1 dependent manner [75]. However, a direct relation between hypoxia-induced oxidative stress and γ-secretase functionality exists. This was later demonstrated in zebrafish by showing that HIF-1α induces increased mRNA expression levels of zebrafish related PSEN1 [292]. In line with these observations, PSEN1 -/- fibroblasts demonstrated impaired induction of HIF-1α [293], suggesting an apparent bi-directional interaction between psen1 and HIF-1α. Importantly, this factor plays a crucial role in the regulation of oxygen homeostasis and the expression and stability of one of the HIF-1α domains is regulated by oxygen levels [205, 210]. Anterior pharynx-defective-1 (APH1) is another component of γ-secretase and it was shown that Hela cells express increased levels of APH1α in response to chemical hypoxia induced activation of HIF-1α resulting in increased AβPP and Notch processing [294]. NF-kB has been identified as important regulator of HIF-1α expression [295]. NF-κB was shown to become activated and translocated to the nucleus in response to oxidative stress by addition of metformin, a pro-oxidative biguanide, to LAN5 neuroblastoma cells. This directly induced transcriptional activation of AβPP and psen1 and ultimately into increased AβPP cleavage, and intracellular accumulation of Aβ which promoted Aβ aggregation [296].
β-secretase is upregulated at a transcriptional level by oxidative stress
BACE1 is an integral part of the amyloidogenic processing pathway of AβPP and the functionality of this enzyme was, similar to γ-secretase, reported to be affected by oxidative stress. Low micromolar concentrations of HNE produced by pro-oxidant stimuli ascorbic acid/FeSO4 or H2O2/FeSO4 or by direct addition to cell medium of NT2 cells in culture were reported to induce BACE1 activity. This induction leads to an increased production of APP C-terminal fragments without affecting AβPP synthesis. Pretreatment of these NT2 cells with α-tocopherol prevented BACE induction and CTF generation demonstrating direct involvement of oxidative stress in inducing BACE activity [81, 297]. Also in APP/PS1 mice, it was shown that administration of an antioxidant compound, tricyclodecan-9-yl-xanthogenate (D609), which is a GSH-mimetic compound, leads to decreased levels of oxidative stress as well as a reduction in BACE1 levels resulting in decreased AβPP processing into Aβ [298]. Various other publications similarly reported that BACE1 protein expression levels were increased in response to oxidative stress [80, 299–305]. A similar observation has been reproduced in various model systems. For example, a developing (48-h post fertilization) zebrafish animal model exposed to hypoxia showed that the mRNA levels and activity of zebrafish bace1, the zebrafish orthologue to human BACE1, were affected by oxidative stress. At the same time, the level of CAT was found to be increased upon exposure of zebrafish to hypoxia [292]. Also, in murine primary cortical neuronal cultures, severe and cytotoxic levels of oxidative stress lead to an increased BACE1 expression. Mild oxidative stress conditions were found to result in subcellular redistribution of BACE1 that promoted amyloidogenic processing of AβPP [306]. Apart from in various animal and cellular models, increased BACE1 levels and activity were also found in brains of sporadic AD patients [307–309]. To understand the cellular signaling pathways involved in oxidative stress-regulated expression of BACE1, an NT2 cell-based assay was used. In this system, pharmacological based inhibition of C-Jun N-terminal Kinase (JNK) and p38MAPK, involved in the stress activated protein kinase (SAPKs)/JNK signaling pathway, but not Akt signaling, was found to inhibit transcriptional regulation of BACE1 [274]. P38MAPK was also reported to be active and identified in Aβ deposits of AβPP tg mice [310]. These observations are consistent with an earlier report that showed that Sp1 regulates transcription of BACE1, where expression levels of Sp1 were positively correlated with the generation of BACE1 and AβPP processing [311]. In line with this, using a lipofuscinfluorphore A2E-mediated photo-oxidation model to investigate the role of BACE1 in age-related macular degeneration, it was shown that BACE1 expression is competitively regulated by Sp1 and DNA methyltransferase 1 (DNMT1) after photo-oxidation [313]. DNMT1 levels were reportedly decreased resulting in demethylation of specific loci within the BACE1 gene promotor [312]. Moreover, members of the SAPK family were also found to be upregulated in AD patient brains [30, 313] and are activated by various stress signals including oxidative stress [314, 315]. The oxidative-stress regulated involvement of JNK has been further demonstrated in studies using transgenic mouse models. For example, JNK was found to be significantly activated in mutant AβPP tg mice with extensive oxidative damage but not in mutant AβPP tg mice with little oxidative damage [100].
