
Abstract
The production of soluble amyloid-β oligomers (AβOs) and the activation of inflammation are two important early steps in the pathogenesis of Alzheimer’s disease (AD). The central role of oligomers as responsible for the neuronal dysfunction associated with the clinical features has been extended to the other protein misfolding disorders definable, on this basis, as oligomeropathies. In AD, recent evidence indicates that the mechanism of inflammation as a consequence of neurodegeneration must be assessed in favor of a more direct role of glial activation in the alteration of synaptic function. Our own experimental models demonstrate the efficacy of anti-inflammatory treatments in preventing the cognitive deficits induced acutely by AβOs applied directly in the brain. Moreover, some promising clinical tools are based on immunological activation reducing the presence of cerebral Aβ deposits. However, the strategies based on the control of inflammatory factors as well as the amyloid aggregation show poor or non-therapeutic efficacy. Numerous studies have examined inflammatory factors in biological fluids as possible markers of the neuroinflammation in AD. In some cases, altered levels of cytokines or other inflammatory markers in cerebrospinal fluid correlate with the severity of the disease. Here we propose, according to the precision medicine principles, innovative therapeutic approaches to AD based on the patient’s inflammatory profile/state. The earlier intervention and a multifactor approach are two other elements considered essential to improve the chances of effective therapy in AD.
Keywords
INTRODUCTION
In Alzheimer’s disease (AD), together with the specific neuropathological features, cerebral amyloid deposits and neurofibrillary tangles, the glial reactivity testify to the involvement of inflammation in the pathological process [1]. However, the loss of synapses is the structural change that correlates best with the cognitive decline in AD patients [2]. The neuronal dysfunction induced by soluble small aggregates of amyloid-β oligomers (AβOs)—considered the real toxic species in AD [3, 4]—might explain the positive correlation of the total cerebral Aβ burden with clinical features [5]; the clinical correlation no longer holds when limited to the amyloid plaque distribution, and is only partial when associated with tangle distribution [6].
According to the latest version of the amyloid cascade hypothesis [7], the pathological scenario of AD identifies the soluble small aggregates of Aβ, AβOs, as the main species responsible for the neuronal dysfunction [8, 9], and this concept has been extended to all the other protein misfolding disorders. We have coined the term oligomeropathy to illustrate the close relationship between neurodegeneration and these heterogeneous formations of soluble aggregates [10, 11]. The mechanism of disease spreading described in AD models, but also proposed in Parkinson’s disease or frontotemporal dementia, might employ the oligomers as the species responsible for cell-to-cell transmission [12, 13]. Although several aspects of this mechanism are still elusive, the intercellular passage might employ small aggregates to spread the pathology [14, 15]. Penetration of the oligomers into the cells could be associated with a seeding mechanism and prion-like replication, followed by their release by themselves or combined with vesicle structures, such as exosomes [16]. Thus, the oligomeric structures might play a central role for both neuronal dysfunction and disease propagation.
Gliosis is generally considered a consequence of amyloid deposition and neurodegeneration, activating the production of toxic factors in a vicious circle that amplifies the neuronal dysfunction. This concept has been recently flanked by a more direct influence of inflammatory factors in the early phase of the pathological process [17]. Physiological role of surveillance exerted by microglia and astrocytes for the presence of pathogens and cellular debris is combined with the synaptic modeling, formation, and elimination during development, but also in adulthood [18–20]. This later activity is an important element in favor of the early participation of glial cells in AD pathogenesis.
The intimate contact between microglia and astrocytes with synapses has been investigated from the functional point of view, inspiring several hypotheses on the positive or detrimental roles of immune factors in physiological and pathological aging. It is very possible that the alteration of the glial cells dynamic relationship with synaptic structures and function contributes with the aberrant pruning to the neuronal dysfunction mechanisms [21–22].
The genetic evidence also helps support an essential, rather than secondary role, of the inflammation and several genes involved in the immune pathway have been associated with AD. In particular, TREM2 (triggering receptor expressed on myeloid cells 2) and CD33 polymorphisms have been recently linked to the risk of AD [23–26]. The expression of these factors in the brain is restricted to microglial cells, indicating the direct involvement of these cells in AD pathogenesis.
AD AS AN OLIGOMEROPATHY
In vitro models proposed a direct neurotoxic effect of Aβ at the beginning of the 1990s [27]; the effect was initially related to the fibrillogenic capacity of Aβ peptides [28, 29]. The critical role of oligomeric forms in the pathogenesis of AD emerged from observations that, in contrast with the common belief at that time, the fibrillogenic capacity of the Aβ peptides did not always correlate with their toxicity [30, 31]. The soluble species of Aβ aggregates were purified from AD brain and CSF and induced neuronal dysfunction in experimental conditions [3].
