
Abstract
A growing body of evidence supports a clear association between Alzheimer’s disease and diabetes and several mechanistic links have been revealed. This paper is mainly devoted to the discussion of the role of diabetes-associated mitochondrial defects in the pathogenesis of Alzheimer’s disease. The research experience and views of the author on this subject will be highlighted.
INTRODUCTION
Mitochondria are fascinating organelles. They are highly dynamic and plastic organelles and can adapt to different surroundings and particularities of each type of cell. Mitochondria are essential for the viability and proper function of cells, being involved in the generation of energy (ATP), metabolism of reactive oxygen species (ROS), buffering of cytoplasmic calcium (Ca2 +) and apoptosis [1], among other things. Considering neuronal cells, mitochondria are crucial for the maintenance of membrane ion gradients, and for neurotransmission and synaptic plasticity [2]. Taking into consideration that neurons are highly differentiated cells (cell body, dendrites, axons, and synaptic terminals) with a high metabolic rate that requires a constant supply of energy substrates (particularly glucose) and oxygen to survive, it is not surprising that alterations in mitochondria will deeply impact the function and viability of neuronal cells. In fact, a delicate balance between mitochondrial fission, fusion, biogenesis, turnover, and transport must exist to ensure a healthy mitochondrial pool and, consequently, viable and healthy cells. This balance is lost in several diseases including Alzheimer’s disease (AD) and diabetes [3–7]. AD is the most common cause of dementia in the elderly and despite the tremendous research efforts in the last decades, no cure or effective treatment exists [3]. Nevertheless, the mechanisms underlying AD pathophysiology have been successfully unveiled and accumulating evidence demonstrates the key involvement of mitochondrial anomalies in the development of the disease. The metabolic defects that characterize (sporadic) AD can be triggered or exacerbated by several risk factors including diabetes [6, 8].
Diabetes is a major public health problem that is reaching epidemic proportions around the world. It is a complex metabolic disorder mainly characterized by chronic hyperglycemia and associated with progressive end-organ damage. Besides the commonly associated chronic complications such as nephropathy, angiopathy, retinopathy, and peripheral neuropathy [9], it was also observed that people with diabetes perform poorly on cognitive tasks examining memory, attention, and verbal learning [10, 11]. The long-term effects of diabetes on the brain are manifested at structural, neurophysiological, and neuropsychological level [12]. Although both type 1 diabetes (T1D) [13, 14] and type 2 diabetes (T2D) [15, 16] are associated with alterations in brain structure and function and increased risk of dementia [17], the association between AD and T2D is stronger than that with T1D. In fact, several lines of evidence demonstrate that T2D is associated not only with AD [15, 19], but also with vascular dementia [19], Parkinson’s disease [20], and Huntington’s disease [21]. At the mechanistic level, it has been shown that mitochondrial defects play an important role in diabetes-associated brain alterations [8, 22, 23] contributing to neurodegenerative events.
In this paper, I will present evidence from epidemiological and clinical studies that support a clear association between AD and diabetes. Then, the role of diabetes-induced mitochondrial defects as triggers and/or accelerators of AD (like) pathology will be discussed. The research experience and views of the author will be emphasized.
EPIDEMIOLOGICAL AND CLINICAL STUDIES HAVE BOOSTED THE STUDY OF THE MECHANISTIC INTERRELATION BETWEEN DIABETES AND AD
The effects of diabetes on the central nervous system (CNS) were described for the first time almost 100 years ago. In 1922, Miles and Root [24], reported that diabetic individuals perform poorly on cognitive tasks examining memory and attention. And, in 1950, Dejong named the diabetes-related CNS complications as diabetic encephalopathy [25]. Subsequent longitudinal and cross-sectional studies confirmed the negative impact of T1D [26–28] and T2D [29–32] on cognitive function. T2D has also been associated with 50% increased risk of dementia [33]. Whether such an association is true for people T1D is not yet clear.
