
Abstract
Aging is the leading risk factor for idiopathic Alzheimer’s disease (AD), indicating that normal aging processes promote AD and likely are present in the neurons in which AD pathogenesis originates. In AD, neurofibrillary tangles (NFTs) appear first in entorhinal cortex, implying that aging processes in entorhinal neurons promote NFT pathogenesis. Using electrophysiology and immunohistochemistry, we find pronounced aging-related Ca2 + dysregulation in rat entorhinal neurons homologous with the human neurons in which NFTs originate. Considering that humans recapitulate many aspects of animal brain aging, these results support the hypothesis that aging-related Ca2 + dysregulation occurs in human entorhinal neurons and promotes NFT pathogenesis.
Keywords
INTRODUCTION
The hallmark neuropathological lesions of Alzheimer’s disease (AD), comprising somatodendritic neurofibrillary tangles (NFTs), amyloid-β deposits. and synaptic loss, along with their downstream consequences, have been studied intensively and are well-characterized [1–3]. In contrast, relatively few studies (e.g., [4–6] have focused on the age-dependent pathophysiological processes that induce these hallmark lesions in idiopathic AD.
Nevertheless, given that advanced age is the leading risk factor for idiopathic AD [7], it seems highly probable that brain aging processes play a central role in generating the pathophysiological conditions that induce vulnerability to AD. Such AD-inducing aging processes presumably operate upstream of the early stages of AD pathogenesis and are present in the same neurons/regions as those in which AD pathology initially appears. Of the hallmark pathological lesions of AD, NFT density exhibits the most consistent anatomical patterns of origination and progression [1]. During early-stage AD, somatodendritic NFTs first appear in the transitional entorhinal cortex (EC) and then spread through layers II and III of the lateral EC (LEC) and medial EC (MEC) to pyramidal neurons of hippocampal subfield CA1. NFTs then spread to the subiculum and other limbic structures and, in later-stage AD, to neocortical neurons [8, 9]. Consequently, a novel strategy for finding candidate NFT-inducing aging processes appears to be the identification of aging processes that are present specifically in the EC and CA1 neurons in which early NFT pathogenesis develops.
However, a major obstacle to this approach is the lack of appropriate animal models of NFT formation. Idiopathic AD does not occur in non-human species [1, 2], precluding mechanistic studies of NFT pathogenesis in normally aging animals. Furthermore, although transgenic tau mouse models exhibit some pathological changes resembling those of AD-like NFTs [10, 11], such genetically-modified animals are not suitable models for studies on the normal aging processes that promote NFT formation in idiopathic AD.
In the present study, we address this problem by assessing aging processes in normally-aging rat neurons that are homologous with the human entorhinal cortex neurons in which NFTs originate. Most mammalian species recapitulate major features of human brain aging, including neuronal and synaptic loss/atrophy, glial-inflammatory activation, oxidative stress, extensive transcriptional changes, decreased motor abilities, and impaired hippocampal-dependent cognitive function [4–6, 12–24]. Moreover, the topography and connectivity of the entorhinal-hippocampal regions in rodents are highly homologous with those present in other mammals, including humans [25–27]. Accordingly, it seems likely that normal aging changes in the rat entorhinal cortex are relatively similar to those that occur in human entorhinal neurons, where such changes may induce NFT pathogenesis.
Here, we employ this strategy for the first time, to test the proposition that aging-related Ca2 + dysregulation appears early in the rat EC-hippocampal circuits that are homologous with the human NFT progression pathway. Brain Ca2 + dysregulation during aging appears to be a strong candidate for a possible role in AD vulnerability, as it has been observed across a wide range of cell types/regions and in multiple animal models of aging [12, 28–38]. There also is considerable evidence of disturbed Ca2 + signaling in postmortem AD samples and transgenic AD mouse models [39–45]. Additionally, elevated intracellular [Ca2 +] potently degrades cytoskeletal structure and induces neurodegeneration [46, 47].
However, the specific manifestations (and possibly the underlying mechanisms) of aging-related Ca2 + dysregulation differ substantially across brain cell types. In the present study, we focus on a frequently-validated electrophysiological marker of aging-related Ca2 + dysregulation, the enlargement of the post-burst Ca2 +-dependent, K+-mediated slow afterhyperpolarization (sAHP). An aging-related increase in the sAHP has been seen consistently in hippocampal CA1 pyramidal neurons [12, 48], and in other pyramidal cells [14] Although magnitude of the sAHP can be modulated by several pharmacologic or behavioral treatments [49], the aging-related increase in sAHP magnitude appears to result predominantly from increased Ca2 + transients, arising from disinhibited Ca2 + channels and ryanodine receptors [28, 50].