The origin and size of the AβO species involved in the neurodegenerative mechanisms and the cellular pathways mediating their effects have been widely debated [32, 33]. Although numerous biological alterations are associated with their presence, their biological activities can be triggered essentially by three mechanisms: the interaction with specific entities on cell membranes [34]; the unspecific membrane perturbation [35] and the capacity to form a pore channel inside the membrane [32]. These basic mechanisms involving oxidative stress, mitochondrial alterations, glial activation, and glutamate receptors are common to virtually all oligomers, regardless of the initial misfolded protein sequence [36–38]; other more specific aspects are associated with sequence, size and conformation [13].
In an AβO preparation, species of the same size can be found that are neither toxic nor recognized by oligomer-specific antibodies, indicating that AβOs may also show size-independent differences in toxicity [39], and differences between two AβOs of similar size and dissimilar toxicity may be seen [40]. These studies suggest that the toxicity of small amyloidogenic oligomers is governed primarily by the degree of solvent exposure of hydrophobic residues and is weakly influenced by their secondary structures [41]. Chaperones play an important part not only in the oligomer conformation and assembling but also in their biological interaction. Chaperons bind to protein oligomers, interfering with amyloid fibril formation, but can also directly inhibit the toxicity of these species [42]. Considering the size of oligomers and their hydrophobic exposure as the main determinants of their neurotoxicity [43, 44], molecular chaperons increase the size of the oligomers and mask hydrophobic patches exposed on their surface [45].
The main components of senile plaques and oligomers are the peptides Aβ1-40 and Aβ1-42. Both have self-aggregation capacity but with different kinetics; the longer sequence can spontaneously aggregate within minutes, while in similar conditions Aβ1-40 takes hours and days to assemble in fibrils. Once the AβOs are formed, the biological effects are relatively independent from the initial sequences [4]. Other Aβ peptide species in the brain are Aβ1-43, and Aβ1-37 or Aβ1-38, and although most part of the experimental studies have been used using Aβ1-42 solutions, the natural substrate of oligomers is a mixture of peptides.
The N-terminally truncated pyroglutamylated form of Aβ (AβpE), also identified in AD brain with high self-aggregation capacity, has been indicated as the seed of nucleation of the Aβ1-42 solution [46–48]. The synergistic effect of Aβ/AβpE hetero-oligomers results in a species with a high level of toxicity, stabilizing the oligomeric structures and retarding fibril formation [49]. Active and passive immunization based on AβpE have given beneficial effects in experimental models of AD [50, 51].
Amyloid deposition is considered an early pathological event in AD [52]; as recently indicated by a meta-analysis, high levels of Aβ were associated with a small cognitive impairment and decline in cognitively normal older adults, suggesting a possible dementia prodromal condition in these subjects [53]. However, the lack of correlation between amyloid plaque distribution and the cognitive decline argues against a direct pathogenic role of Aβ deposits in AD. Furthermore, the amyloid plaques do not correlate with the clinical progression of the disease: after the clinical onset, the Aβ plaque burden was substantially stable [54]. Positron emission tomography (PET) analysis in longitudinal studies showed that in mild cognitive impairment (MCI), an AD prodromal condition, and in AD subjects the cerebral fibrillar Aβ levels reached a plateau early and remained unchanged during the disease progression. Structural changes detected by MRI and cognitive impairment, measured by neuropsychological tests, proceed independently of amyloid accumulation [55]. The presence and the toxic role of AβOs might explain these findings: the early neuronal dysfunction in the preclinical phase of AD is due to the soluble aggregates released by the Aβ plaques.
The burden of AβOs, which unfortunately are not visualized by PET probes, is generally associated with high levels of fibrillar Aβ; the senile plaques are considered to be in dynamic equilibrium with the oligomers, and sequester soluble aggregates which are then released from the deposits [30]. However, in a small percentage of subjects the development of AD is not strictly related to the high level of fibrillar Aβ in the preclinical phase; in this case the production of AβOs is independent of the accumulation of Aβ plaques [56]. This is possible because the passage from the monomeric form of Aβ to the insoluble fibrils is not an invariable, linear process that reaches the fibrillar structures through oligomeric and protofibrillar species. In some cases, oligomers did not generate fibrils (off pathway), and the toxicity of these stable products should be analyzed case by case [57, 58]; in other conditions, it has been proposed that oligomers form fibrils through metastable intermediates [59].
In transgenic models, cognitive decline but also synaptic alterations were evident in plaque-free conditions [60]. Single-transgenic mice (APP23) in a pre-plaque stage displayed memory deficits and long-term potentiation impairment, with synaptic hippocampal damage. APP23 tg mice have hippocampal alterations in the trafficking of synaptic NMDAR subunits NR2A and NR2B and ultrastructural analysis shows AβO localized intracellularly in the synaptic compartments. Importantly, the behavioral and biochemical alterations of NMDAR signaling are associated with the inhibition of long-term synaptic potentiation and inversion of metaplasticity at CA1 synapses in hippocampal slices from another transgenic mouse (Tg2576) [61].
