
Abstract
More than half of the patients with Alzheimer’s disease (AD) have comorbidities including TDP-43 and Lewy bodies, which are also associated with frontotemporal lobar degeneration and dementia with Lewy bodies, respectively. These comorbidities may help explain the overlapping neuropsychiatric symptoms between AD and other dementias. Data on 221 AD patients with Neuropsychiatric Inventory-Questionnaire were obtained from the National Alzheimer’s Coordinating Center. TDP-43 was associated with aberrant motor activity, whereas Lewy bodies were associated with anxiety, irritability, sleep behavior, and appetite problems. The associations between these comorbidities and neuropsychiatric symptoms were more significant for patients with sparse diffuse plaques.
INTRODUCTION
Current criteria for the pathologic diagnosis of Alzheimer’s disease (AD) recommend assessment and classification based on the presence and distribution of amyloid-β deposits and neurofibrillary pathology [1]. However, more than half of the patients clinically diagnosed with AD have comorbid pathologies in addition to plaque and tangle pathology [2]. These include pathologies shared with other neurodegenerative disorders. For example, 30–57% of individuals with AD pathology have comorbid 43 kDA TAR DNA-binding protein (TDP-43) pathology in the medial temporal lobe, which eventually spreads to occipitotemporal cortex, basal ganglia, and frontal neocortex [3, 4]. Up to 50% of individuals with AD have limbic or neocortical Lewy bodies (LB) [5].
Abnormal TDP-43 accumulation is associated with frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis [6], whereas LB pathology leads to Parkinson’s disease (with and without dementia) and dementia with Lewy bodies (DLB) [7]. These different dementia types are associated with different neuropsychiatric profiles. Neuropsychiatric symptoms common in AD include mood fluctuations, agitation, apathy, irritability, aberrant motor behavior, and sleep disturbances [8]. FTLD is associated with disinhibition, apathy or inertia, loss of sympathy or empathy, perseverative, stereotyped, or compulsive behavior, hyperorality, and dietary changes [9]. Common neuropsychiatric symptoms of DLB include sleep disturbances, visual hallucinations, depression, and delusions [10]. Although neuropsychiatric phenotypes of AD, FTLD, and DLB differ from one another, many features also overlap.
In this study, we investigated whether comorbid pathologies in AD are associated with FTLD- or DLB-like neuropsychiatric symptoms.
MATERIAL AND METHODS
Study sample
Data were obtained from the National Alzheimer’s Coordinating Center (NACC) Neuropathology Data Set (NP) and The Uniform Data Set (UDS) for visits conducted between September 2005 and September 2017. Detailed descriptions of these datasets can be found elsewhere [11, 12].
Participants: 1) clinically diagnosed as AD, 2) meeting the pathologic criteria for at least intermediate probability of AD from the revised National Institute on Aging- Alzheimer’s Association (NIA-AA) criteria, 3) with pathological assessment for TDP-43 immunoreactive inclusions in the amygdala, 4) with pathological assessment for LB, and 5) evaluated with Neuropsychiatric Inventory-Questionnaire (NPI-Q) [13] were included. Exclusion criteria was a clinician diagnosis of FTLD or DLB as the primary cause of cognitive impairment. In total, 221 participants were included. Dementia severity was assessed by CDR® Dementia Staging Instrument [14].
TDP-43 and LB pathology
In the NP data set, distribution of TDP-43 immunoreactive inclusions are evaluated in the spinal cord, amygdala, hippocampus, entorhinal/inferior temporal cortex, and neocortex. Patients were grouped as TDP-43 positive or negative based on the TDP-43 immunoreactivity within the amygdala as TDP-43 in AD typically appears first in the amygdala [15]. LB pathology is assessed in brainstem, limbic or amygdala, neocortex, the olfactory bulb, and unspecified brain regions. Individuals were considered LB-positive or negative based on limbic or amygdala predominant, or neocortical (diffuse) LB pathology.