Over the past three decades, many epidemiological and clinical studies have shown a clear association between diabetes and an increased risk of developing AD. Our interest on the interrelation between diabetes and AD started shortly after the publication of the results of the Rotterdam Study [34, 35] showing that patients with T2D are at an increased risk to develop dementia and AD. Further evidence showed that individuals with T2D have nearly a twofold higher risk of AD than nondiabetic individuals [36–42]. Furthermore, the risk of AD associated with the APOE ɛ4 allele has been suggested to be exacerbated by diabetes, as patients with diabetes who are ɛ4 allele carriers are twofold more prone to develop AD than nondiabetic individuals who harbor the ɛ4 allele [43]. Notably, a study of the Mayo Clinic Alzheimer Disease Patient Registry reported that greater than 80% of AD patients exhibit T2D or abnormal blood glucose levels [44], suggesting that AD patients are more vulnerable to T2D and the possibility of a linkage between the processes responsible for loss of brain and pancreatic β-cells in these disorders. However, it must be said that other studies did not find a clear link between AD and diabetes [45–47].
Considering the resemblances found between AD and T2D, and due to the lack of effective treatments for AD, it has been hypothesized that anti-diabetic drugs can help treat AD patients. Promising effects of intranasally administered insulin or insulin analogues have been observed in AD and subjects suffering from amnestic mild cognitive impairment (MCI) [48–50], although insulin administration in APOE ɛ4 carriers seems to exacerbate cognitive deficits [51]. Studies have also shown that the chronic treatment of diabetics with the antidiabetic agent metformin reduces the risk of cognitive decline [52]. However, conflicting effects of metformin treatment have been reported [53].
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists and potent insulin sensitizers. Pioglitazone and rosiglitazone are the best characterized PPAR-γ agonists. Rosiglitazone was associated with an early improvement of whole brain glucose metabolism, but not with any biological or clinical evidence for slowing progression over a 1-year follow up in the symptomatic stages of AD [54]. Mild and moderate AD patients who are APOE ɛ4 non-carriers treated with rosiglitazone during 6 months improved cognitive function, an effect not observed in APOEɛ4 carriers [55]. Also, a systemic review and meta-analysis concluded that pioglitazone may be useful in treating AD patients with comorbid diabetes [56]. A recent longitudinal study show that pioglitazone might provide a protective effect on dementia risk among individuals with T2D [57].
A recent prospective and observational study aimed at evaluating the effect of sitagliptin, a dipeptidyl peptidase-4 inhibitor (DPP-4I), on cognitive function of elderly diabetic patients with and without cognitive impairment revealed that elderly diabetic patients with and without AD treated with sitagliptin during 6 months show improved of cognitive function [58].
Concerning the effects of glucagon-like peptide-1 (GLP-1) analogues, a recent randomized, placebo-controlled, double-blind clinical trial involving 18 AD patients treated with the GLP-1 analogue liraglutide and 20 AD patients treated with placebo revealed that treatment with liraglutide during 6 months prevented the decline of brain glucose metabolism [59]. More clinical trials are ongoing, namely a pilot clinic trial of exendin-4 in MCI and early stage AD subjects (NCT01255163) and a phase II clinical trial assessing the safety and efficacy of liraglutide in mild AD (NCT01843075).
The above evidence demonstrates that AD and diabetes are connected and instigated the research community to investigate the mechanisms linking both disorders.
THE QUEST FOR THE MECHANISTIC LINKS BETWEEN AD AND DIABETES: A FOCUS ON DIABETES-RELATED MITOCHONDRIAL DEFECTS
Based on a strong body of evidence demonstrating a clear connection between diabetes and AD, several pre-clinical studies have been carried out in order to uncover the mechanistic basis of this connection. Several mechanisms shared by diabetes and AD have been identified, namely impaired insulin signaling, inflammation, the accumulation of advanced glycation end-products, oxidative stress, and mitochondrial dysfunction [7, 60]. Here, I will discuss how diabetes-related brain mitochondrial anomalies contribute to cognitive defects and predispose to or exacerbate neurodegenerative events, particularly AD (like) pathology.