In turn, this disinhibition apparently results from declining function of FK506 binding protein 12.6/1b (FKBP1b), a negative regulator of intracellular Ca2 + transients in neurons [13] and myocytes [51]. Since FKBP1b expression declines in hippocampal subfield CA1 during normal aging in rats [13, 52] and during incipient AD in humans [53], we use its immunohistochemical staining here as a validating second marker of Ca2 + dysregulation.
METHODS
All protocols and procedures were performed in accordance with institutional guidelines and were approved by the Animal Care and Use Committee.
Electrophysiological studies
Young-mature (3–5 months old), mid-aged (8–10 months old), and aged (20–22 months old) male Fischer 344 (F344) rats were obtained from the aging rodent colony maintained by the National Institute on Aging. To assess sAHP magnitude in layers II and III neurons of oxygenated 350μm-thick MEC slices, we used intracellular sharp-electrode current clamp methods similar to those used in our previous studies [13, 54]. Layer III primarily contains pyramidal neurons that project to hippocampal subfield CA1 pyramidal cells, whereas layer II contains multiple neuron types, of which the principal neurons are stellate cells that project to dentate gyrus and subfield CA3 and non-dentate projecting pyramidal-like neurons [26, 55–60].
In pilot work we found that the stellate cells do not generate a significant sAHP, whereas the pyramidal cells of layers II and III manifest post-burst sAHPs that resemble those seen in CA1 pyramidal neurons and are Ca2 +-dependent [57]. Because we sought to test the prediction that EC neurons exhibited aging-related Ca2 + dysregulation of the CA1 type (enlarged sAHP), we focused our electrophysiological analysis on the pyramidal-like neurons of MEC layers II and III. Our identification of pyramidal cells was based on electrophysiological criteria developed in extensive morphological-electrophysiological correlation studies, conducted previously by other investigators, which showed high correspondence between a recorded neuron’s electroresponsive pattern and its morphology (pyramidal vs. non-pyramidal-primarily stellate) [55, 57–59] (Fig. 1).
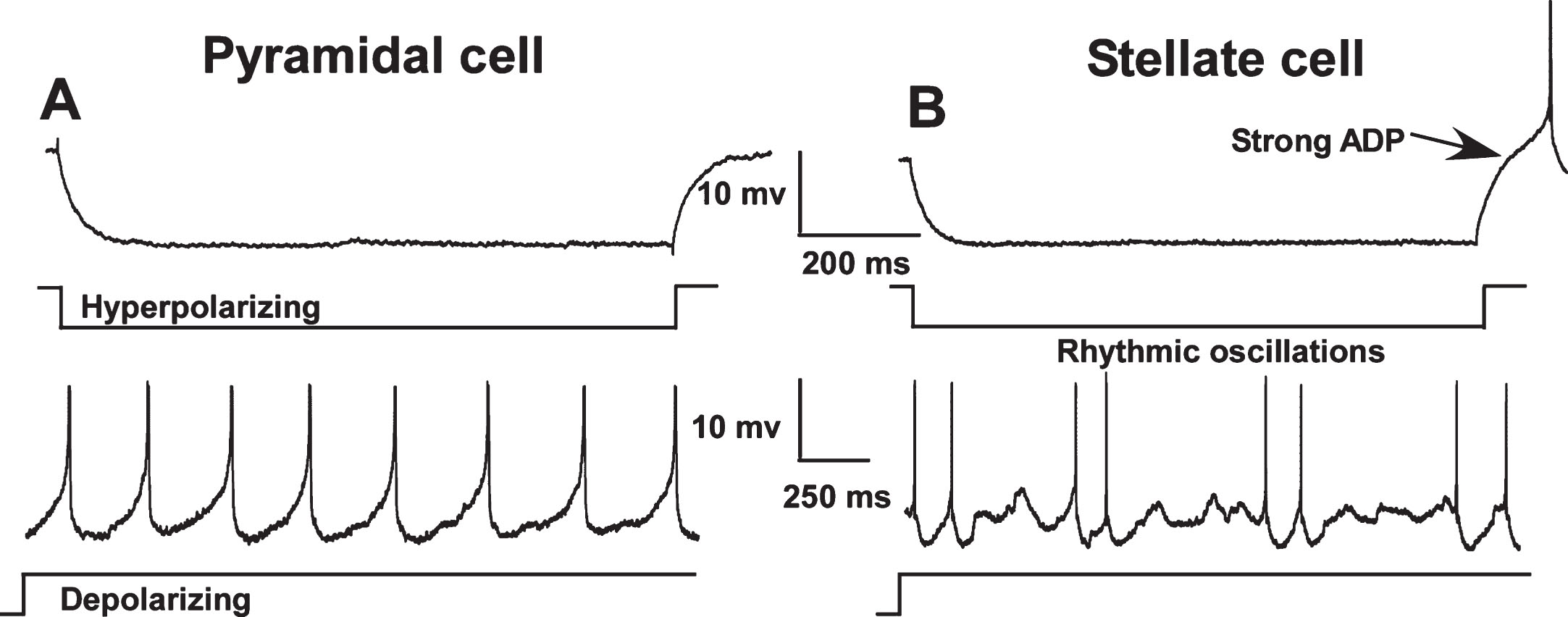
Electrophysiological differences between pyramidal (A) and stellate cells (B) in the medial entorhinal cortex (MEC). In contrast to pyramidal cells, stellate and other non-pyramidal cells exhibit a pronounced afterdepolarization (ADP) following repolarization from a hyperpolarizing pulse and show rhythmic oscillations during sustained depolarization. Neuronal types were distinguished using these characteristics and only pyramidal-like cells were included in this study. (Note: action potentials have been truncated).
