
Abstract
Neurons consume the highest amount of oxygen, depend on oxidative metabolism for energy, and survive for the lifetime of an individual. Therefore, neurons are vulnerable to death caused by oxidative-stress, accumulation of damaged and dysfunctional proteins and organelles. There is an exponential increase in the number of patients diagnosed with neurodegenerative diseases such as Alzheimer’s (AD) as the number of elderly increases exponentially. Development of AD pathology is a complex phenomenon characterized by neuronal death, accumulation of extracellular amyloid-β plaques and neurofibrillary tangles, and most importantly loss of memory and cognition. These pathologies are most likely caused by mechanisms including oxidative stress, mitochondrial dysfunction/stress, accumulation of misfolded proteins, and defective organelles due to impaired proteasome and autophagy mechanisms. Currently, there are no effective treatments to halt the progression of this disease. In order to treat this complex disease with multiple biochemical pathways involved, a complex treatment regimen targeting different mechanisms should be investigated. Furthermore, as AD is a progressive disease-causing morbidity over many years, any chemo-modulator for treatment must be used over long period of time. Therefore, treatments must be safe and non-interfering with other processes. Ideally, a treatment like medicinal food or a supplement that can be taken regularly without any side effect capable of reducing oxidative stress, stabilizing mitochondria, activating autophagy or proteasome, and increasing energy levels of neurons would be the best solution. This review summarizes progress in research on different mechanisms of AD development and some of the potential therapeutic development strategies targeting the aforementioned pathologies.
Keywords
ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is a debilitating disease which can be characterized by severe memory loss and confusion, and can lead to stark changes in mood and personality which may lead to depression [1]. This disease presumably makes up an estimated 75–90% of all dementia cases in Canada [1]. It has been projected that by the year 2030, the number of dementia patients is expected to double. With this in mind, it is now imperative that effective agents are developed, and new therapeutic strategies are employed to combat the progression and devastating outcomes (or aftermath of) AD.
The etiology of this disease is not well understood but is distinguished by the presence of neurofibrillary tangles (NFTs) and amyloid-β (Aβ) plaques in the brain which lead to loss of synapses and death of neurons found in the hippocampus and cerebral cortex. The lesions seen in these areas seem to precede the clinical manifestations of the disease [2]. There are many risk factors associated with the progression of this disease, most being sporadic in origin and are related to both external factors and genetic predispositions [3]. It has been postulated that genetic factors may be contributing to familial type AD, but only make up approximately 1% of all cases [4].
GENES ASSOCIATED WITH AD
There are three autosomal dominant genes associated with familial forms of AD, including genes coding for: amyloid precursor protein (APP), presenilin-1 (PS-1), and presenilin-2 (PS-2). The APP gene is on chromosome 21q21, and when the gene product, amyloid-β protein precursor (AβPP), is proteolytically cleaved, it yields the Aβ protein [5]. In the absence of any mutation in the APP gene, the Aβ protein functions without adverse effects and may be a regulator in synapse formation [4]. Several other studies have suggested possible functions of Aβ including reduction of oxidative stress and a pro-inflammatory response during microbial invasion, although further validation is required [6]. The mutated protein (Aβ40 and Aβ42) arises through missense mutations, most of which are located in the secretase cleavage sites or the AβPP transmembrane domain [4]. Mutant Aβ is able to form aggregates that are translocated inside the membrane of the mitochondria leading to mitochondrial dysfunction [7]. The 32 discovered mutations in these areas are responsible for 10–15% of early onset familial AD [8]. PS-1-related AD is the most common form of familial AD and is responsible for 20–70% of cases [8]. The PS-1 gene, found on chromosome 14q24.3, produces a major component in atypical aspartyl protease complexes that forms the catalytic core of the membrane bound γ-secretase. This γ-secretase in turn cleaves AβPP [4, 9] and is found in small amounts in the endoplasmic reticulum, Golgi, and mitochondrial membranes [9]. Missense mutations found along one of the 12 exons on this gene were found to produce amino acid substitutions which translate to an increase in the Aβ42 isoform [4]. The last of the familial type AD genetic variants is the PS-2 mutation. This gene is found on chromosome 1q42 and similar to PS-1, its function is to form a portion of the γ-secretase responsible for splicing AβPP [10].
There are other genetic predispositions that can lead to an increased incidence of AD that are not familial in nature. Sporadic forms of AD may be attributed to mutations in the genes coding for apolipoprotein E (APOE) and microtubule-associated protein tau. Tau is a protein that assists in microtubule assembly and stabilization [11]. Its gene is on chromosome 17q21 containing 16 exons with 2, 3, and 10 which is capable of being alternatively spliced generating 6 different isoforms [12]. Disease related to tau abnormalities (tauopathies) seem to be localized but not limited to exon 10 (some are on exon 19 and others between 8–12), which encode for protein sites that allow for hyperphosphorylation [12]. The hyperphosphorylated tau proteins are no longer able to bind to microtubules and form aggregates known as NFTs [11]. Although NFTs are a classic marker in the diagnosis of AD, their role in the disease remains poorly understood. It has been suggested that they may be involved in a protective mechanism during times of high oxidative stress, as these tangles have been observed in healthy long-lived neuronal cells [13]. The brain has the highest expression levels of APOE in the body aside from the liver [14]. Its gene is located on chromosome 19q13.2 which contains 5 exons that encodes a single protein [4]. The APOE gene contains three different alleles; ɛ2, ɛ3, ɛ4, which leads to four different potential isoforms [4]. The normal function of this protein is related to the metabolism of lipids and their transport within and between cells [15]. It has recently been elucidated that the cause of APOE related AD is due to the isoform APOE ɛ4 which appears to promote the Aβ aggregation and the hyperphosphorylation of tau [16]. The effects that these genes have on the neural cells and tissue are very widespread but one notable target that is evidently influenced by these pathologies is the mitochondria. In addition, genes involved in the regulation of autophagy and those involved in detoxification mechanisms could also be potentially involved in the development neurodegenerative diseases including AD [17].
AD-RELATED MITOCHONDRIAL DYSFUNCTION
The mitochondrion is a double membrane bound organelle within the cell which is vitally responsible for the generation of the majority of the cells’ energy reserves. This work is achieved through various intracellular pathways which rely on proton gradients named mitochondrial membrane potential (MMP) formed along the inner membrane of the organelle. A way that mitochondrial function is regulated is through intracellular levels of Ca2+ which also aids in generating pH gradients for proper MMP [18]. When the aberrant forms of Aβ are present, they are able to traverse the mitochondrial membrane and block the function of complex 1 thus decreasing the mitochondria’s ability to produce ATP [19]. Another more devastating effect of pathogenic Aβ, through mechanisms not well understood, is the large influx of calcium ions into the cell cytoplasm by activation of voltage gated ion channels or excitatory amino acid channels, such as glutamate activated channels [20]. This influx of excessive or non-physiological levels of Ca2+ leads to a destabilization of Ca2+ homeostasis and mitochondrial Ca2+ overload [21]. Furthermore, increased activity of mitochondria associated endoplasmic reticular (ER) membranes and ER-mitochondrial communication have been observed within PS-1 mutated fibroblasts leading to excess ER calcium and further contribution to breakdown of Ca2+ homeostasis [22]. This overload triggers the formation of a transition pore leading to the release of cytochrome C, and MMP collapse [21]. In addition, it seems that Ca2+ increases the amount of reactive oxygen species (ROS) by two main pathways: generation of nitric oxide which inhibits complex IV of the electron transport chain, and increase of the rate of turn over for the citric acid cycle and electron transport chain leading to increased leakage of ROS [21]. These pathological cellular events of AD are summarized in Fig. 1. A notable feature in AD is the accumulation of defective mitochondria and rapid degradation of healthy mitochondria. As mitochondrial injury escalates, they enlarge due to improper fission and take up more space in the cell contributing to cellular hypertrophy [23]. Consequently, this enlargement renders nutrient transport to the cell’s center inefficient leading to starvation and death [23]. Collectively, these mitochondrial events generate a chaotic intracellular condition propelling cell death.