Type 1 diabetes and AD (like) pathology
Previous studies from our laboratory revealed that brain mitochondria isolated from streptozotocin (STZ)-induced diabetic rats present defects in the antioxidant system defenses, ATPase activity and ability to accumulate Ca2 + [61]. In accordance with our observations, others reported that brain mitochondria isolated from STZ-induced diabetic rats have a deficient respiratory chain [62, 63] that may contribute to oxidative and nitrosative injury of brain cells [64–66], tau hyperphosphorylation [65, 68], amyloidogenesis [69, 70], and cognitive defects [63, 72]. More recently, we observed that insulin treatment modulates mitochondrial dynamics and biogenesis, autophagy, and tau protein phosphorylation in the brain of STZ-induced diabetic rats [73].
Besides hyperglycemia, hypoglycemia, the most serious side effect associated to insulin therapy in T1D, also causes cognitive dysfunction [74, 75] in part by affecting mitochondria [76]. McGowan and collaborators [77] demonstrated that a mitochondrial substrate limitation following hypoglycemia increases mitochondrial free radical production in brain cortical mitochondria from newborn pigs. It was also shown that hypoglycemia in STZ-induced diabetic rats decreases mitochondrial respiratory chain efficiency [78]. Accordingly, we observed that insulin-induced acute hypoglycemia affects the antioxidant defenses of brain cortical mitochondria isolated from STZ-induced diabetic rats causing oxidative damage [79] and increasing the capacity of cortical synaptosomes to release excitatory amino acids [80]. Taking into consideration that mitochondria function is associated with neurotransmitters synthesis and release, our observations support the idea that mitochondrial dysfunction, oxidative stress, and excitatory neurotransmitters release are interconnected factors that may underlie the cognitive impairment observed in T1D patients under insulin therapy. Subsequent studies from our laboratory showed that recurrent hypoglycemia and long-term hyperglycemia affect the antioxidant defenses of brain cortical and hippocampal mitochondria contributing to oxidative stress [81]. Nevertheless, only hippocampal mitochondria showed altered efficiency of the respiratory chain and phosphorylation system affecting ATP production [81]. Similarly, Dave and collaborators [82] reported that recurrent hypoglycemia exacerbates cerebral ischemic damage in T1D rats through the increased generation of mitochondrial ROS.
Besides their effective antioxidant defense system, mitochondria have specific proteins that regulate the rate of ROS production named uncoupling proteins (UCPs). The dissociation of ATP production from ROS generation mediated by UCPs has been pointed as a key defensive mechanism against brain damage [83, 84]. In this line, we observed that recurrent episodes of hypoglycemia render brain cortical mitochondria more susceptible to UCPs-mediated uncoupling as compared with hyperglycemia [85] and the pharmacological inhibition of UCP2 exacerbates glucose fluctuations-mediated neuronal damage, namely mitochondria dysfunction and oxidative stress [86]. Interestingly, UCP2 gene variants have been associated with a reduced risk for diabetic neuropathy in T1D patients [87], suggesting that the increased expression of UCPs related to specific gene polymorphisms can limit neuronal death.
Type 2 diabetes and AD (like) pathology
An increased oxidative stress and mitochondrial dysfunction was observed in the Zucker diabetic fatty rat, a genetic model of T2D [88]. Accordingly, we found that brain vessels and synaptosomes from Goto-Kakizaki rats, a spontaneous model of non-obese T2D, present an age-dependent redox imbalance [89], which increases the vulnerability of brain structures to degenerative events. We also observed that T2D-like mice and triple transgenic AD mice (3xTg-AD) present a similar profile of brain mitochondrial anomalies (defects in function, biogenesis, and turnover), redox imbalance, increased amyloid-β (Aβ) and phosphorylated tau levels, central and peripheral vascular alterations, and loss of synaptic integrity [90–93]. Accordingly, 3xTg-AD and T2D-like mice show similar behavioral and cognitive anomalies characterized by increased fear and anxiety and decreased learning and memory abilities [91]. Such findings prompted us to suggest that the metabolic alterations associated with diabetes contribute to AD-like pathologic features [90–92]. Accordingly, it was recently suggested that the higher hippocampal susceptibility to synaptic injury and cognitive dysfunction is linked to mitochondrial defects [94]. These authors observed that db/db mice, a model of obese T2D, present altered brain mitochondrial morphology, reduced ATP production, and impaired mitochondrial complex I activity. These mitochondrial abnormalities seem to result from an imbalanced mitochondrial fusion and fission via a glycogen synthase kinase 3β (GSK3β)/dynamin-related protein-1 (Drp1)-dependent mechanism [94]. It was also reported that high glucose and Aβ oligomers cause aberrant S-nitrosylation of insulin-degrading enzyme and Drp1 inhibiting insulin and Aβ catabolism as well as hyperactivating mitochondrial fission machinery, which results in dysfunctional synaptic plasticity and synapse loss [95]. A recent study from Petrov and collaborators [96] also showed that T2D induced by high fat diet affects brain mitochondrial function contributing to cognitive decline and AD pathology, which are ameliorated by the anti-diabetic drugs dipeptidyl-peptidase-4 inhibitors [97] and metformin [98]. Also, the use of GLP-1 analogues (e.g., liraglutide and exendin-4) increase brain insulin, insulin-like growth factor 1, and GLP-1 signaling and decrease phosphorylated tau levels and apoptosis [99–102].