Downregulation of non-amyloidogenic AβPP processing
While it was consistently reported that the amyloidogenic processing pathway of AβPP is upregulated by various direct and indirect mechanisms, the non-amyloidogenic pathway, involving sequential cleavage of AβPP by α-secretase and γ-secretase, was found to be downregulated under conditions of oxidative stress [280]. Multiple lines of evidence have shown that γ-secretase is upregulated under conditions of oxidative stress. As such, it was anticipated that the net lowering of the non-amyloidogenic processing of AβPP should be accommodated for by a decrease in α-secretase activity. It was indeed shown that human neuroblastoma cell line SH-SY5Y exposed to hypoxic conditions decreases the expression of disintegrin and metalloproteinase domain-containing protein 10, or ADAM-10, also called α-secretase, in an O2-dose dependent manner [316]. Similarly, human MSN cells exposed to oxidative stress induced by H2O2 or FeCl2 were shown to downregulate the active form of ADAM10 [303]. Mechanisms that explain the downregulation of α-secretase under conditions of oxidative stress have not been explored in great detail. One of the hypotheses that has been postulated involves the JNK3-dependent phosphorylation of Thr668 of AβPP which is considered to be a direct target for BACE1 [303, 317].
Circadian clock genes
Even though the connection between circadian clock, oxidative stress, and AD has been little investigated and may not be directly related, the findings that have been reported on this topic show that a potential interaction between these features may well exist and warrants further investigation. This paragraph summarizes the experimental evidence in supports of such a connection. Circadian rhythm disturbances and associated disorders of the sleep-wake cycle, i.e., fragmentation of sleep and decreased duration of rapid eye movement sleep (REM), occur at various stages of AD-related neurodegeneration and symptoms are generally more severe with increasing Aβ burden and tau pathology [318–321]. One report shows that extensive loss of sleep reduced the activity of SOD and the production of ATP in rat hippocampi [322], features that are strikingly similar in the AD brain. In a follow-up report, apart from reduced SOD activity, it was found that the activity of glutathione peroxidase was also decreased while liver malondialdehyde levels were increased with the extent of sleep deprivation [323]. On the other hand, brain and peripheral tissues were shown to differ in their peroxiredoxin oxidation rhythms [324] as well as in other clock components [325]. This means that peripheral observations cannot be automatically extrapolated to brain processes. During periods of REM sleep firing rates of wake-active noradrenergic locus coeruleus neurons, cells that display high sensitivity to metabolic stress [326–328], are profoundly reduced [329]. Extensive wakefulness induces loss of locus coeruleus neurons and sirtuin type 3 (SirT3) was observed to be involved in this neurodegenerative process [330]. NAD+-dependent deacetylase SirT3 is an important regulator of energy production and redox response by reduction of GSH [331, 332], mediating the upregulation of SOD2 and catalase [333], and activation of SOD2 [334]. Extensive deprivation of sleep is related to reduced levels of SirT3 in young adult wild type mice locus coeruleus neurons while oxidative stress levels increase presumably as a result of a combined increase in metabolic activity and decline in antioxidant response [330]. Other circadian clock related factors associated with redox homeostasis of NAD cofactors include transcriptional activator complex BMAL1 and its binding partners CLOCK and NPAS2 [335]. These clock genes were observed to be involved in glucose metabolism and redox homeostasis in peripheral tissues [335–337]. Expression levels of the master circadian clock regulator genes Bmal1 and Clock are significantly decreased in the cerebral cortex of aged mice [338] although expression levels of these genes in AD brains have not been published. Mice generated with a deletion of Bmal1 demonstrated increased systemic [337, 339] and low levels of brain oxidative stress [340] mediated by a disturbance of its transcriptional targets, including Period Circadian Regulator 2 (Per2) and albumin D-element binding protein (Dbp) [341] as well as neuropathologies and synaptic degeneration [337, 341]. For example, the kinetic occipital region in the brain of these Bmal1 knock-out mice showed a three-fold increase in level of F4-neuroprostanes when measured indicative of increased lipid peroxidation levels in neuronal cells [341]. The deletion also resulted in increased neurodegeneration caused by mitochondrial 3-nitropropionic acid which was suggested to be a direct consequence of the decrease in BMAL1 transcription [341]. Both SirT3 and BMAL1 levels are tightly regulated by circadian rhythmicity of oxidoreductase factor nicotinamide adenine dinucleotide (NAD+) [342], while, in turn cellular redox status affects the activity of clock transcription factors [335]. Proteasome expression levels and activity were observed to follow circadian oscillations that correlated with the level of carbonylated proteins [343] demonstrating that clearance of oxidized proteins are also showing circadian rhythm. It would be of interest to investigate Bmal1 and Clock expression levels in AD patient brains to establish a possible connection between the observed symptoms of sleep deprivation in AD, oxidative stress status and cognitive dysfunction.