Intracellular recordings were obtained from identified pyramidal-like cells in MEC layer II (10 neurons from 4 young, 5 neurons from 6 mid-aged and 8 neurons from 4 aged animals) and layer III (20 neurons from 8 young, 13 neurons from 9 mid-aged and 14 neurons rom 8 aged animals).
After cell-type identification and cell health assessment, sAHP duration and amplitude were measured following four depolarization pulse-induced Na+-action potentials. Amplitude of the sAHP was measured at 300 ms following the end of the depolarizing step and sAHP duration was determined as the interval from the end of the current step to the time point at which the membrane potential returned to pre-stimulus baseline. Recordings were alternated among animals of the three age groups.
Immunohistochemical studies
In a separate cohort of 30 male F344 rats, we performed semi-quantitative immunohistochemical (IHC) analyses of FKBP1b expression in the MEC across the adult lifespan (Fig. 3). Methods were similar to those reported previously [52] Coronal sections (30μm) were incubated with the primary antibody, monoclonal mouse anti-FKBP1b (1:100; sc-376135, Santa Cruz, Santa Cruz, CA), for one week at 40°C. Sections were then rinsed and transferred to solution containing biotinylated secondary antibody for 2 h. The sections were rinsed and transferred to a solution containing ExtraAvidin for 2 h, after which the sections were incubated for 3 min with Ni-enhanced DAB solution. The primary antibody, monoclonal mouse anti-FKBP1b, used here required one week incubation at 4°C to achieve staining topographically comparable to that yielded by the polyclonal anti-FKBP1b previously available from this vendor [13, 54]. Western blot analyses and staining specificity controls were also performed to validate this antibody. To allow semi-quantitative comparisons, sections from all animals were stained simultaneously in the same staining tray. The FKBP1b stained sections of the entorhinal cortex were digitized using an Olympus DP73 camera, and the resulting images were analyzed using the ImageJ (NIH Image) program. For optical densiometric analysis of FKBP1b immunohistochemistry of the medial entorhinal cortex, two sections per brain, each containing layers II and III (Fig. 3), were measured by an investigator blinded to groups. All photomicrographs were taken with the same settings of the DP73 camera.