Neuropsychiatric assessment
Neuropsychiatric symptoms were evaluated using the NPI-Q, a brief questionnaire assessing the presence and severity of neuropsychiatric symptoms commonly observed in dementia [13]. These symptoms are delusions, hallucinations, agitation, depression, anxiety, euphoria, apathy, disinhibition, irritability, aberrant motor behavior, disturbances of sleep and nighttime behavior, and abnormalities of eating and appetite. Scores for each item are rated by the caregiver from 0 to 3. Non-present symptoms are rated as 0. The severity ranking for each item ranges from 1 to 3 (1 = mild; 2 = moderate; and 3 = severe). Total score ranges from 0 to 36, with higher scores indicating worse symptoms. If a participant had longitudinal NPI-Q data, the assessment closest to time of death was used to better reflect the neuropsychiatric profile closest to the pathology findings.
Statistics
SAS (SAS Institute Inc., Cary, NC) and IBM SPSS 25.0 (Armonk, NY) were used. Comparisons were made between TDP-43 positive and negative, between LB-positive and negative groups using Chi-square and t-tests. Main effects of TDP-43 and LB on NPI-Q scores; interactions of TDP-43 and LB; TDP-43 and diffuse plaque density; and LB and diffuse plaque density were investigated with demographics (age, years of education, gender) and AD pathology stage (Thal phase, Braak stage, CERAD score) as covariates with Bonferroni correction. p < 0.05 was considered statistically significant.
RESULTS
Out of 221 eligible individuals, 121 (54.8%) were TDP-43 positive and 98 (44.3%) were LB-positive. Sixty-four (29.0%) had both TDP-43 and LB; 57 (25.8%) had only TDP-43; 39 (17.6%) had only LB; 61 (27.6%) did not have either pathology. LB was more common in individuals with TDP-43 and vice versa (χ2 = 5.152, p = 0.023) (Fig. 1).
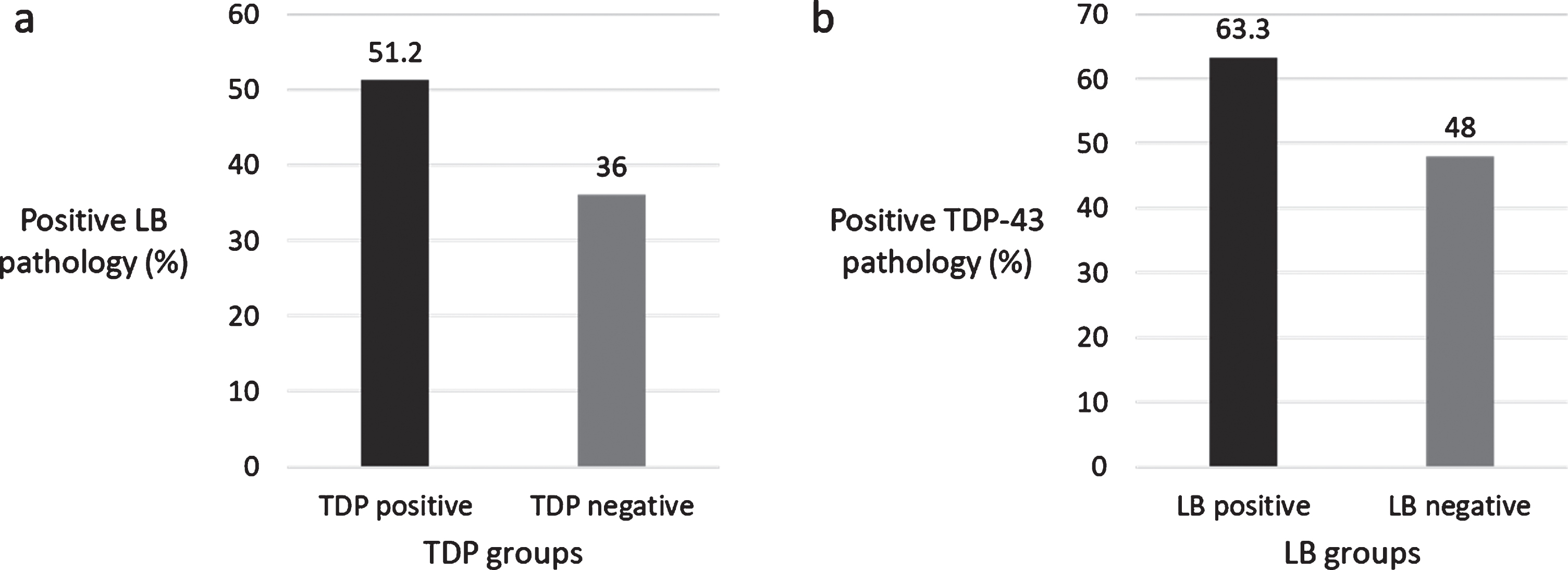
a) Percentage of LB pathology in TDP-43 groups, b) Percentage of TDP-43 pathology in LB groups.