Klotho, the aging suppressor gene, regulated by oxidative stress
Klotho is a single-pass transmembrane protein hormone containing a long type I transmembrane domain and a short secreted domain [344]. The latter is released into the extracellular space upon insulin-mediated release by ADAM family members ADAM10 and ADAM17 [345]. Mutations in the KLOTHO gene were observed to induce a human aging resembling phenotype in a transgenic mouse model which could be genetically rescued by exogenous expression of klotho cDNA [346]. Consistent with this, aging was found to be suppressed in a klotho overexpressing mouse model [347]. Single nucleotide polymorphisms of KLOTHO affect trafficking and catalytic activity of klotho which was associated to onset of aging in a human population based study [348]. Expression of the protein declines with age as was demonstrated by microarray analysis of the aging brain of a rhesus monkey model [349]. Subsequently, the klotho gene has been dubbed an aging suppressor gene which acts by regulating oxidative stress [350] because it was shown to effectively reduce urinary excretion of 8-OHdG upon renal overexpression in mice [351]. This hypothesis was further supported by the finding that different cell types incubated with Klotho protein were protected from oxidative stress and apoptosis induced by paraquat [350], hydrogen peroxide [352], glutamate and oligomeric Aβ [353]. Apart from in the kidney, the protein is also expressed in the choroid plexus in the brain with low levels of expression in the hippocampus [346, 354]. In this brain region Klotho plays a role in hippocampus-dependent memory by regulating adult hippocampal neurogenesis [355]. Mutation of klotho in a transgenic mouse model demonstrated increased levels of 8-OHdG and malondialdehyde in the hippocampus in an age-dependent manner [356]. One of the molecular models by which secreted Klotho has been suggested to regulate oxidative stress includes the insulin/IGF-1 growth signaling pathway [357]. Klotho inhibits this pathway leading to activation of Forkhead box O (FOXO) transcription factor. This, in turn, enhances the expression of ROS scavenging enzyme mitochondrial manganese SOD2 [350] indicating that Klotho may perhaps lend its anti-aging capacity by indirect regulation of the generation of an antioxidant enzyme.
PUTATIVE MOLECULAR MECHANISMS OF ALZHEIMER’S DISEASE-RELATED OXIDATIVE STRESS
One of the striking observations is that microglia in close proximity to amyloid plaques are often found to be activated and release O2·– and H2O2 [358]. Clearly, the multicellular organization of the brain may be a relevant determinant in outcome of oxidative stress in AD, and potentially also other, neurodegenerative diseases. For both Aβ and tau a number of potential contributory as well as inhibitory pathways in the process of oxidative stress generation have been proposed. For example, Aβ can reduce Cu2+ to Cu+ in a catalytic cycle that uses O2 and biological reducing agents as substrates while generating neurotoxic H2O2 [359, 360] and a peptide radical inducing oxidation and dityrosine cross-linking of Aβ were observed [93, 361]. Figure 1 provides an overview of the various cellular factors and mechanisms that are thought to associate oxidative stress and AD to date.