Gandy et al. [62] showed that single-transgenic mice (APP E693Q) present a significant oligomer-dependent delay in acquisition of the Morris water maze task at 12 months of age, which it is not dependent on the development of AD-like plaque pathology or macrohemorrhage. The presence of AβOs in the pre-plaque period of APP tg mice and in other transgenic models that never develop Aβ plaques has been proved and might explain the pathological alterations in the absence of the fibrillar Aβ formations. Although other hypotheses have been put forward to explain the neuronal dysfunction independent of the plaques, i.e., imbalance of amyloid-β protein precursor (AβPP) metabolism [63], or intraneuronal accumulation of AβPP [64], a specific approach depleting AβOs affected the neuronal dysfunction observed in plaque-free animals [65].
In humans, the diffuse presence of AβOs may possibly precede or follow the formation of plaques; the variable production of toxic species and the neuronal vulnerability can differentiate the dynamic of the pathological process and the functional consequences. The recent distinction between the toxicity of oligomers according to their aggregation size and the bidirectional connections between the large and small oligomers confirmed the complexity and the non-linear trend of AβOs formation [66].
We recently developed a simple method to analyze the effect of AβO in vivo by direct intracerebroventricular (ICV) injection in wild type mice of a low-concentration solution of AβOs (1μM) followed by a behavioral test, novel object recognition test (NORT), histological and biochemical analysis [4, 67]. We realize that this approach can cover only a few aspects of AD complexity, but our purpose was to dissect the biological effects of AβOs without any confounding factors. We took particular care about two aspects: the chemicophysical characteristics of the AβOs and the behavioral test. As our own experience and numerous studies indicate, the preparation of AβO as a tricky step that needs to be efficiently controlled to ensure the quality of the product and keep their chemicophysical features constant over time.
We used the synthesis of the water-soluble depsi-Aβ1-42 peptide, from which the native sequence is easily obtained in aggregate-free conditions. In basic conditions, the switching of the depsi-peptide means its aggregation can be triggered only when required [67]. AFM analysis of the AβO solutions is routinely done to confirm the structural characteristics of the species prepared. NORT is a behavioral test with some undoubted advantages: the procedure is relatively easy, the results can be recorded, and it is based on spontaneous behavior without any training. We have developed the test within the home cage to reduce any interference from anxiety induced by placement in a new space. The animals’ motor behavior was constantly monitored.
The memory impairment induced by the AβOs was specific, and Aβ fibers and monomers were not active, were reversible, and involved glial activation in a complex interaction with neurons. We were able to clarify the potential role of prion protein (PrP) as a ligand of AβOs with no direct functional consequences. Although we confirmed the previous reports of the high-affinity binding of AβOs to PrP, the application of AβOs in PrP/KO mice showed the same memory impairment as in the wild type, arguing against a functional consequence of PrP-AβO interaction [4]. The role of this interaction is still debated [68, 69]. However, our paradigm is adequate to determine the biological consequences of AβO exposure in vivo.
INFLAMMATION IN AD
The concept of inflammation in AD and other neurodegenerative disorders describes a particular biological process different from the classical inflammation outside the CNS; although the factors involved are often the same, the timing and the activities are quite distinct [70, 71].
Inflammation within the brain is a typical double-edged sword: it involves the positive activation of surveillance with phagocytic activity of both microglia and astrocytes to eliminate the debris and the pathogenic elements including protein aggregates, while on the other hand, the glial activation induces the production of factors with harmful consequences for the neuronal system. The neuroinflammation in AD is persistent with no resolution, a sort of chronic reaction of the innate immune system that causes neuronal alterations. The immune response is terminated with removal of the stimulating pathogen, and the resolution is an active process regulated by specialized pro-resolving mediators [72, 73]. This mechanism seems to be altered in AD and there are fewer specialized pro-resolving mediators in autoptic samples [74, 75]. This defect may also possibly be associated with the continued presence of Aβ aggregates, which aliment the chronic inflammation [76].
In the last few years, the role of inflammation in AD pathogenesis has gained importance with the accumulation of experimental evidence that immune factors can interfere in the early phase of the disease [77, 78]. As mentioned in the introduction, these results are now backed by genetic findings that indicate the immune system, together with lipid metabolism and vesicle traffic, are the main biological pathways where genetic alterations are associated with AD [79]. Not only the polymorphisms TREM2, CD33, or CR1 but several other missense mutations on genes encoding for immune factors have been identified as influencing the risk of developing AD.