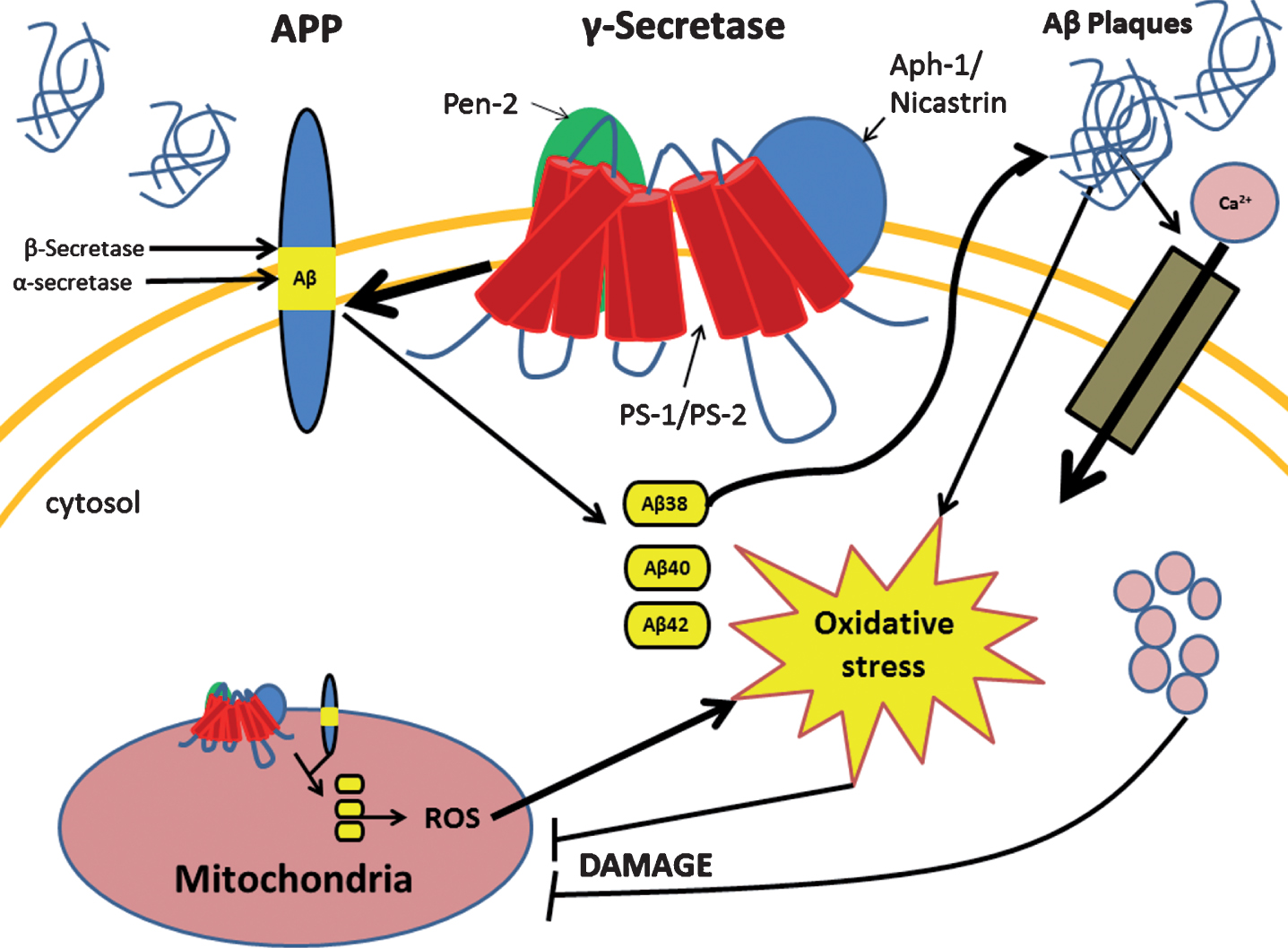
Presenilin-1 (PS-1) mutation leads to several changes in the cell due to improper cleavage of the amyloid-β protein precursor (AβPP). PS-1 mutation causes the improper cleavage of AβPP leading to aberrant forms of amyloid-β (Aβ) which occurs in both the cell and mitochondrial membrane. Accumulation of aberrant Aβ proteins form Aβ plaques which cause increased intracellular oxidative stress and influx of calcium ion. Both of these events lead to mitochondrial dysfunction which in turn increases oxidative stress and cellular damage.
AD-RELATED OXIDATIVE STRESS
Oxidative stress is a cellular pathological phenomenon produced when cell detoxification mechanisms are unable to compensate with increases in oxidative free radicals. This is especially important in AD as studies have suggested that the increase in free radicals precedes any other hallmark of AD [24, 25]. The origin of the initial increase in ROS in AD is still unknown, but in some cases, the initial influx has been seen to originate in the mitochondria [25 –27]. It has been shown that mutations of the CO1 and CO2 genes, which encode subunits I and II of cytochrome c found in mitochondrial DNA, are responsible for mitochondrial dysfunction in some late onset AD patients [27]. Such dysfunction was seen to yield increases in ROS which preceded any of the markers attributed to AD. Enhanced ROS production leads to the oxidation of major biomolecules including nucleic acids, proteins, and lipids, thus creating increased amounts of dysfunctional mitochondria. This creates a loop which amplifies oxidative stress as these damaged mitochondria continue to produce ROS [28]. The brain is extremely vulnerable to oxidative imbalances due to its high demand for energy, large amounts of oxygen consumption, relatively low amounts of antioxidants, and its high levels of easily oxidizable polyunsaturated fatty acids and ROS catalyst iron [29]. It has been demonstrated that when there are high concentrations of H2O2, ROS interacts with iron (II) and converts it to iron (III) and (OH·) via the Fenton reaction pathway [30]. The newly formed iron (III) has been associated with formation of senile plaques, NFTs, and neuropil threads [30]. Another metal that seems to play a role in AD is Cu2+, which associates with Aβ. When in high concentrations, the Aβ-Cu2+ complex is able to convert phospholipids in to phospholipid hydroxides which then get degraded to reactive aldehydes [31]. These aldehydes also contribute to ROS insult and can lead to cell death [31]. Another marker in AD produced by oxidative stress is the formation of protein carbonyls (aldehydes and ketones) [32]. These carbonyls are most often formed on the side chains of Pro, Arg, Lys, and Thr and resulting in aberrant protein function [32, 33]. Another cause of oxidative stress-associated protein dysfunction is protein glycation, considered a hallmark of AD. Protein glycation is a modification that occurs when proteins are in contact with reducing sugars and become oxidized when unmediated by appropriate enzymes [34]. Each of these processes all seem to affect common proteins, these being: β-actin, β-tubulin, p-Tau, ApoE, and most often Aβ [30 –34]. Speculations suggest Aβ and NFTs to be compensatory mechanisms as neurons seen with more plaques had succumbed to less oxidative damage than those that had fewer deposits [24]. This positive feedback mechanism produced by oxidative stress and the change in macromolecular function leads to devastating changes in cell compensatory mechanisms and cell cycle eventually leading to the induction of cell death. It has also been observed that hyperhomocysteinemia is another risk factor that contributes to ROS damage in AD. Individuals with AD and mild cognitive impairment had increased levels of total homocysteine (tHcy) with a negative correlation of total antioxidant capacity (TOC). Furthermore, there was a negative correlation observed between disease duration and TOC in patients. Levels of tHcy, TOC, and ROS were also measured in patients with vascular dementia. While tHcy and ROS were similar to AD patients, the same decrease in TOC in AD patients was not observed in patients with vascular dementia. Thus, it is possible that oxidative imbalance in AD could be due to a decrease in TOC [35].