A protective or advancing effect of Aβ on oxidative stress may be subtly defined
A potential anti-oxidant effect of Aβ has been attributed to the reported ability of Aβ to sequestrate redox-active metals [54, 361–367]. As a result of this binding, it has for example been proposed that Aβ may quench Cu2+ preventing Cu2+ from generating H2O2. In contrast, other reports have shown that the binding of Aβ to such transition metal ions actually results in enhanced formation of ROS [368] by the reported ability of Aβ to reduce Cu2+, upon binding, to Cu1 + [369]. A subsequent publication questioned the role of Cu2+ binding in generation of ROS as it was shown that rodent Aβ1-42, in which Cu2+ binding is ameliorated, results in similar levels of oxidative stress as human Aβ1-42 [369]. Heme-a, an essential component of mitochondrial complex IV, was shown to interact with Aβ1-40, resulting in decreased assembly of this complex into a functional electron transport chain complex [90]. These in vitro observations are supported by the finding that mitochondrial whole brain levels of cytochrome-a are decreased with 25% in the AD brain [370, 371]. Both these protective and toxic roles of Aβ-metal complexation in oxidative stress receive ample support in the scientific field and perhaps the actual outcome whether metal ion binding is toxic is more subtly defined by factors such as Aβ level, Aβ aggregation state, co-occurring factors such as the availability of a reducing agent [359], redox kinetics [372], or disease stage. Supportive of such a hypothesis is the observation that Aβ was found to act as a neurotrophic agent selectively at low nM concentrations while at higher peptide concentrations neurotrophic functionality was abolished [363, 374]. Even though the functional role of Aβ has been heavily debated, it has been argued that antioxidant activity may be the primary role of this peptide in the brain [374]. However, a compelling finding arguing against an anti-oxidant function as primary role for Aβ was that oligomeric human Aβ, but not rodent Aβ, can bind two molecules of heme with sub μM affinity which results in the generation of a peroxidase [362]. Moreover, rat Aβ1-40 was reported to interact with zinc with lower affinity than human Aβ1-40. Such species specificity directly argues against a primary anti-oxidant role of Aβ although it does not rule out the potential of the peptide to also, next to its yet to identify primary role, demonstrate anti-oxidant activity via indirect routes.
Tau plays an indirect role in oxidative stress via organelle transport
Upon overexpression tau inhibits kinesin-dependent transport of mitochondria and peroxisomes into neuronal processes while the microtubular network remains intact [375]. Lack of transport of these two organelles was shown to deplete neurites from ATP and protection mechanisms against oxidative stress as was illustrated by an increased vulnerability of tau overexpressing differentiated N2a cells upon exposure to H2O2 [375]. Another parameter proposed to play a role in tau-induced oxidative stress includes tau aggregation into paired helical filaments [376]. Addition of synthesized HNE at micromolar concentrations to retinoic acid differentiated P19 embryonal carcinoma cells induced tau to crosslink into high molecular weight species [376]. Crosslinking of tau into paired helical filaments was shown to be driven by abnormal levels of phosphorylation, or hyperphosphorylation, of tau, which renders the protein insoluble and dysfunctional [377, 378]. The role of phosphorylation of tau in this process and the link to oxidative stress were highlighted by three subsequent publications showing that 1) phosphorylation of tau is partly regulated by the extracellular signal-regulated protein kinase (ERK2) which becomes activated upon exposure to H2O2 [379], and also, 2) by rapid and potent activation of transcription factor NFκB by reactive oxygen intermediate-mediated release of inhibitory factor IκB from NFκB [181], and 3) acrolein, a peroxidation product of arachidonic acid, induced p38 stress-kinase-mediated tau phosphorylation [380]. An in vitro follow up study demonstrated that co-incubation of a pseudo-phosphorylation mimicking form of tau with acrolein and methylglyoxal induced the formation of tau dimers and high molecular weight oligomers [381]. Other than phosphorylation, also the glycation of tau was observed to connect tau tangle formation with oxidative stress. Using SH-SY5Y cells, advanced glycation end product-recognizing antibodies, increased HO-1 and malondialdehyde reactivity were shown to colocalize with tau paired helical filaments [111].
Direct generation of ROS by Aβ fragments
A pioneering publication in 1994 used mass spectrometry and electron paramagnetic resonance spin trapping to demonstrate that Aβ in vitro under cell free conditions itself can fragment into free radical peptides in an oxygen-dependent but metal-independent manner [382]. The generated Aβ25-35 fragment was capable of inactivating the enzymes glutamine synthetase and creatine kinase. The authors suggested that methionine 35 may be capable of reacting with oxygen to produce sulfoxide, which, in turn, can result in radical generation [382, 383] although this hypothesis has not been experimentally verified.
Aβ aggregation state
Aβ was demonstrated to accumulate into various aggregation states ranging from monomeric to larger assemblies into amyloid plaques found upon postmortem analysis of AD patient brains. In-between these two states an apparent continuum of oligomeric aggregates with different aggregation numbers, e.g., the number of monomers per oligomer, exists, and many studies have attempted to identify and characterize the most toxic species within this range. Detailed reviews have been published on this specific topic highlighting the potential toxic role of these intermediates and their relation with the clinicopathological features of AD [384–387].