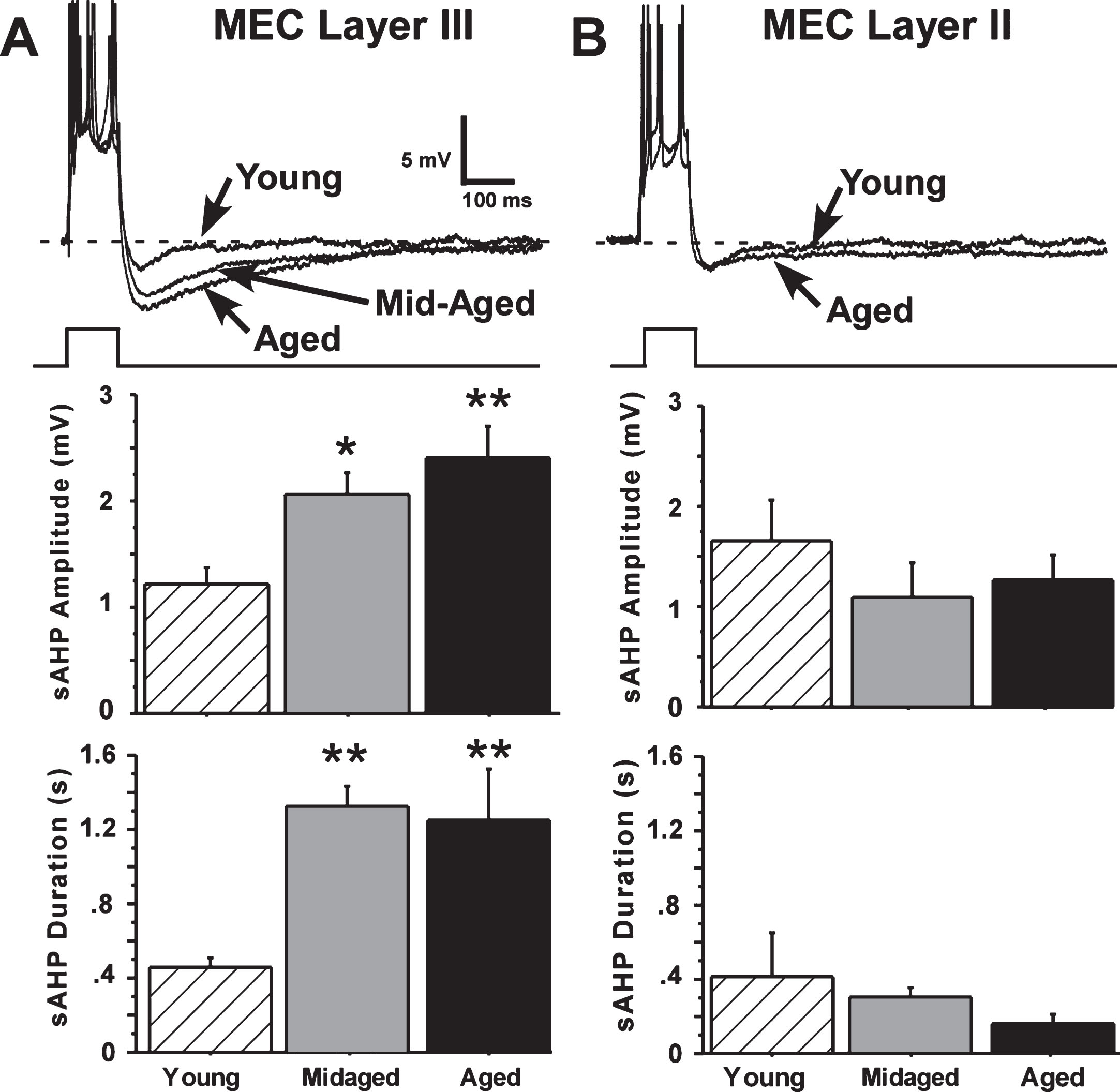
The slow afterhyperpolarization (sAHP) is altered by aging only in layer III. A) Representative examples and age-group means of sAHPs measured in layer III of the MEC. Significant age differences in slow AHP amplitude and duration were found for layer III neurons, with sAHPs in aged (21 months old) and mid-aged (10 months old) animals increased compared to sAHPs in young (4 months old) rats. B) Representative examples and age-group means of sAHPs measured in layer II of the MEC. No aging differences were observed in layer II neurons. (Means ± S.E.M. for group values; *p < 0.01; **p < 0.001 ANOVA pLSD significant pairwise contrast versus young).