Demographics and disease features are summarized in Table 1. NPI-Q scores and statistical results are given in Table 2. The interval between ages during NPI-Q and death (timepoint for pathological assessment) was similar within TDP-43 (TDP-43 positive: 6.06 (2.74) versus TDP-43 negative: 5.73 (2.81); t(219) = –0.875 p = 0.382) and LB groups (LB-positive: 6.09 (2.96) versus LB-negative: 5.76 (2.61); t(219) = –0.873, p = 0.384).
Demographics and disease features
All variables are reported as mean (standard deviation) or percentage where stated. Statistical significance is marked with *.
NPI-Q scores of the groups and statistical results
All variables are reported as mean (standard deviation) or percentage where stated. Statistical significance is marked with *. n/a, not applicable; multivariate analysis of covariance was only run for quantitative variables.
TDP-43 group comparisons
The TDP-43 positive group had a higher frequency of hippocampal sclerosis, more advanced Thal phase and CERAD score. Symptom presence or total NPI-Q score did not significantly differ. Depression was more severe in the TDP-43 negative; aberrant motor behavior was more severe in the TDP-43 positive group.
LB group comparisons
The LB-positive group was younger and more educated. The LB-positive group had a higher frequency of APOE ɛ4 carriers, more advanced Thal phase and CERAD score. Anxiety was more frequent in the LB-positive group. Apathy was more severe in the LB-negative; total NPI-Q score was higher and anxiety, irritability, sleep behavior, and appetite problems were more severe in the LB-positive group.
Interactions between TDP-43, LB, and diffuse plaque density
Significant interactions are depicted in Supplementary Figure 1. TDP-43 and LB had an interaction for aberrant motor behavior. LB pathology was associated with more severe aberrant motor behavior within the TDP-43 negative group.
TDP-43 and diffuse plaque density had interactions for total NPI-Q score, depression, anxiety, irritability, aberrant motor behavior, sleep behavior, and appetite problems. TDP-43 pathology was associated with higher total NPI-Q score, depression, anxiety, irritability, aberrant motor behavior, sleep behavior, and appetite problem severity for patients with sparse diffuse plaques, and lower scores for patients with moderate plaques.
LB and diffuse plaque density had interactions for total NPI-Q score, apathy, and irritability. LB pathology was associated with increased total NPI-Q scores; the increase was greatest for patients with sparse diffuse plaques and least for individuals with frequent diffuse plaques. LB pathology was associated with increased apathy severity for patients with sparse diffuse plaques; and increased irritability severity for patients with sparse and moderate diffuse plaques.
DISCUSSION
In this study, we investigated whether TDP-43 and LB in AD are associated with specific neuropsychiatric symptoms. TDP-43 was associated with increased severity of aberrant motor behavior; while LB was associated with increased anxiety, irritability, sleep behavior, and appetite problems. For the TDP-43 negative group, LB positivity was associated with more severe aberrant motor behavior. Both TDP and LB had several interactions with diffuse plaques, with more apparent deterioration associated with TDP-43 and LB observed in individuals with sparse diffuse plaques.
More common TDP-43 pathology in the LB-positive group (and vice versa), as well as more advanced amyloid deposition in both TDP-43 and LB-positive groups compared to their negative counterparts suggest that comorbidities are more common in more advanced AD. This is not surprising, as amyloid has been shown to promote both LB and TDP-43 pathology in AD [16, 17]. The different hippocampal sclerosis frequency between the TDP-43 groups was also expected, as hippocampal sclerosis is the most significant predictor of comorbid TDP-43 pathology in AD [18]. Significant differences between the LB groups included age and APOE ɛ4 status. Similar to a previous study [19], the LB-positive group was younger although they had more advanced amyloid deposition. Additionally, having at least one APOE ɛ4 allele was more frequent in the LB-positive group. Considering that APOE ɛ4 is associated with AD onset at a younger age [20], age differences between the LB groups may be due to higher frequency of APOE ɛ4 carriers in the LB-positive group.