AUTOPHAGY INHIBITION IN AD
Autophagy is a cellular mechanism of recycling old or damaged cytoplasmic constituents and organelles through a degradative pathway [36, 37]. This is generally a pro-survival response in the cell that is activated during times of stress such as starvation and pathogenic invasion but is also imperative in the maintenance of normal cellular function via elimination of potentially hazardous materials including damaged organelles and misfolded proteins [37]. There are three main classifications of autophagy: macroautophagy, microautophagy, and chaperone-mediated. Microautophagy consists of a lysosome surrounding its target followed by target degradation. In contrast, chaperone-mediated autophagy occurs when specific chaperones complex with proteins containing the peptide sequence KFERQ. This complex then binds with lysosome-associated membrane protein (LAMP) which brings the protein into the lysosome for degradation [36]. In AD, macroautophagy is the most prominent dysfunctional form of autophagy, either operating abnormally or totally nonfunctional [23 , 38]. During optimal functioning, macroautophagy first involves the formation of a double membraned vesicle surrounding the targeted cytoplasmic constituents (usually organelles) followed by a decrease in pH and, ultimately orchestrating fusion of the vesicle with a late endosome or lysosome. Subsequently, the acidic pH and degradative enzymes are able to break down the vesicular contents which can be recycled or disposed of [36, 37]. As discussed earlier, oxidative stress plays a prominent role in AD and its effects also influence autophagic processes. It seems many of the regulatory proteins of autophagy such as mTOR, which is responsible for inhibition of autophagy. In addition, Atg3, an upstream activator for the conversion of LC3-I to LC3-II plays a crucial role in the expansion of the autophagosome and are susceptible to changes in function due to redox reactions caused by oxidative stress occurring at cysteine residues present in their active sites [36]. Proteins similar to these as well as other cell constituents that have been negatively affected by oxidative stress lead to a breakdown of the chaperone mediated autophagy, which leads to an even greater burden on the macroautophagic mechanism [36]. Another factor, possibly leading to the decrease in function of autophagy, was presented by Brunk & Terman (2002), which showed that over time lysosomes which play a critical role in autophagy will begin to accumulate a substance called lipofuscin which is a substance that cannot be degraded and cannot be removed from the cell leading to lysosomal dysfunction [23]. The accumulation of lipofuscin is presumably exacerbated during cellular stress. An interesting suggestion provided by De Grey (2005) states that mutant mitochondria have a reduced proton gradient which would lessen the oxidative damage they would be susceptible to [39]. In contrast, healthy mitochondria would be more susceptible leading to their preferential degradation by autophagic processes, thus increasing the overall number of defective mitochondria found in the cell. V-ATPase is a protein located in the membrane of autophagosomes which function to increase the proton concentration in the lumen [38]. The decrease in pH of the autophagosome appear to direct its targeting to the lysosome for degradation of its cargo [38, 40]. Lee (2010) has demonstrated that PS-1 mutation leads to a failure of v-ATPase to target the lysosome which leads to stagnancy of the autophagic vesicles within the cell. It was shown that autophagic vesicles exhibit AβPP secretase activities and other reactants necessary to generate Aβ [41]. Yu (2004) had proposed that because of the accumulation of autophagic vesicles present in AD leads to an increased accumulation of Aβ within these cells, this would further exacerbate the problems associated with AD.
Our studies have shown that autophagy is reduced in fibroblast containing the PS-1 mutation as indicated by reduced LC3-II to LC3-I ratio [42]. This observed reduction in the LC3-II/LC3-I ratio may be a contributing factor to the increased levels of ROS that was also observed. As stated earlier, this increased oxidative stress may be affecting mTOR and Atg3 resulting in changes in the LC3-II/LC3-1 ratio [36]. Furthermore, we have also observed that PS-1 mutated fibroblasts also exhibited greater amount of superoxide generated by mitochondria as indicated by increased MitoSOXTM red mitochondrial superoxide indicator [42]. This may be suggestive of increased dysfunctional/mutant mitochondria, which are less targeted compared to healthy mitochondrial as suggested by De Grey [39].
The role of transcription factor EB (TFEB) in AD is another topic that has recently drawn considerable attention. TFEB is a major transcriptional regulator of autophagy highly expressed in the CNS which is involved in promoting the expression of genes involved in the formation of autophagosomes and lysosomal biogenesis and function [17]. Based on the experimental evidence, it has been proposed that there are a number of important changes in TFEB in various models of AD. Subcellular fractions of brain samples of AD patients displayed selective loss of nuclear TFEB which was inversely correlated with levels of hippocampal TFEB and progress of AD pathology [43]. A similar observation of nuclear TFEB was seen in double presenilin knock-out cells [44]. Patients with AD also showed a significantly decreased amount of TFEB expression in lymphocytes, and monocytes (cells involved in migrating to damaged areas of the CNS and possibly regulate AD expression) showed decreased TFEB expression, which may suggest that dysregulation of TFEB may be involved in dysfunction of lysosome function in AD [45]. Similar to AD patients, accumulation of Aβ and paired helical filament p-Tau was observed in the brains of mice with neuron-specific TFEB excision [44]. Interestingly, observations have also been made in contrast to TFEB upregulation. Fibroblasts from AD patients containing the familiar presenilin-1 A246E mutation displayed increased TFEB expression [46]. Similar observations were also made in CA1 pyramidal neurons of AD patients [47]. Even though these two observations are contradictory, they may not be mutually exclusive as AD presenilin mutations can reduce lysosome function via lysosomal alkalinization, which can result in TFEB activation as a compensatory mechanism [17].
Another autophagy protein we have observed in our laboratory to be implicated in AD is beclin-1. Beclin-1 is a major regulator of autophagy [48] and was shown to be downregulated at the genetic and protein level in fibroblasts with the presenilin-1 mutation compared to non-mutated fibroblasts [42]. One possible explanation for this activation of beclin-1 could be activation of beclin-1 via JNK-1. JNK-1 has been observed to be involved in a number of regulatory and stress response pathways with some including autophagy (via activation/phosphorylation of beclin-1). It is possible that the reduced JNK-1 activity may be involved in the reduced activity of beclin-1 and autophagy overall [49, 50]. It is possible that Ubisol-Q10 could be involved in activating JNK-1 which in turn activates beclin-1 but more work is required to investigate/confirm this mechanism. Current work is being done in our laboratory to elucidate the mechanism of beclin-1 upregulation due to Ubisol-Q10.