The initial condition that indicated the glial reaction as merely a consequence of the neurodegeneration and Aβ deposits has progressively evolved. The activation of glial cells with the production of cytokines, radicals, and other immune factors, not necessarily detrimental, shifts from a secondary phenomenon to become an essential aspect of the pathological condition. Microglia as well as astroglia can model synaptic pruning and function, and in several experimental conditions it has been proven that the loss of glial cells or their alteration clearly influence neuronal function not only during development, but also in adults [18, 80–82]. This does not mean that AD and the other neurodegenerative diseases can be thought of as inflammatory disorders, but we have to consider that the non-neuronal components are potentially able to induce synaptic loss or damage in the absence of amyloid plaques or neurodegeneration [83], confirming the multifactor characteristics of AD.
In this complex scenario, peripheral conditions like diabetes, obesity, or metabolic syndrome can substantially contribute to creating an inflammatory milieu that facilitates the detrimental effects on neuronal function [84, 85]. Clinical studies have suggested that patients with AD who experienced an infection have accelerated cognitive decline, positively related with peripheral levels of TNFα [86]. Patients suffering from chronic periodontitis are also at higher risk of AD. Plasma concentrations of TNFα and antibodies to periodontal bacteria are higher in patients with AD than in non-demented control subjects and are independently associated with AD [87–89]. This sustained release of proinflammatory cytokines may affect the brain in much the same way as systemic inflammation. The gut microbiota has also been indicated as a possible source of elements that can influence—once again from the inflammatory point of view—AD pathogenesis [90].
TREM2 AND AD
Since genetic evidence become available on the rare mutations of genes encoding TREM2 associated with AD [22], numerous investigations have focused on the biological mechanism responsible for this relationship [91]. TREM2 is expressed in several myeloid lineages but in the CNS, the expression of the receptors is limited to microglial cells [92, 93]. This feature has been considered particularly attractive from the biological point of view because, for the first time, a non-neuronal component is directly involved in AD pathogenesis.
In general, TREMs are a signal relevant to the clearance of cellular debris and pathogens, with no excessive inflammation and tissue destruction. TREM2 is a glycoprotein of 230 amino acids, belonging to the immunoglobulin (Ig) superfamily. On the functional level, TREM2 inhibits cytokine and chemokine expression, acts as a negative regulator of toll-like receptor (TLRs)-mediated response, and promotes microglial phagocytosis [94]. Apparently TREM2 can drive the microglial state from the inflammatory (M1) to anti-inflammatory condition (M2); TREM2 overexpression induces the expression of all the immune markers that characterize the M2 phenotype like ARG1, IL-10, and IL4 with reparative meaning [95, 96]. Microglial knock-down for TREM2 exposed to apoptotic neurons increased the expression of pro-inflammatory cytokines, but reduced phagocytic activity [99]. Since apoptotic neurons express TREM2-specific ligands, the absence of TREM2 production by microglia would result in the reduction of its phagocytic activity. Mazaheri et al. [97] used a trascriptomic approach to show that the differently expressed messenger RNAs in wild type and TREM2–/– cells had impairment of the genes involved in chemotaxis, migration, and motility. Their deficit in chemotaxis was confirmed with functional analysis and the defect was rescued with the re-expression of TREM2.
In AD, transgenic mice the expression of TREM2 in the brain was higher than in wild type mice and the action on its expression can influence the AD phenotype [98]. The knock-down of TREM2 or the reduction of its expression increased cerebral Aβ deposition, synaptic loss, and cognitive decline in various AD models [99, 100]. While overexpression of TREM2 resulted in a reduction of AD neuropathology and improved behavior. Only in one specific model (APP/PS1-21) were there positive consequences induced by the TREM2 knock-down [101].
After the initial missense mutation (R47H) identified as a risk factor in AD, a second polymorphism (R62H) was associated with the disease [102–103]; TREM2 variants confer a similar risk for AD as one copy of ApoE ɛ4 [104]. Apparently the R47H mutation is associated with a reduction of the function of TREM2. The partial deletion of TREM2 in APP23 mice did not affect the number of plaques but reduced the number of microglial cells surrounding the amyloid deposits. Thus the mutation might alter the chemotactic activity of microglial cells toward senile plaques [105]. In addition, these variants might abolish the capacity of microglia to recognize and/or bind specific ligands. New functional data support this view, demonstrating that TREM2 variants may have different expression and either in boost or reduce its ligand binding ability on microglia [106]. Investigation in autoptic material revealed a low level of expression of TREM2 in healthy control brain and a clear increase in the microglia surrounding the senile plaques in AD brains [107]. However, the neuropathological results at the late stages of disease gave limited information on the role of TREM2, which at a certain point may lose its function, depriving microglia of its regulatory surveillance.