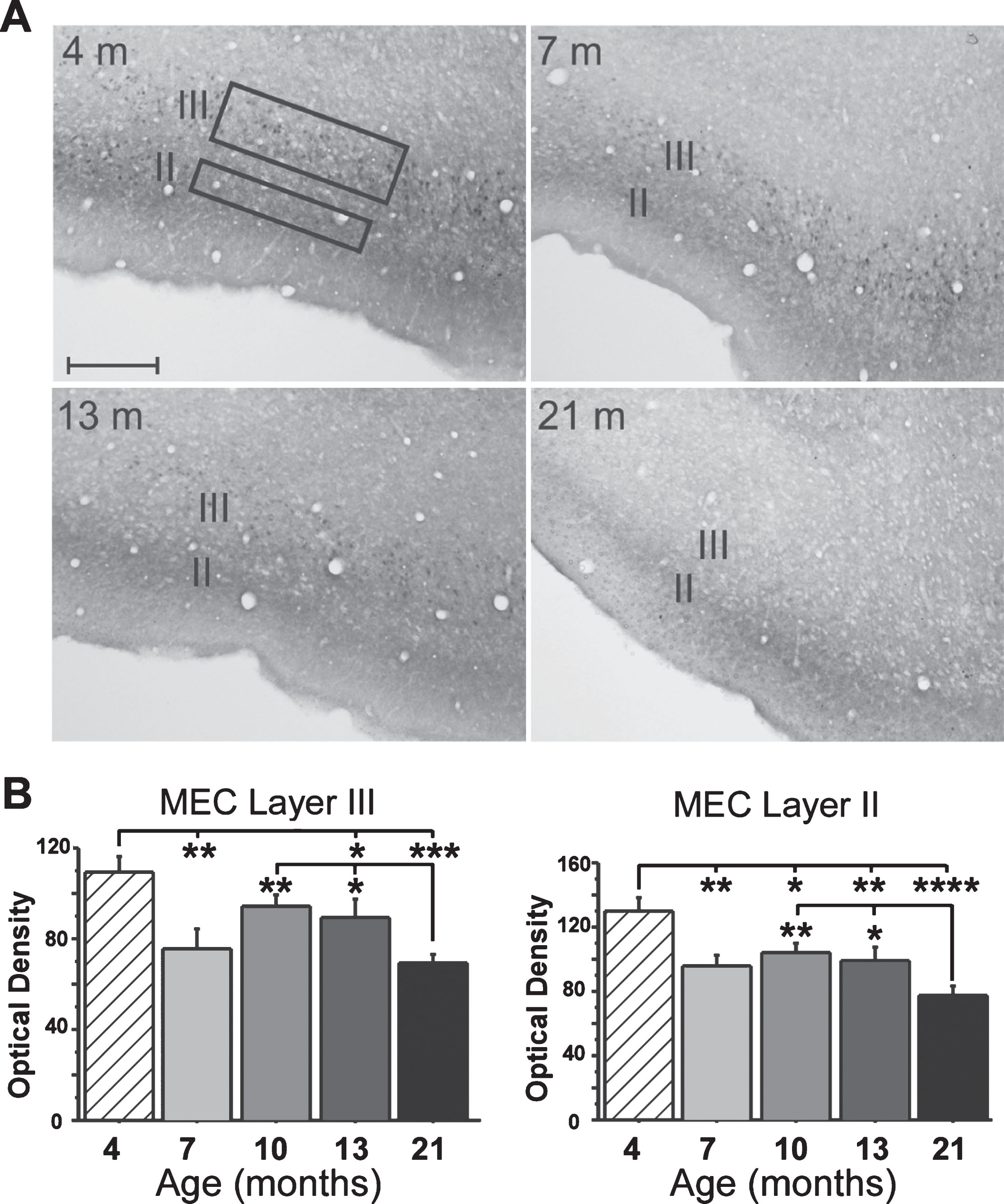
Immunostaining for MEC FKBP1b expression. A) Representative micrographs of MEC FKBP1b immunostaining at 4, 7, 13, and 21 months-of-age. B) Means ± S.E.M. for FKBP1b immunostaining at 4 (n = 5), 7 (n = 5), 13 (n = 6) and 21 (n = 7) months-of-age. Significant reduction in FKBP1b occurs as early as 7 months in both layers II and III of the MEC. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.000; ANOVA pLSD significant pairwise contrast versus 4 months and versus 21 months).
RESULTS
It was apparent that the sAHP in layer II cells did not differ in amplitude or duration across young, mid-aged and aged rats. In contrast, the sAHPs in layer III pyramidal cells from aged and mid-aged rats were substantially increased compared to sAHPs in young-adult rat cells (Fig. 2).