TRANSGENIC ANIMAL MODELS OF AD
Since AD is a fairly slow progressive neurodegenerative disease, various transgenic models of AD have been developed in rodents to simulate the pathology and behavioral symptoms of AD. A couple examples of the more common transgenic animal models of AD include double (TgAPEswe, PSEN1dE9 mice) and triple transgenic mice. Double transgenic mice express chimeric mouse/human APP (Mo/HuAPP695swe) and a mutant human PS-1 (PS1-dE9) which is moth directed to CNS neurons. Both mutations are associated with early onset AD, and the mice develop Aβ deposits in the brain around 6-7 months of age. Triple transgenic mice are homozygous for the following three mutant alleles: Psen1 mutation and APPswe and tauP301L transgenes. These mice exhibit amyloid-plaque and tau tangle pathology as soon as 3-4 months. Of the two rodent models, triple transgenic mice are more representative of an acute model of AD, while double transgenic mice are more representative of a chronic model of AD. Overall, double transgenic mice seem to be a more precise model of AD as mentioned before since AD is a slow progressive disease. Also, it has been observed that there are more confounding complications (which have been seen in both double and triple transgenic mice) when more foreign genes are inserted into an animal’s genome [51]. Furthermore, along with the above stated biochemical pathologies, it has been shown that both transgenic models of mice experience behavioral changes as well with one of the main pathologies being memory impairment. It is important that both biochemical and behavioral changes are measured when working with animal models of AD as there could be behavioral changes but no biochemical changes and vice versa. Therefore, it is important to be able to measure behavioral differences as well in transgenic mice.
The first signs of memory loss associated with early stage of AD in humans involve spatial reference and working memory attributable to initial neurodegeneration in the hippocampus [52 –56]. As AD progresses, non-spatial working and reference memory become noticeably poorer due to expansion of neurodegeneration into other cerebral areas as well in the hippocampus that is also involved in long-term memory consolidation, episodic memory, and non-spatial working memory [57 –63]. Many studies involving animal models of AD depend on robust behavioral measures of spatial memory [64]. Two most frequently used preparations for studying spatial memory, the Morris water maze (MWM) [65] and the radial arm maze [66], however, involve strong, negative (escape from drowning) or positive (finding food) motivational/emotional factors during trial and error acquisition. Any ancillary changes with such underlying factors from possible neurodegeneration in subcortical limbic structures (e.g., the amygdala) [67] or their connections to higher cortical areas could affect motivational/emotional factors producing a secondary disruptive impact on spatial cognition performance [68 –72]. In view of these issues, some researchers have utilized rodents’ (rats, mice) spontaneous, non-reinforced, unlearned preferences for investigating an unfamiliar (novel) object [73] (Novel Object Recognition – NOR) or a familiar but moved object (Novel Location – NL) among previously encountered (familiar) unmoved objects [74] or a combination of both [75] in the open field. Although such preparations eliminate positive motivational (food seeking) factors, their testing in open field environments promotes negative emotional escape-like tigmotaxtic behavior in mice [76, 77] of running near distal walls. Such locomotor behavior can obscure rodents’ spatial cognition [70] especially in those with compromised hippocampal-amygdala linkages [78]. The same problem has been pointed out concerning poorer MWM performance in transgenic mice [79]. Badour (2016) [80] attempted to replicate Benice et al.’s work [75] by exposing wild-type mice to three objects either in the open field chamber or in an enclosed transparent 4-arm radial (X) maze within it. Both groups appeared to explore an object more when it was moved on the 4th trial to a corner previously without any object, but this difference was only significant in the open field group. On the 5th trial when a novel object replaced one of the remaining two unmoved objects, only the X-maze group explored it more than the other two familiar objects. Thus, open field mice primarily attended to which corner previously had no object but not to any object’s specific non-spatial features while they thigmotaxically ran around the periphery of the chamber. Our most recent study with a transgenic mouse model of AD [81], animals were only exposed to an enclosed Y-maze in a 2-object variation of the NL/NOR task. Only unprotected transgenic mice failed to explore the familiar but moved object any more than the other two unmoved objects (trial 2) but showed heightened exploration of the novel object (trial 3) as well as the wild type and protected transgenic mice. Thus, as in earlier and perhaps less controlled studies [61 , 82–90], we also demonstrated that hippocampal degeneration decreased rodents’ spatial but spared their non-spatial working memory. Even though we may have eliminated or reduced anxiety-induced behavior in NL/NOR by running mice in an enclosed maze rather than in an open field, we found in a subsequent research [91] that wild type mice could not be repeatedly tested on their NL/NOR performance. Thus, this preparation presents a major challenge to study the progression of the severity and nature of the cognitive disintegration over the lifespan of transgenic mice that are necessary for comparisons with human AD [92]. In a current study we have modified Badour’s preparation [79] to solve this problem by allowing mice to search for treats (sunflower seeds) while they were being repeatedly tested for NOR in the X-maze. Under these conditions, rats demonstrated reliable working memory across several trials.
IMPLICATIONS OF PRESENILIN-1 MUTATION IN FIBROBLASTS FROM AD PATIENTS
Since mature neurons are found in the G0 phase and those affected by AD do not get diagnosed until after neuronal maturity, it is not possible to do studies directly on the neurons as they will not divide in culture. Thus, a model system is needed in order to get a better understanding of the disease processes. Fibroblasts obtained from AD patients harboring key AD mutations are such a model system and have been used for many years as these cells are highly abundant and are robust [93]. The use of these cells hinges on the assumption that the disease being studied is a systemic one, which is so for some cases of familial AD [94]. Benefits of this system include the ease of storage and culture, their expression of related genes to neurons is similar, and they accumulate damage in a similar way. Fibroblasts also make more intimate contact with each other similarly to neurons [93, 95]. However, there are some drawbacks of this system mainly as it is not able to precisely mimic the physiological conditions of the neurons in the brain in a living system, and they are a different cell type and will have slightly different expression levels of various genes [93 , 96]. One interesting discovery discussed by Wray [96] was that these cells can be reprogrammed to be pluripotent through the use of exogenous transcription factors allowing them to obtain gene expression profiles of more similar neurons in physiological conditions.
We have previously used PS-1 mutated skin fibroblasts from AD patients (PSAF) as a model for AD, as these cells have lower levels of autophagy as seen in AD neurons [42]. These AD fibroblasts demonstrated increased levels of endogenous ROS in comparison to skin fibroblasts from healthy individuals harboring wild type PS-1. This is consistent with the mounting evidence of publications demonstrating elevated levels of oxidative stress in AD neurons [6 , 97]. Moreover, we found that these AD fibroblasts, with respect to their counterparts from healthy individuals, exhibited a small increase in the expression of p53, as well as marked increases in p21, p16Ink4A, and Rb. These cells are unique because they have the ability to detoxify higher levels of MnSOD, an enzyme that facilitates the conversion of superoxide to the less reactive H2O2. Interestingly, MnSOD has been linked to the onset of senescence [98]. Collectively, our findings suggest the elevated levels of endogenous ROS in PSAF contribute stress-induced premature senescence (SIPS), a phenomenon well characterized in fibroblast models [42 , 99–101].
Using cells of peripheral tissues from AD patients, such as lymphocytes and fibroblasts, provides an efficient model to study the effects of the putative gene mutations of AD etiology and how they are linked to increases in oxidative stress. However, in the place of cell death that occurs in neurons of AD patients as a result of oxidative insult, we have shown AD fibroblasts to undergo SIPS from the exposure of sublethal doses of endogenous ROS [42]. Such an observation can be a result of the increased expression of p21 in AD fibroblasts which is linked to the onset of senescence and inhibition of apoptosis [102 –104]. Furthermore, fibroblasts possess more potent antioxidant defense mechanisms to that of neurons [105] and can tolerate oxidative insult by elevating expression of vital proteins such as MnSOD, which has been linked to the onset of senescence [98]. Additionally, neurons depend heavily on oxidative phosphorylation to satisfy its higher energy requirements [106] that can lead to higher levels of ROS generation and exacerbation of oxidative insult.