In AD-related TREM2, the surface membrane expression of receptors was the same as in control or other pathologies, but the signal has less efficacy [108]. The mutations can alter the folding kinetics, which might imply slower replenishment at the cell surface or the inability to bind properly to their signaling sustainers. Specific investigations have confirmed a small conformational change in the R47H TREM2 mutation but no changes in the R62H one. Investigations on TREM2 binding capacity to its ligands [104] confirmed that the presence of R62H and R47H mutations affected ligand engagement, probably because of a defect in ligand recognition that consequently impairs microglial activity.
In a recent paper, Ulland et al. [109] reported that microglia in AD patients carrying TREM2 risk variants and in TREM2-deficient mice crossed with 5xFAD mice had abundant autophagic vesicles, metabolic alteration, and energy failure, reflecting defective activation of mTOR signaling. Activation of the alternative pathway and signaling with cyclocreatine restored the energy metabolism, reduced the autophagy and improved the viability of TREM2–/– macrophages. The treatment of TREM2–/– 5XFAD in vivo by supplementation with cyclocreatine in drinking water restored mTOR signaling and the energy metabolism and reduced the autophagic markers in microglia. Cyclocreatine, while not affecting the total plaque number, significantly reduced plaque-associated neurite dystrophy, compared to untreated TREM2–/– 5XFAD mice, to the levels observed in 5XFAD mice. The authors suggest that these data indicate that cyclocreatine improves microglial metabolism and boosts the protective response to Aβ plaques in TREM2-deficient 5XFAD mice. The functional consequences of the beneficial effects observed on histological examination were not analyzed, so we do not know how cyclocreatine affected the neuronal dysfunction. Furthermore, the complete absence of TREM2 expression and the very aggressive AD model (5XFAD), are extreme experimental conditions, hardly to be compared to AD conditions. However, the data do indicate a precise pathway to improve the neuroprotective activity of microglia.
Another specific target in neurodegenerative conditions was proposed by Keren-Shaul et al. [110], who identified a new type of microglial cell closely involved in neurodegeneration (DAM) with a specific panel of receptor expression. The activity of this subpopulation was beneficial and its activation was partially TREM2-dependent. Apparently DAM is needed to mitigate the disease by supporting phagocytosis but it occurs at relatively late stages of the disease [111].
CR1 AND AD
Together with TREM2, recent large genome-wide association studies have identified single nucleotide polymorphisms in the C3b/C4b receptor (CR1) [105, 112]. CR1 is a transmembrane protein expressed by astrocytes involved in phagocytosis enhancement by C3b-, C4b-, C1q-, mannose-binding lectin-, and ficolins-opsonized particles. The complement cascades driven by C1 and C3 are both fundamental in brain development and synaptic pruning [113–115], but their overactivation might be detrimental. Recently it has been shown how excessive microglial activation can culminate in significant loss of synapses. C1q protein steeply accumulates at the synapses in normal mouse and human aging brains [116] and is highly upregulated in most or all neurodegenerative diseases, including AD [117] and dementia [118], where complement cascade components have emerged as significant enhancers of synapse vulnerability. Synapse loss was evident in these mice at the ages investigated but prevented, together with a decrease in the phagocytic activity of microglia, by inhibiting C1q, C3, and the complement receptor CR3.
An essential contribution has also been proposed of astrocyte phagocytosis in this mechanism dependent on the ApoE genotype [119]. According to these findings, ApoE isoforms might control the rate at which C1q protein accumulates in the aging brain. ApoE ɛ2, a protective allele against AD, enhances the phagocytic capacity of astrocytes. The decrease in C1q accumulation in aged knock-in expressing ApoE ɛ2 animals supports the hypothesis that ApoE ɛ2 helps keep the synaptic environment clean from senescent synapses and aberrant immune reactions. In contrast, ApoE ɛ4 reduces the overall phagocytic capacity of astrocytes, leading to faster accumulation of senescent synapses and their debris. Similarly, ApoE isoforms may strongly control the rate at which astrocytes and microglia help clear amyloid plaque accumulation [120]. The same authors noted that C1q accumulation at synapses is not enough to trigger their degeneration because another signal is required to activate the complement cascade, culminating in removal of the complement-coated synapses by microglia [121]. The presence of AβOs is sufficient to trigger this cascade. Accumulation of senescent synapses and their debris in the aged brain may induce reactive gliosis and neuroinflammation, with eventual synapse loss—an early pathophysiological aspect of AD. This was confirmed in AβO-treated mice, showing reductions in synaptic markers, though this was no longer detectable when AβOs were injected in C1qa knockout mice, suggesting cooperation between C1q and AβOs to trigger synapse loss via CR3 [121]
INFLAMMATION AND MEMORY
Using our acute experimental model, we showed the early involvement of inflammation in the memory impairment induced by ICV application of AβOs from depsi-peptide. A single ICV injection of AβOs (1μM) in wild type mice before the initial exposure to the objects in the NORT, nullified the animals’ ability to distinguish the familial from the novel object. Similar results were obtained with Aβ1-40 or Aβ1-42 oligomers [4] and, with a distinct mechanism, oligomers from α-synuclein, the essential component of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies, impaired the memory [24]. A toll-like receptor 4 (TLR-4) antagonist completely rescued the AβOs effect and in TLR-4-KO mice AβOs lost their ability to induce memory impairment [122].