The dynamic nature of Aβ assembly hampers studies into oxidative stress
Generally, experimental studies indicate that particularly the intermediate soluble aggregated forms of Aβ are highly toxic and these species are commonly referred to as oligomers or pre- or protofibrils [388, 389]. More specifically, soluble SDS-stable dimers, extracted from AD brains [390], up to 56 kDa soluble Aβ assemblies that are capable of inducing cognitive impairment in Tg2576 mice [391] have been identified as potential toxic species acting upon AD progress. While most of these studies merely investigate the association between assembly state and loss of cellular viability, synaptic function, or cognition, consistent with these observations, a study investigating the effects of different Aβ aggregate species on oxidative stress showed that specifically prefibrillar and oligomeric Aβ1-42 potently increased levels of oxidative stress in NT2 cells as detected by HNE and H2O2 generation [392]. Using a combination of electron spin resonance spectroscopy coupled to spin trapping, a short burst of H2O2 generation was observed during early aggregation stages of Aβ1-40 [393]. Recently, incubation of Aβ with Cu2+ and H2O2 were shown to modulate Aβ self-assembly. Depending on the Cu2+ to H2O2 ratio, up to hexameric species of Aβ1-42 could be detected using SDS-PAGE with little propensity to develop into ThT positive fibrils upon prolonged incubation [394]. These Aβ1-40 and Aβ1-42 oligomers demonstrated prolonged disruption of phospholipid vesicles which is one of the proposed cytotoxic mechanisms of Aβ oligomers [394]. However, the precise aggregation number of such Aβ oligomers is difficult to pin down using most standard biophysical and biochemical techniques as a result of their heterogeneous, dynamic, and interconverting nature.
Interaction of Aβ with mitochondrial factors
Several mechanistically indirect pathways have been suggested by which means Aβ can influence mitochondrial function.
By interaction of Aβ with mitochondrial alcohol dehydrogenase
Yeast-two-hybrid based screening of the human brain and a HeLa cell model demonstrated that Aβ and mitochondrial alcohol dehydrogenase may interact [183]. This interaction was shown to be specific involving residues 12–24 of Aβ1-40 and disturbs the NAD-binding pocket and the catalytic triad of the enzyme leading to functional inhibition of nicotinamide dinucleotide binding required for the function of the enzyme [183, 396], resulting in oxidative stress and neurodegeneration [183]. Given the fact that Aβ is primarily an extracellularly generated peptide it is questionable whether Aβ and mitochondrial alcohol dehydrogenase may ever reside in close proximity of each other. However, by means of co-immunoprecipitation assays, immunogold electron microscopy and confocal microscopy it was demonstrated that Aβ can be detected in the mitochondria within the cerebral cortex of AD patients and mutant APP (mAPP) as well as mAPP/ABAD transgenic mice. Also, in these in vivo animal models Aβ was reported to co-localize with the N-terminus (residues 98–101 and 108–110) of alcohol dehydrogenase while mutations within this site abrogated Aβ binding to alcohol dehydrogenase [197]. Strengthening the suggestion that alcohol dehydrogenase may play a role in AD, degenerating neurons in the brains of AD patients were found to express up-regulated levels of alcohol dehydrogenase [197], particularly those in close proximity to Aβ deposits [183]. Although it cannot be ruled out that this observation represents an Aβ-unrelated compensatory mechanism to counteract the impaired energy homeostasis generally observed in AD neurons [313, 397]. The role of alcohol dehydrogenase in AD has been more extensively covered in a review [398].