Using a rat model of sporadic AD induced by the intracerebroventricular (icv) administration of a sub-diabetogenic dose of STZ (icvSTZ), a model firstly described by Hoyer’s team [103–105], we found that the insulin-resistant brain state that characterizes the pathological course of the disease is accompanied by mitochondrial abnormalities [106]. We observed that icvSTZ promotes a significant decline in both brain cortical and hippocampal mitochondrial bioenergetics as reflected by impaired mitochondrial respiration and phosphorylation system, increased susceptibility to Ca2 +-induced mitochondrial permeability transition pore opening, and oxidative stress. Importantly, increased levels of Aβ and phosphorylated tau and cognitive defects accompanied those mitochondrial defects [106]. Similarly, Paidi and colleagues [107] observed that the cognitive defects observed in icvSTZ rats were associated with an increased mitochondrial fragmentation. In monkeys, icvSTZ caused a pronounced ventricular enlargement and parenchymal atrophy, Aβ deposition, hippocampal cell loss, tauopathy, astrogliosis, and microglial activation [108]. The Chinese herbal medicine geniposide ameliorates learning and memory deficits, reduces tau phosphorylation, and decreases apoptosis via GSK3β pathway in icvSTZ rats [109]. Also, the GLP-1 analogue exenatide [110] and intranasal insulin [111] improve cognitive function, attenuate the levels of hyperphosphorylated tau and inflammation, and enhance neurogenesis in icvSTZ rats.
Diabetes/hyperglycemia exacerbate(s) AD (like) pathology
All the above studies demonstrate that diabetes induces AD-like changes supporting the notion that diabetes increases the risk of developing AD. However, there is also evidence that diabetes exacerbates AD progression.
Early studies from our laboratory [112] showed that brain mitochondria isolated from Goto-Kakizaki rats present an age-related decline of the respiratory chain and oxidative phosphorylation system efficiency and a higher susceptibility to oxidative damage; those age-dependent effects being highly exacerbated by the neurotoxic peptides Aβ25-35 and Aβ40 [112] and ameliorated by coenzyme Q10 treatment [113]. Also, brain mitochondria isolated from STZ diabetic rats are highly susceptible to Aβ40, which cause an impairment of the respiratory chain and phosphorylation system and an increased production of ROS [61]. Interestingly, Aβ40-mediated mitochondrial defects were attenuated by insulin treatment [61]. Consistently, brain endothelial cells under chronic hyperglycemia are more susceptible to Aβ40 toxicity, an effect mediated by mitochondrial ROS [114]. In this line, Gou and colleagues [115] observed that chronic hyperglycemia induced via the heterozygous knockout of Pdx1 worsens AD-like neuropathology in mice. Similarly, Hayashi-Park and colleagues [116] reported that diabetes exacerbates neuropathology, but not cognitive dysfunction, of middle-aged 3xTg-AD mice. It was also reported that hyperglycemia exacerbates mitochondrial defects, synaptic injury, and cognitive dysfunction in the brains of transgenic AD mice [63], which suggest that the synergistic interaction between diabetes (hyperglycemia) and AD on mitochondria may be responsible for brain alterations characteristic of both disorders.