In a similar model, the depressive effect associated with the inflammatory reaction induced by AβOs was also abolished in TLR-4-KO mice [123]. The involvement of TLR-4 indicated the possible involvement of inflammation in this acute model confirmed by the protective effect of indomethacin, a classical NSAID: injected intraperitoneally the drug antagonized the amnesic effect of AβOs. Within 24 hours, after the AβO injection transient gliosis was observed with a peak effect at 4–8 hours, quantified by western blot analysis and histopathology of GFAP and Iba-1, markers of astroglia and microglia, respectively, in hippocampus and cortex. With a parallel time course, AβOs also induced the expression of a panel of cytokines, including IL1β, TNFα, IL-6, and IL-10. The essential role of neuroinflammation in this experimental model was confirmed by the antagonism of the memory impairment and gliosis by IL-1RA [122].
The information provided by this relatively simple approach is the intimate relationship between neuronal dysfunction and neuroinflammation, both activated by AβOs through TLR-4, indicating the need for treatments that interfere with different aspects of the pathological mechanism (Fig. 1). These results are in line with various other findings [123, 124]. An interesting example is the study by Xu et al. [125] where the AβOs from human AβPP-expressing cultured cells or purified from AD brain tissue, applied ICV in wild type mice, induced microglial activation in the hippocampus determined by morphological analysis and by measuring mRNA changes in multiple inflammatory genes. The environmental enrichment, a well-documented approach to induce cognitive and neuronal activation, silenced the innate immune response activated by AβOs. Similar results were obtained by Beauquis et al. [126]. In this case, environmental enrichment prevented astroglial pathological changes in the hippocampus of plaque-free AβPP transgenic mice. Here again it was hard to distinguish the glial activation from the neuronal dysfunction when the brain was exposed to AβOs. PET analysis that combined the identification of Aβ deposits with the microglial activation using 11C-(R)-PK11195 binding in MCI subjects confirmed a positive relation between amyloid load and levels of microglial activation. 11C-(R)-PK11195 binding by PET revealed increased inflammation in a majority (85%) of amyloid-positive MCI cases, its cortical distribution overlapping that of amyloid deposition [79].
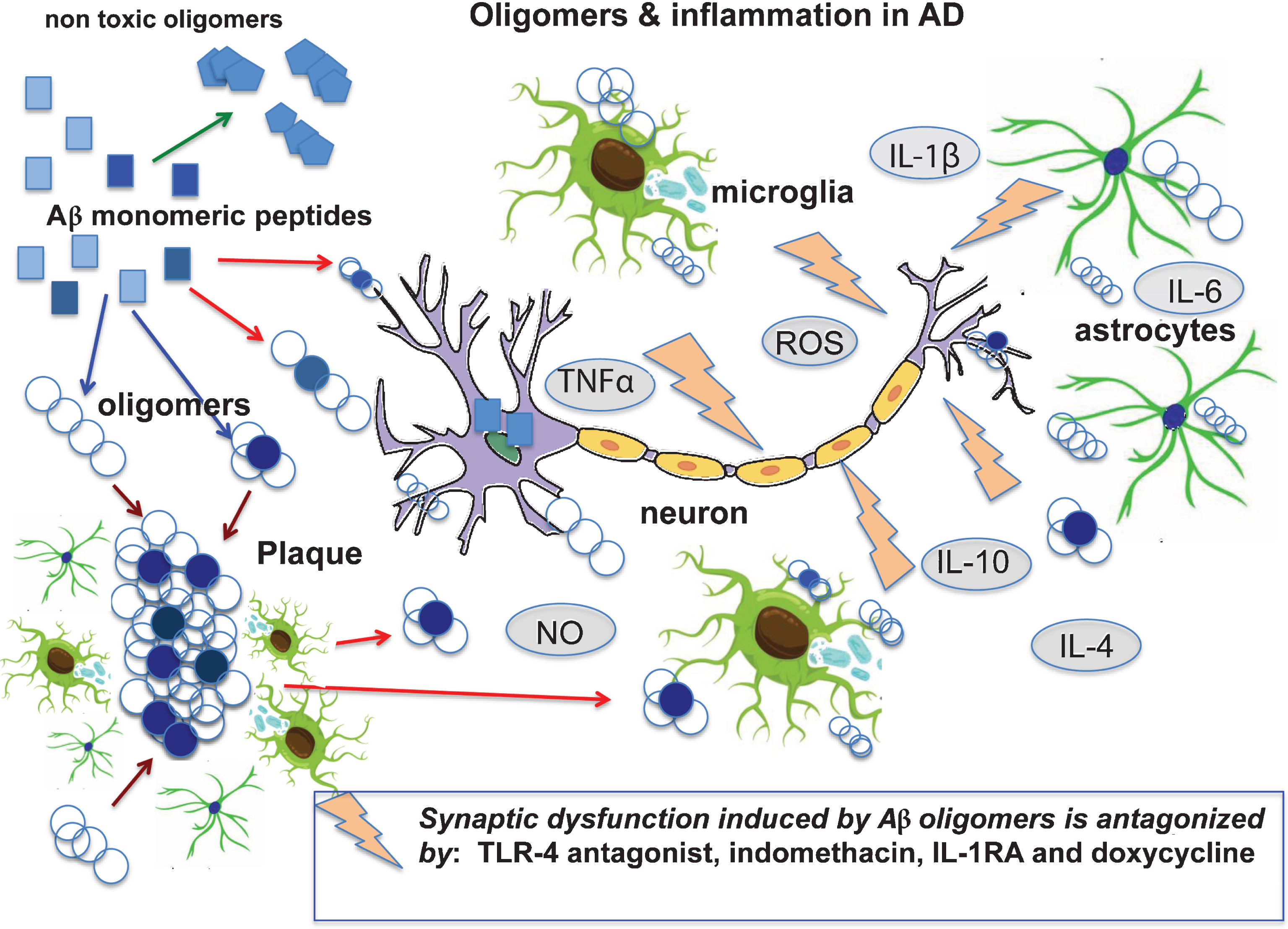
The cartoon illustrates several aspects of AD pathogenesis starting from the formation of Aβ oligomers, a variety of entities with different sizes, peptide combinations and conformations (in light blue and blue); the seeding passage with conformation change (from square to circle) may also occur intracellularly. The oligomers in dynamic equilibrium with senile plaques can act directly at the neuronal level or induce neuronal dysfunction through glial activation by the production of cytokines radicals and other factors. In our experimental studies [133], we found that anti-inflammatory drugs, doxycyline and a TLR-4 antagonist antagonized the cognitive decline induced by direct intracerebral injection of Aβ oligomers.
THERAPEUTIC OPPORTUNITIES