Amplitude of the sAHP in layer III neurons was significantly increased with aging (main effect of age, F(44,2) = 7.54, p = 0.0015, ANOVA), and both mid-aged and aged rat neurons differed from young in pairwise comparisons (Fisher’s protected least-squares difference (pLSD), p < 0.05 and 0.001, respectively). sAHP duration was also highly significantly increased with age (F(44,2) = 13.91, p = 0.000021, ANOVA) and both mid-aged and aged rat cells differed from young (p < 0.0001 and p < 0.0001, respectively) (Fig. 2). Power analyses predicted that neither increasing the number of layer II sAHP observations from 27 to 43, nor reducing the number of layer III observations from 43 to 27, would alter the present results. (Preliminary studies indicate that layer III neurons of the LEC also exhibit a large aging-related increase in sAHP magnitude—data not shown.)
Results for the IHC analyses showed a sharp decline in FKBP1b expression in layer III neurons very early in adulthood (between 4 and 7 months-of-age). Expression then remained relatively stable through mid-life, after which it declined further by 21 months (Fig. 3). Although layer II neurons did not exhibit electrophysiological evidence of Ca2 + dysregulation, a similar pattern of decrease was found for FKBP1b expression in layer II (see Discussion).
DISCUSSION
Our findings show that rat MEC layer III pyramidal neurons, which project to CA1 neurons, selectively develop a type of aging-related Ca2 + dysregulation similar to that found in CA1 pyramidal cells. Moreover, the EC neurons appear to develop Ca2 + dysregulation at a slightly earlier age (8–10 months-old, Fig. 2) than do CA1 cells (12 months old) [48], mirroring the anatomic sequence of NFT progression during early-stage AD [8]. Considering that rat layer III neurons are highly homologous with human layer III EC neurons [26, 60], and that animal and human brains exhibit highly similar patterns of normal aging changes (Introduction), it seems likely that early aging-related Ca2 + dysregulation also develops in human EC neurons, possibly generating conditions that promote NFT pathogenesis. Consistent with this view, some biomarkers of aging-related Ca2 + dysregulation are estrogen-dependent [38] while aging changes in estrogen function may contribute to sex differences in AD vulnerability [5].
One potential mechanism through which aging-related Ca2 + dysregulation might promote NFT pathogenesis is by degrading the cytoskeleton, a well-recognized effect of elevated Ca2 + [46, 47]. Moreover, we recently found that FKBP1b overexpression counteracted age-related downregulation of cytoskeletal gene expression [61]. Thus, the cytoskeleton of entorhinal neurons may be disrupted by aging-related Ca2 + dysregulation. In human EC neurons, the cytoskeleton may be particularly sensitive to such disruption, resulting in NFT pathogenesis.
It seems intriguing that the layer III EC neurons that show pronounced Ca2 + dysregulation project to CA1 neurons, which exhibit a similar form of Ca2 + dysregulation [30, 50]. These findings raise the possibility that Ca2 + dysregulation can be transmitted trans-synaptically. Moreover, during early-stage AD, NFTs track a similar EC to CA1 progression pathway [8], consistent with the view that Ca2 + dysregulation precedes and promotes NFT pathogenesis. On the other hand, NFT progression may also require transmission of toxic tau molecules, as several studies have suggested that tau proteins may be transmitted trans-synaptically [10, 63].
Somewhat surprisingly, layer II showed a pattern of FKBP1b decline similar to that in layer III, even though layer II pyramidal neurons did not manifest electrophysiological signs of aging-related Ca2 + dysregulation. It is unclear what the effects of the age-dependent FKBP1b decline in layer II may be, or whether stellate or pyramidal cells are the primary locus of the decline. Alternatively, the decline could reflect decreased FKBP1b expression in layer III pyramidal cell dendrites, which ramify extensively in layer II [57, 58]. Clearly, considerable additional work will be needed to resolve these questions.
Overall, our results support the hypothesis that aging-related Ca2 + dysregulation in entorhinal-hippocampal neurons is a key factor in age-dependent vulnerability to NFT pathogenesis. Many aspects of this hypothesis remain to be tested, but if further work continues to support this view, counteracting aging-related Ca2 + dysregulation in the EC-hippocampal NFT pathway [eg.,13, 61] may eventually become a novel therapeutic approach for preventing AD progression.