BIOMARKERS OF AD
There are a number of difficulties in diagnosing AD as clinical diagnosis can only be done when patients already experience cognitive decline. Therefore, it is imperative that there be reliable biomarkers for early diagnosis of AD before symptoms begin to show. A possible diagnosis tool for AD has been the use of positron emission tomography (PET) with radiotracers to detect the presence of fibrillar deposits of amyloid in the brain that are greater than 1 mm in size. PET scans are quite expensive and there are issues in correlating deposits of amyloid with progression of AD, as its been found that plaques also deposit in the brains of healthy individual [107]. Detecting Aβ via antibodies in serum and cerebrospinal fluid (CSF) have also shown to be promising biomarkers but the predicative power of these biomarkers has not been particularly encouraging [108, 109]. Furthermore, attempts to correlate serum/CSF with PET are not very convincing as of yet [107]. A potential minimally invasive variant to measuring amyloid without the use of expensive radiotracers or immune-based probes was developed by Tiiman et al. [107]. This technique measures amyloidogenic oligomeric aggregates (called nanoplaques) in blood via a specialized ThT (Thioflavin T) fluorescence assay [107]. Tiiman et al. was able to show that there was a significantly higher amount of larger nanoplaques in the blood of AD patients compared to control subjects. While collection of CSF is highly invasive compared to blood collection, biomarkers other than Aβ in CSF have been implicated in AD as well. Levels of neurofilament light chain and visinin-like protein (VILIP-1), markers of neurodegeneration were elevated in the CSF of AD patients. Increased levels of inflammatory proteins YKL-40 and monocyte chemoattractant protein-1 were also observed in the CSF of AD patients. Indicators of amyloidosis (apolipoproteins) and synaptic dysfunction (neurogranin) were also observed to be implicated in AD. These biomarkers were shown to indicate the stage and state of AD progression and using a combination of these biomarkers could aid pre-clinical diagnosis and identify early changes in the brain before onset of the disease [110].
CLINICAL TRIALS OF AD THERAPEUTICS
While great strides have been made in understanding the pathologies of AD, there have been many failed attempts at developing effective disease modifying therapies for AD. A multitude of therapies have been developed that target various different aspects of AD such as BACE inhibitors, immunotherapy, gamma/beta-secretase inhibitors, Aβ aggregation inhibitors, and alpha-secretase activators [111]. Many of these types of therapeutics are either undergoing clinical trials or have failed due to various reason such as toxicity or lack of efficacy. Some drugs that target production of Aβ such as BACE inhibitors were actually found to accelerate cognitive decline, possibly due to off-target effects [111]. Furthermore, Aβ and enzyme associated with it do have a physiological role in facilitating neuronal function and therefore targeting them may have adverse side effects. Another possible reason that many clinical trials have failed may be due to many of them only targeting a single pathology such as Aβ. It is critical to remember that AD should be considered a multifactorial disease and targeting only a single pathology of AD may not be enough to halt the progression of the disease.
NOVEL THERAPEUTICS FOR AD
Current treatments for AD, such as cholinesterase inhibitors, are directed at softening the symptoms of this age-related disease and do not deal with the cause of the disease [112]. With the increased number of people entering their later years in life, it is imperative that precise therapeutics targeted at preventing late onset of AD are investigated and developed. The treatments listed are still being evaluated for their efficacy and are geared toward targeting the probable causes of AD.
Curcumin and other ayurvedic herbs
Curcumin is a polyphenolic compound found in turmeric spice [113]. Traditionally it has been used to treat swelling, which in later studies was revealed to be true as curcumin provides its action by inhibiting nuclear factor kappa B mediated transcription of inflammatory cytokines [113]. More recently, curcumin has captured the attention of numerous investigators as a potent antioxidant where it has been found to effectively quench nitric oxide-based radicals [113, 114]. A study in 2004 by Yang et al. showed in a mouse model that curcumin was able to inhibit Aβ oligomerization and aggregation and thus, reduce the deleterious effects associated with Aβ plaques [115]. Currently it has been hypothesized that its mode of action is to influence salt bridges causing global rearrangements in the structure of Aβ [116]. It is also important to note that this was done with a low concentration. Although this compound is able to pass the blood-brain barrier, it has low bio-availability and a short half-life in the body. To overcome these shortcomings of curcumin, Mulik et al. [117] designed a vector to transport curcumin directly to the brain. ApoE3 mediated poly(butyl) cyanoacrylate nanoparticles were made that surrounded the curcumin in a vesicle like structure [117]. This structure utilized the strong affinity between ApoE3 and low-density lipoprotein receptors which mediated endocytosis of the vesicle. Mulik et al. reported that this process increased the stability of curcumin and provided sustained drug release [117]. It was also reported that Aβ and oxidants were significantly reduced in comparison to the use of curcumin alone although, the increased effects may be partially attributed to the use of ApoE3 which has been shown to be an antioxidant and competes with Aβ for entry into the cell.
Withania somnifera (ashawagandha) root extract has been used for thousands of years in ayurvedic medicine as a promoter of brain health and neurological functions, and thus has been proven to be at least well tolerated and non-toxic for human use. Interestingly, several groups have recently used various forms of ashwagandha extract as a neuroprotective agent in vitro and in vivo [118]. Ashwagnadha extract has shown to ameliorate key inflammatory cytokines and modulated stress response [119]. In particular, Seghal et al. in 2012 demonstrated that ashwagandha reversed AD pathology by enhancing low-density lipoprotein receptor related protein in the liver of transgenic mice [120]. Furthermore, the extract was shown to reduce the amount of oxygen species. Their results showed that the extract did indeed enhance reduction of amyloid plaques as well as improving behavior. However, the doses of the extract administered were unrealistically high (1 g/kg/day) to be translated for use in humans [120]. Of course, better formulations need to be developed to increase the bioavailability so that the extract can be used in humans. Alternatively, lower doses of ashwagandha extract could be combined with other natural compound/extracts to enhance the neuroprotective potential as a combination. Based on these recent findings, ashawagnahda should prove to be a potential therapeutic to halt the pathogenesis of AD, but further research is required.
Similarly to ahswagandha, Bacopa monnieri (brahmi) has also been used as a nerve tonic in Ayurveda. Extract of brahmi was shown to protect neurons from Aβ-induced cell death, and the mechanism was thought to be via inhibition of acetylcholinesterase activity suppression [121]. In the same study done by Limpeanchob et al. [121], brahmi extract was shown to promote cell survival as well as reduce ROS and lipid peroxidation, leading to cellular longevity. Brahmi was also shown to significantly improve the memory in amnesic mice treated with diazepam, scopolamine, or phenytoin. In a clinical trial, brahmi improved cognition in patients with senile dementia of the Alzheimer-type. Alongside the improved cognition, various biochemical markers showed reduced inflammation (homocysteine, C-reactive protein, and tumor necrosis factor alpha) and oxidative stress (glutathione peroxidase, glutathione, thiobarbituric acid reactive substances, and superoxide dismutase) [122].