TDP-43 follows a distinct spread pattern in AD compared to that observed in FTLD [15], which may explain why it does not appear to produce FTLD-like neuropsychiatric symptoms in AD. Our findings are consistent with studies reporting that TDP-43 in AD does not affect the total NPI score [21] or neuropsychiatric symptom occurrence [22]. However, TDP positivity was associated with more severe aberrant motor behavior, and less severe depression. Similar results have been shown in FTLD patients compared to AD, with more severe apathy and disinhibition in FTLD [23]. Although our TDP-43 positive group did not exhibit a complete FTLD-like profile, more severe aberrant motor activity and less severe depression suggest a potential TDP-43 effect in AD.
The association between comorbid LB and neuropsychiatric symptoms in AD have been inconsistent with some reporting increased delusions, hallucinations, depression, confusion, and motor disturbances in LB-positive AD [19, 25], and some reporting no differences between LB-positive and negative AD [26–28]. Compared to AD, higher NPI total, delusions, hallucinations, anxiety, aberrant motor behavior, sleep behavior, and appetite problem scores in DLB have been shown [29]. Our findings were similar, suggesting a partially DLB-like profile in LB-positive AD. Both frequency and severity of anxiety was increased in the LB-positive group, in line with studies showing that anxiety is more common in DLB than AD [30, 31]. However, as delusions and hallucinations are important aspects of DLB [32], the lack of delusion and hallucination differences prevent the full scope of DLB-like profile to be associated with LB comorbidity in AD in our study.
There were also several interactions between TDP-43, LB, and diffuse plaques. LB was related to more severe aberrant motor behavior without TDP-43 positivity, supporting previous reports on more severe aberrant motor activity in DLB compared to AD [29]. TDP-43 pathology was associated with increased severity of multiple symptoms in AD patients with sparse diffuse plaques; and decreased severities in patients with moderate diffuse plaques. LB pathology was associated with increased NPI-Q total score as well as apathy, and irritability severity especially in AD patients with sparse diffuse plaques. This association decreased as diffuse plaque frequency increased. These patterns of interaction suggest that effects of comorbid pathologies in AD may be more pronounced in the earlier stages.
Compared to the 1997 NIA-Reagan neuropathologic criteria [33], the sensitivity of clinical AD diagnosis ranges from 70.9 to 87.3%, while the specificity is 44.3–70.8% [34]. Primary pathologies in misdiagnosis are tauopathy, cerebrovascular disease, LB, and hippocampal sclerosis. Utilizing neuropathologic criteria for the accurate diagnosis may help determine the mechanisms behind clinical symptomatology better. Multisite studies with pathological data such as NACC allow for the assessment of a large number of patients with neuropathologically confirmed AD diagnosis, while earlier studies relied on clinical diagnosis or small numbers of individuals with neuropathologically confirmed AD. Nevertheless, evaluation for TDP-43 pathology was only recently included in the NACC, which restricted our sample size. In AD, focal amygdala involvement represents a very early stage of TDP-43 pathology [4]. Involvement of multiple regions may be associated with more neuropsychiatric symptoms, leading to different neuropsychiatric profiles for advanced TDP-43 stages. Although our sample size was not small, it was not large enough to allow stratification by dementia stage, TDP-43, or LB location. Additionally, the majority of the patients did not exhibit symptoms on the NPI-Q. Future studies may benefit from assessing the effects of comorbid pathologies in a bigger sample with longitudinal visits.
Although the interval between age when the NPI-Q was assessed and age at death was not different between the groups, the interval was around six years, which is a long gap to represent the accompanying pathology during the time of NPI-Q. Evaluation closer to death can yield more reliable results. Future studies may also benefit from using NPI, or other scales for more detailed assessment of neuropsychiatric symptoms, as the NPI-Q is a brief scale.
In conclusion, TDP-43 and LB comorbidities are partially associated with common FTLD- or DLB-like neuropsychiatric symptoms. Although both comorbidities are associated with advanced amyloid deposition, their neuropsychiatric effects may be more pronounced in the earlier stages of AD. Elucidating the pathophysiology can help establish new and more efficient treatment targets for AD-related neuropsychiatric symptoms.
Footnotes
ACKNOWLEDGMENTS
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG005131 (PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).