AUTHOR VIEWS AND CONCLUSIONS
Mitochondria are crucial organelles for life and death of cells. Recent discoveries show that mitochondria have a key role in regulating synaptic transmission, brain function, and cognition in aging [117] and, most probably, age-related disorders such as diabetes (namely T2D) and AD. Although mitochondrial dysfunction affects all organs, the brain appears most vulnerable to mitochondrial defects suggesting that mitochondria regulate fundamental aspects of brain function. With glucose oxidation the most relevant source of energy in the brain, neurons rely almost exclusively on the mitochondrial oxidative phosphorylation system to obtain ATP to fulfill their high energy needs. Since diabetes and AD are characterized by defective brain glucose metabolism, it is not surprising that mitochondrial anomalies are a defect shared by both disorders and that mitochondrial alterations caused by diabetes can contribute to neurodegenerative events such as AD.
As discussed above, a strong body of evidence from our laboratory and others support a mechanistic role for mitochondria in the connection between diabetes and AD (Fig. 1). However, it remains uncertain whether defective mitochondria are the initiating defect or secondary to altered insulin signaling. As stated above, altered insulin signaling is another mechanistic link between AD and diabetes. The existing literature shows that both insulin signaling and mitochondria defects can affect each other (Fig. 1). Peng and colleagues [118] observed that neurons exposed to high glucose develop mitochondrial defects, which affect 5’ AMP-activated protein kinase (AMPK)/AKT signaling contributing to insulin resistance. It was also shown that resveratrol, which activates peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) that stimulates mitochondrial function [119, 120], improves AMPK/AKT signaling increasing insulin sensitivity [118]. In skeletal muscle and liver mitochondrial dysfunction and ROS overproduction activate c-Jun N-terminal kinase (JNK) leading to insulin resistance [121]. A similar process can occur in the brain since JNK activation has been observed in AD [62, 123].
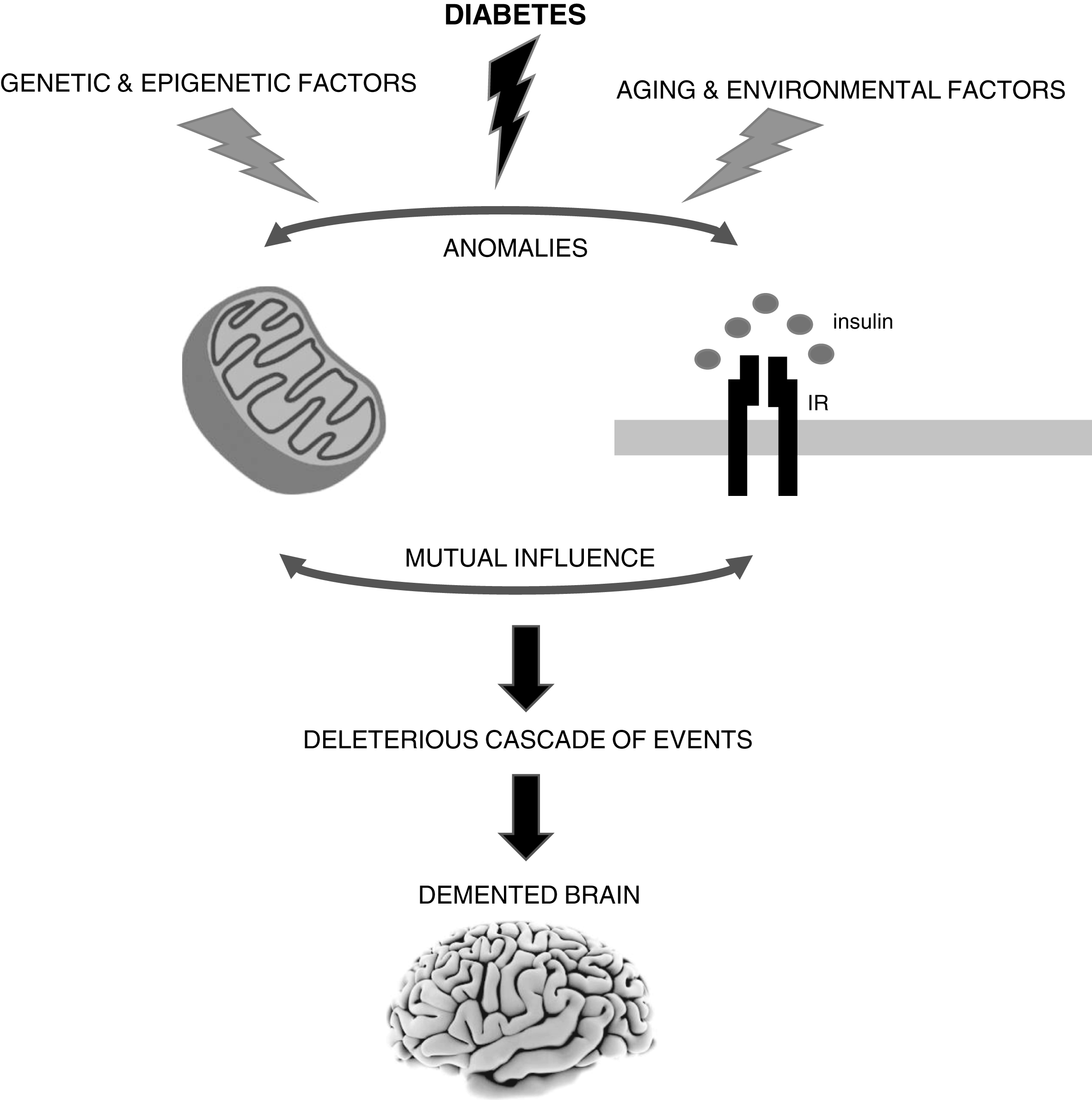