Methylene blue
First discovered as a biological stain, Methylene blue (MB), has been used to treat several diseases such as malaria, drug-induced methemoglobinemia [123], and cyanide poisoning. Several recent discoveries strongly suggest that this drug may be very useful in the combat against AD. Xie et al. [123] demonstrated that MB is able to induce autophagy in the hippocampus and cortex in vivo through the AMPK signaling pathway during serum starvation. Interestingly, they found that these effects were mainly prevalent in mature neurons and not in neural progenitor cells. MB has also been shown to be a potent antioxidant but not in the conventional sense. Instead of quenching ROS immediately, it must accept electrons from NADH reducing MB to leuco-MB allowing it to then scavenge free radicals reverting it back to MB [124]. Additionally, MB acts as an electron carrier between NADH and cytochrome C systems in the mitochondria increasing oxygen consumption and reducing the levels of ROS that are produced [124, 125]. Callaway et al. [125] showed that the assistance provided by MB during oxidative phosphorylation was able to improve memory retention in rats. MB has also been shown to prevent oligomerization of Aβ as well as its extracellular accumulation forcing aggregates to go through nucleate fibril formation [126, 127]. This is done by targeting beta secretase-1 (BACE1) reducing its stability and reduces β-secretase activity which decreases the number of Aβ species [127]. Additionally, MB’s efficacy has been shown to minimize tau fibrilization through the oxidation of disulfide bonds of 4-R and 3-R tau promoting disulfide formation [128]. Mori et al. have demonstrated the ability of MB to increase the chymotrypsin and trypsin like activities of the proteome allowing the degradation and removal of protein aggregates [127]. Finally, MB has been shown to be an acetylcholinesterase inhibitor which is helpful for AD patients as the cholinergic system has been shown to be a regulator of learning and memory [127].
Acetyl-L-carnitine
There is little literature surrounding the benefits of Acetyl-L-carnitine (ACL) as a treatment for AD, and thoughts are divided as to how beneficial it really is. It is worthwhile to include ACL in this category because it appears to have tremendous therapeutic potential in AD. ACL seems to exhibit most of its effects by stabilizing lipid membrane fluidity by regulating levels of certain molecules such as phosphomonoesters and phosphocreatine and decreasing lipid peroxidation [129]. ACL also seems to aid in the function of aged mitochondria by supporting oxidative phosphorylation, increased turnover of mitochondrial membrane proteins, and reverses the impairment of DNA/RNA transcriptase [129, 130]. In addition, it was reported that ACL is able to increase the levels and utilization of growth factors in the CNS.
Dehydroepiandrosterone
The most abundant steroids in the body are dehydroepiandrosterone (DHEA) and its sulphated ester (DHEAS). These work to increase the effects of excitatory neurotransmitters [131]. Part of its neuroprotective effects may come from its anti-glucocorticoid action as high levels of cortisol cause damage to neurons of the hippocampus [131]. Bastianetto et al. has reported many benefits associated with the use of DHEA which include, regulation of Ca2+ channels, and may act as an antioxidant by reducing lipid peroxidation possibly by increasing glutathione levels [132]. The anti-peroxidative property of membrane lipids may help to modulate membrane fluidity which is commonly found associated with neurodegenerative diseases [133]. Additionally, DHEA has been reported to increase levels of acetylcholine, catecholamine, and brain derived neurotropic factor which were thought to have led to increased memory and learning in mice [134]. It should be noted that in a short study done on humans, there seemed to be very little evidence to suggest that DHEA had these effects [131]. However, Malouf and Grimely Evans stated that this may be due to the length of the study and that the effects may become more profound past the 3-month mark; however, it warrants further investigation [131].
Dimebon
Dimebon was initially used in Russia as an antihistamine and has recently been noted to have potential benefits for treating neurodegeneration. It was first noted that its relevance for treating these diseases came from its ability as an inhibitor of acetylcholinesterase, N-methyl-D-aspartate receptors, and voltage gated calcium channels [135, 136]. Only preliminary studies on this particular drug were done but the results appear to be extremely promising. Yamashita et al. reported that dimebon was able to prevent mitochondrial transition pore in experimental models of AD [137]. Additionally, they found that dimebon may act by reducing the production or accumulation of abnormal protein aggregates. More recently it was found that treatment of dimebon in AD transgenic mice lead to a decrease in Aβ aggregates but without any effect on AβPP or soluble Aβ oligomers [138]. This was attributed to the stimulating effect that dimebon has on glial fibrillary acidic protein in astrocytes leading to an increase in their role in Aβ aggregate clearance. Interestingly, Perez et al. [138] observed that dimebon had no effect on tau aggregation. According to Zhang et al. [135], dimebon may act as an anti-uncoupling agent and thus, decreasing proton leakage and would act to increase mitochondrial membrane potential and ATP production. Yamashita et al. [137] noted a delay in cellular senescence with the treatment of dimebon in SH-SY5Y cells. Importantly, it was noted that dimebon only exerted its neuroprotective effects before or during stress but was unable to rescue cells after an insult and after cell death pathways had been triggered [135].
Ginkgo biloba
Ginkgo biloba (GB) is an ancient tree that has been used in Chinese medicine for thousands of years. It has been reported that GB works as an antioxidant and is able to protect neural tissue from lipid peroxidation and general peroxidative events [139, 140]. It has also been reported that GB acts as a monoamine oxidase inhibitor which may be useful in alleviating some of the symptoms associated with AD [141, 142]. Inflammation seems to be a persisting feature in AD, and it seems that GB is able to act as an anti-inflammatory agent by inhibiting platelet activating factor [140]. Luo et al. also discussed the potential for GB to act as an anti-apoptotic agent [143]. GB seems to have some effect in abolishing AD symptoms and aided in improving memory but more studies are needed to affirm such conclusions; preferably with larger sample sizes to truly show GB’s effectiveness in enhancing memory in AD patients [144]. The agents that seem to be responsible for this are flavonoid glycosides and terpenoids (bilobalide, ginkgolides) which are found in the most common GB extract Eb761 but recently fractions of GB extracts were analyzed which identified an unknown compound that may contribute to the benefits of GB [140, 145]. Although GB does have some beneficial effects, there are some side effects that have been noted such as prolonged bleeding, hemorrhage, and ulceration [140]. Further well-controlled multicenter studies are needed to evaluate the bioavailability of GB and to determine the proper dosage for AD patients.
Other nutrients
There seems to be levels of a few nutrients that decrease as age progresses and these nutrients have been consistently found to be reduced in AD patients. In the adult brain, folate is a methyl group donor and plays an important role in normal brain function and DNA repair. In addition, it acts as a coenzyme in the synthesis of serotonin and catecholamines [131]. However, few reports have emerged that suggest decreased levels of folate sensitize neurons to oxidative stress and Aβ toxicity although evidence supporting such effects of folate needs carefully designed studies [146]. Vitamin B12 (cobalamin) is a cobalt containing vitamin that is a cofactor for methionine synthase which converts homocysteine to methionine and 5-methyltetrafolate to tetra folate. Low levels of this compound can lead to fatigue depression and poor memory. Sasaki et al. showed that supplementation of B12 in acetylcholine-deficient mice increased acetylcholine synthesis [147]. Thymine and its phosphorylated derivative serve as important cofactors in brain oxidative metabolism. There is also evidence that suggest thiamine may modulate the cholinergic status of the brain [148]. It was reported that thiamine deficiency leads to memory impairment cholinergic neuronal loss [149]. It was also shown in rodents that thymine deficiency causes accumulation of AβPP. However, treatment of AD patients with thymine only showed cognitive improvement in mild cases [150]. Vitamin E (α-tocopherol) is a lipid soluble molecule that has been known to act as an antioxidant for some time. Its main mode of action is to prevent lipid peroxidation [151]. Some studies have shown that vitamin E may considerably slow the progression, or at the very least, provide some symptomatic relief [152, 153]. These results, however, do not provide very convincing picture of the effectiveness of vitamin E as a supplement for the attenuation of AD. In our laboratory, we have used a water-soluble formulation of vitamin E as a control for our water-soluble co-enzyme Q10 treatments, as vitamin E is a part of the construct built to shuttle co-enzyme Q10 though an aqueous medium. Our results appear to be promising because vitamin E shows ability to alleviate some of the AD pathologies [42].