Despite the accumulation of results in favor of the toxic activity of oligomers and the robust scientific support for the involvement of inflammation in AD pathogenesis, we are still waiting for a disease-modifying drug based on interference with Aβ aggregation and/or inflammation. According to the Aβ cascade, most of the therapeutic approaches in AD has been aimed at amyloid aggregates or AβPP metabolism, and the develop of antibodies against Aβ in particular has produced various anti-dementia candidates. Active anti-Aβ immunization was proposed at the end of 1990s, on the basis of clear evidence in AD experimental models [127] and the vaccination was clinically tested a few years later. Although the first trial was stopped due to intolerable side effects [128], passive or active immunization is still considered one of the most interesting approaches for AD [129, 130]. This concept is supported by the biological efficacy in reducing the Aβ burden, proven in experimental models and confirmed in human neuropathological samples [131, 132]. A recent phase II investigation with aducanumab, an antibody targeting aggregated oligomers and fibers, but not monomer Aβ, gave promising results [133]. This antibody, the last of a long list that have failed to show efficacy in AD, was derived from a lymphocyte library collected from healthy elderly subjects with no signs of cognitive impairment and cognitively impaired elderly people with unusually slow cognitive decline. This human origin might boost the effectiveness and make the difference in favor of this treatment. Currently, aducanumab is in phase III clinical trials that are expected to be completed in 2020. The therapeutic possibilities deriving from the control of inflammation in AD are supported by several epidemiological and observational studies reporting a reduction of the risk of AD in subjects exposed to NSAID in a preclinical period [134]. However, when the investigation moved on from observational studies to a curative approach, virtually all the randomized controlled trials failed to show any benefit effect with NSAIDs [135, 136]. The availability of numerous anti-inflammatory drugs, mostly various chemical species of COX inhibitors, has favored the design of many studies. When the efficacy was formally investigated in a randomized clinical trial, a few studies founds the anti-inflammatory treatment induced a modest improvement. A positive tendency in favor of the treatment has been obtained with indomethacin, but the small size of the populations studied did not permit any firm conclusion [137, 138]. The side effects of indomethacin do not recommend its prolonged or widespread use, and after a quarter of a century since the initial pilot study reported encouraging results in AD subjects treated with this drug, we still have no final opinion on its efficacy.
More specific reviews focused on therapeutic studies in AD [139, 140] have noted that beside the complexity of the disease, there are several potential explanations for these unsatisfactory results: identification of the wrong target; selection of the wrong drug; inappropriate design of the trial or treatment schedules; and, finally, insufficient selection of the subjects. Other aspects referring to the natural history of the disease might suggest starting the treatment earlier, in the preclinical phase. In this case, biological markers are needed, to follow the drug’s effect and the progress of the disease. In addition, the complex AD pathogenesis will presumably need multitarget therapy [141].