The brain is highly vulnerable to mitochondrial defects since neurons rely almost exclusively in the mitochondrial oxidative phosphorylation system to obtain ATP to fulfill their high energy needs. Accumulating evidence shows that mitochondrial alterations caused by diabetes can contribute to neurodegenerative events such as Alzheimer’s disease (AD). However, it remains uncertain whether defective mitochondria are the initiating defect or secondary to altered insulin signaling. In fact, both insulin signaling and mitochondria defects can affect each other. It is also important to note that sporadic AD is a multifactorial condition that depends on the complex interplay between environmental, genetic and epigenetic factors. IR, insulin receptor.
However, insulin resistance can also affect mitochondrial function. Previous studies showed that pancreatic β-cells from β-cell specific insulin receptor knockout (βIRKO) mice [124] and the deletion of insulin receptors in mice cardiomyocytes (CIRKO mice) cause mitochondrial dysfunction [125]. Furthermore, prolonged exposure to insulin affects mitochondrial DNA (mtDNA), biogenesis and mass, and ATP content in hepatocytes due to a decrease in the levels of both nuclear respiratory factor (NRF) and mitochondrial transcription factor A (Tfam) [126]. Insulin also modulates mitochondrial biogenesis through the mammalian target of rapamycin (mTOR)-dependent regulation of PGC1-α, a master regulator of mitochondrial biogenesis responsible for the co-activation of several metabolically significant nuclear and non-nuclear receptor transcription factors such as NRF 1 and 2 [127, 128]. In addition, thiazolidinediones, clinically used to ameliorate insulin resistance in T2D, increase mitochondrial biogenesis in human subcutaneous adipose tissue, human neuronal NT2 cells, and mouse brain [129–131], which suggest that insulin modulates mitochondria.
In conclusion, although mitochondrial defects, particularly those associated with diabetes, seem to play an important and early role in AD development, further studies are needed to clarify if those defects can be the triggers or secondary events. This is of utmost importance since diabetes and AD have become global epidemics and no effective treatments exist for AD. Importantly, we must keep in mind that (sporadic) AD is a multifactorial condition and that depending on the complex interplay between environmental, genetic, and epigenetic factors, it may have distinct or even multiple triggers (Fig. 1).
ACKNOWLEDGMENTS
This work was financed by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Program: project CENTRO-01-0145-FEDER-000012-HealthyAging2020, the COMPETE 2020 - Operational Program for Competitiveness and Internationalization, and the Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, I.P.: project POCI-01-0145-FEDER-007440.
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/17-0931r1).