Co-enzyme Q 10
Co-enzyme Q10 (CoQ10), also known as ubiquinone, was discovered in 1957 by Crane et al. to be a component of the mitochondrial respiratory chain and acts as an electron carrier between NADH, succinate dehydrogenase, and cytochrome c complex [154, 155]. What initially captured the attention to this effect is that, in its reduced state, ubiquinol can serve as an effective antioxidant. Later it was shown that ubiquinol was able to provide protection against lipid peroxidation and oxidation against DNA and proteins [154]. Do et al. later showed in a yeast model that CoQ10 was able to directly target and protect the mitochondria from the same stressors. CoQ10 was also found to preserve mitochondrial respiratory functioning in aged rat skeletal muscle and protect cerebellar neurons from glutamate toxicity [156]. It has been observed that in neurodegenerative diseases there may be a decrease in production of CoQ10 and therefore, exogenous supplementation of CoQ10 may hold promise in ameliorating neurodegenerative diseases such as Parkinson’s disease (PD), Huntington’s disease, and AD [157]. Unfortunately, despite its ability cross the blood-brain barrier, its lipophilic nature makes it a poorly bioavailable compound [157]. To circumvent this, researchers developed vectors that would allow CoQ10 to be soluble in aqueous solutions [157, 158]. The water soluble CoQ10 (WS-CoQ10) is formed by linking a hydrophobic group (α-tocopherol) and a hydrophilic moiety (polyethylene glycol) joined by sebacic acid linker. This molecule, Polyoxyethanyl-α-tocopheryl sebacate (PTS), associates with CoQ10 via its hydrophobic α-tocopherol component via hydrophobic interactions, and this entire complex spontaneously forms nano-micelles in aqueous solutions, thus solubilizing CoQ10. This formulation showed significant protection against MPTP toxicity in a PD model [159]. Additionally, it was shown that this method of solubilization brought down treatment with CoQ10 to a clinically relevant dose of 5 mg/kg/day, as compared to a required dose 40 times higher with an oil-soluble formulation [160]. We have recently shown in PSAF that WS-CoQ10 was able to prevent the onset of SIPS in these cells by lowering ROS levels and ameliorating mitochondrial dysfunction [42]. Additionally, we have shown that in PSAF there are increased levels of MnSOD, p21, RB, and p16Ink4A compared to fibroblasts with wild type PS-1 from healthy individuals. Interestingly, treatment with WS-CoQ10 decreased levels of these proteins. Ma et al. also showed a modest increase of autophagy, a process known to be disrupted in AD [40], with the supplementation of WS-CoQ10. Further studies in our laboratory are being conducted to pinpoint mechanisms as to how WS-CoQ10 is affecting the senescent and autophagic pathways in PSAFs.
Autophagy is a cellular mechanism that involves the removal and recycling of damaged and toxic intracellular components such as harmful protein aggregates, dysfunctional mitochondria, and oxidized proteins [36]. When these materials are detected, the rough endoplasmic reticulum releases a double membrane, also called a phagophore, which surrounds and sequesters these materials in an autophagosome. These vesicles then bind to a lysosome which is thereafter referred to as an autolysosome or phagolysosome which then degrade the engulfed contents with various different lysosomal proteins such as cathepsins and acidic hydrolases [161]. Lysosomes contain a proton pump vacuolar [H+] ATPase (v-ATPase) that is required for the acidification of the autolysosome [38]. The low pH is required for the activation of the degradative enzymes found within the lysosome. Lee et al. [40] found that PS-1 was essential for targeting v-ATPase to the lysosome and thus, mutations in PS-1 lead to defective lysosomal acidification. Autophagy is especially important in long-lived cells like neurons which rely on high basal levels of autophagy for survival [162]. A mutation in PS-1 then would lead to accumulation of hazardous materials and dysfunctional organelles laying the foundation for AD development. In this regard, we found WS-CoQ10 was able to modestly induce autophagy in PSAF. LC31 to LC3 conversion seemed to be insignificant, indicating that autophagic induction may be independent of this phenomenon. However, beclin-1 did show a difference between the untreated and the PSAF treated with WS-CoQ10. Interestingly, this protein may be the link between mitochondrial dysfunction and autophagic insufficiency.
Beclin-1 is a protein that is involved in the initial stages of autophagy but is cleaved by caspases which are induced during times of cellular stress to activate apoptosis [48]. Since the mitochondria are responsible for the activation of caspases and the increase in oxidative stress in PSAF, it may be that treatment with WS-CoQ10 aids in the preservation of autophagy. Additionally, mTOR a protein kinase is activated by several factors but has been seen to be elevated during times of cellular stress as it helps to regulate cell cycle and growth and has been shown to be required for cell senescence maintenance [163]. mTOR has also been shown to act as an inhibitor of autophagy through global transcription regulation and through its hyperphosphorylation of Atg13 which is needed in the initial stages of autophagosome closure [164]. Other possible mechanisms for the inhibition of autophagy may be due to increased levels of calcium. It has been shown that intracellular levels drastically increase in AD due to aberrant Aβ accumulation [20]. It seems that high calcium concentrations inhibit autophagy through the activation of calpain which cleaves Gsα allowing cAMP to be produced, a presumed inhibitor of autophagy [165]. Co-enzyme Q10 has been shown to normalize serum calcium levels [166] and thus, may aid in this process, although this hypothesis needs to be tested. Another possible reason for induction of autophagy with WS-CoQ10 may be due to stabilization of the mitochondria membrane which may allow for surface markers to be presented such as Nix, allowing recognition by autophagosomes [167]. These attributes, as well as other potential AD-related causes of autophagic dysfunction, require further investigation to identify the validity of their role and the likely potential for WS-CoQ10 to act as a therapeutic agent against them. Furthermore, in some of our unpublished results, we have observed the upregulation of various autophagy regulating genes (including beclin-1) in PSAF that were treated with water soluble WS-CoQ10. These results where further confirmed by western blot analysis. We have yet to discern the exact mechanism by which WS-CoQ10 is upregulating these autophagic proteins. In Fig. 2 below, we have provided a map of the possible mechanisms of how WS-CoQ10 might be involved in autophagic reintroduction. Using a cellular model can be very useful in understanding the basic mechanisms of a disease trait, but often they are a far cry from the actual effect of a drug seen in vivo as drugs can be differentially metabolized and lead to a different outcome or even toxicity. Mouse models can provide a better link as to how a drug is going to impact the body when supplemented in humans.