These concepts acquire particular significance in the light of the itemized considerations in this review, since both the accumulation of AβOs and the neuroinflammation are early events in AD pathological process. It is reasonable to assume that at the very outset, the primus movens is the presence of oligomers, released from senile plaques or formed independently. The appearance of AβOs has different biological implications, with different lag periods for each individual the inflammatory cascade is also activated (Fig. 1). The constant presence of AβOs is associated with uncontrolled inflammation, so immune factors and AβOs may well combine deleterious effects on the neuronal system with an external contribution from the periphery. At this point, a single treatment to eliminate the AβOs or an anti-inflammatory intervention would be unlikely to give curative results. Thus, to change the trajectory of the disease early intervention on both flanks is necessary. The contribution of inflammation should be monitored by measuring biological markers in the blood or—though less convenient—in the CSF, in combination with the profiles of genes encoding inflammatory factors [142, 143] and analysis of the microbioma. Furthermore, as reviewed by Knezevic & Mizrahi [144], PET analysis offers the possibility of monitoring microglial activation in the brain of AD and MCI subjects, with a series of ligands.
Several inflammatory markers in plasma and CSF are elevated in AD and MCI subjects compared to age-matched controls [145–147] and in some cases the level of changes correlated with the severity of the disease, including sTREM2 in AD liquor [148]. An interesting approach was to monitor inflammatory parameters in CSF following the treatment in humans given CHF5074, an ibuprofen derivative with microglial modulation activity [149]; there was a constant reduction, possibly indicating drug efficacy. However, the correlation between the inflammatory factors in plasma and the disease progression, determined by clinical markers, is only weak, when it occurs, because of the biological variability of the parameters considered. This variability more than an inevitable subjective oscillation, may describe real differences reflecting the heterogeneity of the disease, especially in its very early or prodromal phase [150]: under the broad umbrella of AD a variety of pathogenic pathways coexist.
The elements of the pathological scenario may have different weight, and inflammation must certainly be considered in relation to the time and the characteristics of the subjects. According to the precision medicine principle, the aim is to identify a common profile in a selected group of subjects, considering all the available parameters and the possibility of distinguishing different conditions. The application of precision medicine principles using the inflammatory profile to orient a personalized approach to AD was proposed by Wilcock’s group [151,156, 151,156]. They indicate M1 and M2 as broad inflammatory profiles, based on the measure of plasmatic proteins like ILRa, CAM1, haptoglobin, and fibrinogen, that can distinguish the AD population in the early phase from the immune point of view [153]. This polarization could be useful to identify subjects requiring a treatment to promote the process of restoration and repair associated with the M2 phenotype [153].
However, correct manipulation of the immune system will not be sufficient to interfere with AD progression, and a multifactorial approach will be necessary, focused on several targets including inflammation, amyloid aggregation and probably neuroprotection, to exert an effective anti-dementia action. The same molecules could share different activities. With intracerebral injection of AβOs and AβPP transgenic animals, we demonstrated that the antibiotic doxycycline had not only a well-defined anti-amyloidogenic profile [154, 155] but also anti-inflammatory activity [156, 157]. Thus, doxycycline might be an interesting candidate for testing in the early phase of AD [158]. The drug has a clear safety profile and is currently under investigation in a preventive trial in subjects at genetic risk of fatal insomnia [159], a rare prion disease, included in the category of oligomeropathies [160].
CONCLUSIONS
The biological complexity of AD emerges from the scientific information accumulated in the last three decades, progress in imaging analysis, and general improvements in the diagnostic tools, which have extended our knowledge about the clinical progression of the disease, though less clearly in the preclinical phase. The aspects covered in this review point to the presence of AβOs and activation of the inflammatory cascade as early steps in the pathogenic scenario of AD, which involves several other important features, like tau phosphorylation, mitochondrial damage, and peripheral components. The control of inflammation and of amyloid aggregation is not a new target for AD therapies; large numbers of studies have been designed to test the effects of anti-amyloid or anti-inflammatory drugs but, until now, with scant results. To improve this approach, we have to move essentially in three directions: 1) improve the schedule and timing of treatment, possibly in a preclinical phase; 2) identify appropriate inflammatory markers that together with the genotype information can individualize treatment according to precision medicine principles; and 3) employ specific tools but also contemplate a multifactorial approach coherent with the distinct alterations seen in the disease.
Some of these aims would in principle be relatively simple to reach, while others call for careful investigation to identify appropriate tools. What is absolutely necessary is close collaboration between all the expertise at various levels—academic, pharmaceutical, and regulatory—to develop innovative trials where the quality of the patients selected is given priority, even at the expense of the numbers.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0819r2).