By disruption of energy generation from mitochondria
The brain requires ATP and its intermediates for the formation of the neurotransmitter acetylcholine [399], and the critical membrane component cholesterol [374]. To accommodate these requirements it was recognized more than a century ago that the vascular system exerts some degree of plasticity to ensure sustained local activity of the neuronal network [400]. Further, astrocytes, expressing GLUT1 type glucose transporters [401], play a central role in neuronal energy supply [402] supporting the notion that the multicellular context of the brain is highly supportive of neuronal energy-consuming activities. Current PET and fMRI functional brain imaging techniques are based on the assumption that brain function is associated with brain energy consumption and from such techniques a wealth of information has been acquired over recent years on brain energy homeostasis in a variety of neurodegenerative disorders. The association between oxidative stress and energy production becomes apparent when considering that the regeneration of GSH from GSSG is an NADPH dependent process, where NADPH is mainly obtained through glucose metabolism. In various neurodegenerative disorders, including AD, a decrease in brain ATP generation is observed. Membrane fluidity changes observed upon iodoacetic acid-induced inhibition of ATP production could be rescued by treatment with anti-oxidants tirilazad and gossypol [403] suggesting that reduced ATP availability may result in oxidative stress and membrane damage. Upon aging, glucose metabolism derails progressively as a result of changes in brain insulin [404], and cortisol [405] levels. Also, activity of synaptic ATPases was significantly decreased in rat frontal cerebral cortex upon aging [406]. This condition seems to worsen in AD patients as it was shown in patients with incipient late-onset AD type dementia, that cerebral glucose utilization had reduced with 45% which progressed to 55% in advanced stages of the disease [407]. Oxidative stress, induced by NO was shown to transiently reduce ATP generation in rat astrocytes while increasing glycolysis rate in an F1F0-ATPase and adenosine nucleotide translocase dependent manner specifically in primary astrocytes to rescue this ATP-depleted phenotype [408]. At the same time, rat primary neurons exposed to NO were shown to progressively become ATP depleted which ultimately lead to cell death [408]. A number of metabolism-related enzymes have been identified to be affected in a progressive manner in AD, including pyruvate dehydrogenase, ATP-citrate lyase and acetoacetyl-CoA thiolase [409]. Activities of glycolysis and citric acid cycle related enzymes aldolase, triose phosphate isomerase, phosphoglycerate kinase, and phosphoglycerate mutase are affected in AD as a result of oxidation or nitration [410–414]. Inconsistent results have been published on phosphofructokinase activity in AD brains, with some researchers suggesting no significant reductions [415]. Subcortical regions of the brain further displayed increased activities of hexokinase, an enzyme involved in the initiating step of glycolysis, and lactate dehydrogenase in AD patient brains while activities of these enzymes in cortical regions were unaffected [416]. As the authors already suggest [416], increased activity of lactate dehydrogenase is suggestive of a metabolic need for anaerobic respiration to compensate for lost ability to generate ATP via aerobic metabolic routes. Taken together, oxidation and nitration processes of many of the enzymes involved in cellular metabolism may reduce activity of such enzymes sufficiently to explain the significantly reduced ATP generation observed in AD brains while the bioenergetic adaptation of the cell towards anaerobic routes for obtaining sufficient quantities of ATP to sustain high levels of ATP generation cannot be sufficiently compensated for. The question remains whether tau or Aβ only play an indirect role in activity reduction by inducing oxidation or nitration of these enzymes or whether a direct role, for example by activity-reducing interaction such as demonstrated for ABAD causes loss of metabolic rate. A study using co-immunoprecipitation assays combined with tandem mass tag multiplexed quantitative mass spectrometry identified glycolysis enriched proteins such as pyruvate kinase and aldolase as potential interactors with tau [417]. Using ELISA and gel filtration assays, it was reported that synthesized Aβ42 and Aβ1-28 can interact in vitro with a KD of 5 nM with rat phosphofructokinase, but not lactate dehydrogenase [418]. It is still debatable what the triad of factors oxidative stress, Aβ or tau and cellular metabolism exactly comprises in terms of molecular interactions and how they reciprocatively interact with each other. One attempt to address this question was published by Casley and colleagues who incubated isolated rat brain mitochondria either with Aβ25-35 or Aβ1-42 with or without NO to determine the relative impact of each of these factors on mitochondrial respiration using oxygen sensitive electrodes [419]. Both Aβ peptides significantly inhibited mitochondrial respiration by specifically affecting the activity of complex IV, while exposure of the mitochondria to NO substantially worsened respirational outcome.