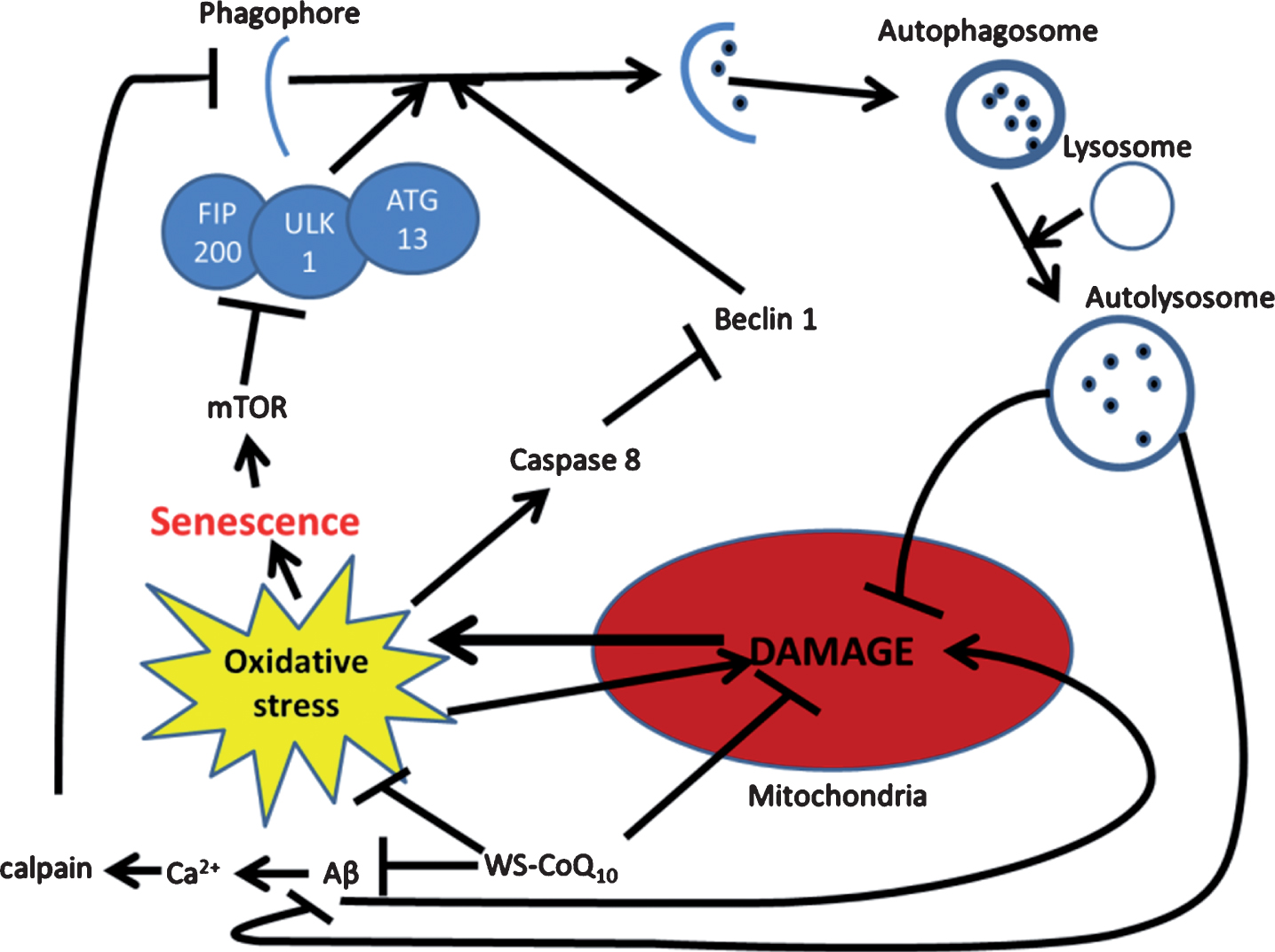
Possible mechanism for autophagic reintroduction by WS-CoQ10 in PS-1 mutated fibroblasts. mTOR is related to the maintenance of cellular senescence as well as the inhibition of autophagy through the induction hyper-phosphorylation of ATG13. Beclin-1 a known inducer of autophagy is degraded by caspase 8 which is activated during times of oxidative stress produced in huge quantities by the damaged mitochondria of PS-1 mutated fibroblasts. Aβ has been seen to lead to mitochondrial damage and can increase the influx of Ca2+ which can activate the antiapoptotic protein calpain. WS-CoQ10 is able to considerably lessen mitochondrial damage and the levels of ROS that lead to oxidative stress. Its possible the reduction of oxidative stress may lead to reduced degradation of beclin-1 via caspase 8 leading to increased autophagy. This may act as a positive feedback loop as the reintroduction of autophagy would allow the degradation of dysfunctional mitochondria and oxidized proteins.
Recently we have reported unprecedented efficacy of Ubisol-Q10 ® (Next Remedies Inc.) (previously called WS-CoQ10) in protecting neurons in in vitro and in vivo models of neurodegenerative diseases. The formulation contains CoQ10 and PEG-α-tocopherol forming jointly water-soluble nano-micelles. We have confirmed bioavailability of this formulation in the brain. A comprehensive behavioral analysis of transgenic animals (PD and AD mice models) fed with this formulation indicated significant improvement in motor activity in PD and long-term memory and emotional reactivity in models of AD compared to untreated animals. These results complemented histochemical analysis that indicated significant protection of neurons in substantia nigra region in PD models, lower Aβ burden (plaques), and increased autophagy in AD mice. This treatment led to the stabilization of mitochondrial functions, decreased oxidative stress in neuronal cells, and increased autophagy and induction of pro-survival astroglial cells. Thus, we optimistically hypothesize that Ubisol-Q10 ® could offer a potential treatment that could halt the progression of disease in AD and PD patients.
WS-CoQ10 is effective in ameliorating mitochondrial dysfunction, increased ROS generation, and defective autophagic processes, all of which are reputed principle abnormalities linked to the AD etiology. Therefore, WS-CoQ10 is a novel potential prophylactic and therapeutic agent for AD and may have implications for other mitochondrial and autophagy-linked disease.
CONCLUSION AND FUTURE PERSPECTIVES
It has been established that multiple factors including production of toxic Aβ peptide, oxidative stress, mitochondrial dysfunction, accumulation of damaged proteins, inhibition of autophagy and proteasome mechanisms, and inflammation are involved in the initiation and development of AD pathology. Many of the pathways discussed in this review appears to be provocative and promising. Some of these mechanisms are related to each other, for example, oxidative stress could cause mitochondrial dysfunction which in turn increases oxidative stress; thus a vicious cycle could start. However, targeting any one of these mechanisms has not been successful in halting the progression of the disease. In AD patients, where the vicious cycle of oxidative stress and related damages have already begun, blocking Aβ production or removal by antibodies could not block the cycle, and the disease progression continued despite clearance of Aβ. Similarly, anti-oxidants alone or anti-inflammatory regimens have not shown to be effective. It would be necessary to stabilize mitochondria and resume autophagy or protein clearance coupled with reduction of oxidative stress and inflammation to successfully stop progression of neuronal cell death. Recent research from cellular and animal models indicate that combination of tocopherol containing water-soluble formulation of CoQ10 and ashwagandha extract together could limit the damage in AD brain by stabilizing mitochondria and reducing oxidative stress and inflammation while resuming autophagy. Therapeutic regimens of this nature (similar to nutritional supplement) could be given to patients over a long period of time (possibly throughout life) without expecting any side effects. There is urgent need to evaluate efficacy of these potential therapeutic agents in clinical studies, as well as investigate novel natural compound and extract-based therapies for AD.
DEDICATION
The authors would like to dedicate this paper in the memory of Mr. Joseph Szecsei who was a great warrior and supporter in the research on neurodegenerative diseases.