Interaction with RAGE
RAGE is a transmembrane receptor widely expressed in all tissue types including the brain [420, 421]. One diverse group of ligands known to interact with this receptor are advanced glycation end products, AGEs. AGEs are the product of non-enzymatic aldose-mediated glycated or oxidized proteins [422], and accumulation of AGEs was shown to be aging-related [423, 424] and accelerates in conditions such as diabetes [425]. Potential clinical relevance of RAGE to AD was demonstrated using ELISA of AD brain homogenates showing a 2.5 fold increase in expression of RAGE compared to age-matched control subjects [176]. The roles of RAGE and AGEs in AD pathogenesis have been covered in a number of reviews [426, 427]. The offspring of a transgenic RAGE overexpressing mouse model crossed with Tg APP animals showed neuronal perturbation already at 3-4 months of age, and astrogliosis and reactive microglia at 14–18 months [428]. One of the general observations is that Aβ can interact with RAGE. For example, Yan and co-workers have shown that endothelial or PC12 RAGE provides a binding site for 125I-labeled synthetic Aβ on cellular surfaces. This interaction was found to result in cellular perturbation and dose-dependent generation of TBARS, NF-κB-mediated microglial activation, and cytotoxicity. TBAR generation could be blocked by pre-treatment of cultures with antioxidants probucol or N-acetylcysteine [176] demonstrating a direct or indirect association with oxidative stress. Indeed, interaction of Aβ with RAGE on the cell-surface exerted localized oxidant activity. The authors further observed that stimulation of RAGE by other ligands which do not themselves generate ROS induces intracellular generation of oxidants in target cells [176] suggesting that it is the interaction between RAGE and its ligand that induces the oxidation effect and not the ligand by itself. Not only Aβ as AD hallmark peptide appeared to show an association with RAGE-induced oxidative stress: using immunostaining it was shown that tau paired helical filaments colocalize with AGEs in AD temporal cortex tissue [116]. Also, tau was shown to be amenable to ribose-mediated glycation and exposure of SH-SY5Y cells to these glycated tau species resulted in oxidative stress without affecting cell viability [116]. It appears that AGE-RAGE interaction with AD hallmark partners results in a variety of cellular responses that can mediate oxidative stress.
Disruption of calcium homeostasis
ROS generation and Ca2+ signaling – a bidirectional paradigm
One important step in the process of synaptic transmission is the Ca2+-regulated fusion of neurotransmitter containing synaptic vesicles with the pre-synaptic membrane which results in the release of neurotransmitters in the synaptic cleft. Apart from neurotransmitter release a plethora of other cellular mechanisms are regulated by Ca2+-dependent signaling, for example regulation of membrane excitability, neuronal growth and differentiation as well as expression of a wide range of genes that are activity-induced and apoptotic processes. Extracellular and endoplasmic reticular Ca2+ levels largely exceed intracellular levels and this sustained Ca2+ homeostasis is an important factor required for cellular functioning. A number of reviews that extensively cover neuronal Ca2+ homeostasis mechanisms have been published [429–432]. To aid Ca2+ homeostasis, Ca2+ levels in neuronal cells are strictly regulated by controlled release from various intracellular and extracellular pools. Release of Ca2+ from the extracellular pools into the cell is mediated by factors such as voltage-gated Ca2+ channels, N-methylo-D-aspartate (NMDA) receptors and transient receptor potential channels while intracellular Ca2+, stored in the endoplasmic reticulum, is released via the inositol triphosphate receptor (Ins[1,4,5, 1,4,5]P3 R) and ryanodine receptor (RyRs) [429]. These factors act in a concerted manner to regulate intracellular Ca2+ levels in a temporal and spatial manner. Reports show that dyshomeostasis of Ca2+ levels and oxidative stress amplify each other in a bidirectional manner. For example, ROS species superoxide anion, hydrogen peroxide and hydroxyl radicals are known to regulate signaling pathways involving Ca2 + . Mitochondrial Ca2+ mediates activity of a number of mitochondrial enzymes involved in the tricarboxylic acid cycle and oxidative phosphorylation and, as such, increased Ca2+ levels were reported to elevate cellular metabolic rate [433–435]. In turn, cellular metabolic rate was shown to be directly proportional to ROS generation in rat and porcine lung [436], and hepatomas [437]. Bidirectionality of the ROS-generation/Ca2+ signaling paradigm was observed by functional impairment of membrane-bound receptors and channels that regulate influx or efflux of Ca2+ mediated by oxidative stress-induced lipid peroxidation. Ca2+ dyshomeostasis was reported in neurodegenerative disorders in general, including AD. For example, whereas wild type presenilin was reported to form Ca2+ permeable channels allowing Ca2+ leakage from the endoplasmic reticulum [438, 439], some, but not all FAD related mutations in Psen1 and 2, show deficiencies in Ca2+ leak function in a mouse embryonic fibroblast model, primary fibroblasts obtained from patients and a planar lipid bilayer [439, 440]. Apart from neurons, astrocytes also play an ion regulating role supporting neuronal activity. Astrocytes act in a concerted effort to regulate Ca2+ levels via release of ATP which subsequently binds to purine receptors on the membrane of adjacent astrocytes [441]. ATP binding then results in release of intracellular Ca2+ via the phospholipase C beta/inositol triphosphate (IP3) pathway [442]. Using a microarray analysis, it was reported that the expression of 32 Ca2+ signaling pathway-related genes were altered in astrocytes at various stages of AD pathology [443]. Aβ was reported to perturb astrocytic Ca2+ regulation indirectly by inducing oxidative stress which impairs membrane Ca2+ pumps and enhances Ca2+ influx through voltage-dependent channels and ionotropic glutamate receptors [444–446].
By potentiating mitochondrial permeability transition pore formation
The mitochondrial permeability transition pore (mPTP) plays an important role in mitochondrial Ca2+ homeostasis [447]. Its molecular composition and mechanism of action have been under debate in the last years. Initially, the mPTP was thought to consist of the voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocase (ANT) in the inner membrane and Cyclophilin D (CypD) in the matrix. In this way, when CypD translocates to the inner mitochondrial membrane to interact with ANT and VDAC [448], the mPTP opens allowing non-selective exchange of calcium [449–451]. More recent findings dismiss this initial idea and indicate that VDAC is not a key component of the mPTP [452, 453] and ANT has only a regulatory function rather than being a core unit of the mPTP [454]. Only CypD, a peptidyl-prolyl isomerase F located in the mitochondrial matrix, remains a critical molecule in the mPTP in both postulations. Consistent with this, studies in animal models have shown that mPTP formation can be efficiently blocked by the addition of a cyclophilin D inhibitor, cyclosporine A (CSA) or by depletion of CypD [448, 456]. Short and transient mPTP opening has been shown to occur under physiological conditions, allowing rapid passage of protons, Ca2+ and other substances of a size up to 1.5 kDa [457, 458]. However, prolonged mPTP leaking may result in mitochondrial disruption. Apart from a high concentration of Ca2+ in the mitochondrial matrix [459], ROS can trigger mPTP opening. Moreover, cytosolic ROS was observed to induce signals leading to mPTP opening resulting in further ROS release and this phenomenon is referred to as “ROS-induced ROS release (RIRR)” [460]. Several lines of investigation have proposed that Aβ-induced ROS and increased Ca2+ levels could well link with mPTP formation. In this regard, increased CypD expression has been identified in neurons of the hippocampus and temporal lobe of AD patients [461]. CypD was shown to specifically bind to Aβ oligomers both in vitro and in vivo in a dose-dependent manner facilitating permeability transition and ROS formation causing mitochondrial dysfunction [461–463]. Removal of CypD improved cognitive and synaptic function in a mouse model for AD [462], protecting cells from Ca2+, oxidative stress or Aβ-induced cell death [448, 465], and restored oligomeric Aβ42-mediated loss of axonal mitochondrial transport in neurons. The role of ROS was highlighted by demonstrating rescue of Aβ-induced loss of axonal mitochondrial movement upon administration of Probucol, an anti-oxidant agent [466]. As a result of these observations, various studies embarked on targeting CypD/mPTP inhibition as potential treatment for neurodegenerative diseases [467, 468].
CONCLUSIONS
Oxidative stress is an early clinical feature of AD, as well as other neurodegenerative disorders. AD-related oxidative stress arises as a result of increased generation of ROS, induced by mitochondrial failure, but also decreased levels of endogenous antioxidants are commonly observed in AD. Many of the biomacromolecules that are part of normal cellular physiology are susceptible to oxidative modification altering their function. Also, Aβ and tau, two AD hallmark proteins are subject to oxidation, while Aβ itself was reported to induce the formation of ROS. A number of genetic factors have been identified to play a role in AD-related failure to maintain physiological ROS levels within strict limits. For example, clusterin, apolipoprotein E, klotho, and enzymes involved in the AβPP processing machinery regulating Aβ generation are related, either directly or indirectly, to oxidative stress. Various molecular mechanisms explaining the association between oxidative stress and AD have been identified. Aβ and its potent interaction with Cu2+ was identified as key feature in AD-related oxidative stress. But also various other mitochondria-associated factors play a role, including mitochondrial permeability transition pore formation, regulation of cellular metabolic pathways and Ca2+ signaling. The association between oxidative stress and AD appears complex, bi-directional, and self-reinforcing.