
Abstract
Subcortical small-vessel disease (SSVD) is a disorder well characterized from the clinical, imaging, and neuropathological viewpoints. SSVD is considered the most prevalent ischemic brain disorder, increasing in frequency with age. Vascular risk factors include hypertension, diabetes, hyperlipidemia, elevated homocysteine, and obstructive sleep apnea. Ischemic white matter lesions are the hallmark of SSVD; other pathological lesions include arteriolosclerosis, dilatation of perivascular spaces, venous collagenosis, cerebral amyloid angiopathy, microbleeds, microinfarcts, lacunes, and large infarcts. The pathogenesis of SSVD is incompletely understood but includes endothelial changes and blood-brain barrier alterations involving metalloproteinases, vascular endothelial growth factors, angiotensin II, mindin/spondin, and the mammalian target of rapamycin pathway. Metabolic and genetic conditions may also play a role but hitherto there are few conclusive studies. Clinical diagnosis of SSVD includes early executive dysfunction manifested by impaired capacity to use complex information, to formulate strategies, and to exercise self-control. In comparison with Alzheimer’s disease (AD), patients with SSVD show less pronounced episodic memory deficits. Brain imaging has advanced substantially the diagnostic tools for SSVD. With the exception of cortical microinfarcts, all other lesions are well visualized with MRI. Diagnostic biomarkers that separate AD from SSVD include reduction of cerebrospinal fluid amyloid-β (Aβ)42 and of the ratio Aβ42/Aβ40 often with increased total tau levels. However, better markers of small-vessel function of intracerebral blood vessels are needed. The treatment of SSVD remains unsatisfactory other than control of vascular risk factors. There is an urgent need of finding targets to slow down and potentially halt the progression of this prevalent, but often unrecognized, disorder.
Keywords
INTRODUCTION AND CLASSIFICATION
Since the 17th century, stroke has been recognized as a cause of age-associated cognitive impairment. However, until early in the 20th century, the prevailing concept of age-associated cognitive impairment causation was the impairment of cerebral blood flow (CBF) resulting from partial blockage of the vessels that supply the brain; thus, arteriosclerotic dementia resulting in global brain hypoperfusion was considered the most common form of major cognitive disorder in the elderly [1]. Early neuropathologists recognized subcortical ischemic white matter injury and lacunes as typical lesions resulting from small-vessel disease [2]. During the 1970s, amyloid-β (Aβ) plaques and neurofibrillary tangles were identified as critical components of the neurodegenerative process occurring in demented elderly patients [3]. Alzheimer’s disease (AD) became recognized as the most common form of major cognitive disorder and the notion of chronic brain ischemia as an explanation of progressive cognitive impairment was abandoned. The claim that vascular disease could lead to cognitive disorders not by means of CBF-related energy deficiency but by repeated stroke episodes resulting in cerebral tissue lesions led to the concept of multi-infarct dementia (MID). Although limited by scarce neuropathological support, the diagnosis of MID grew popular and was applied to a larger group of patients than originally proposed. The demonstration that vascular-related white matter damage (Binswanger disease) [4] contributes to cognitive impairment in the elderly began questioning multiple infarcts as the most important cause of vascular cognitive impairment (VCI) [5]. Aging of the population, along with changes in the panorama of cerebrovascular disease (CVD) in terms of reduced stroke mortality [6], have led to reevaluation and renewal of concepts in this area. Moreover, the role of CVD—particularly small-vessel disease—in the clinical expression and pathogenesis of AD has been increasingly recognized [7–9].
AD and VCI are the most common causes of cognitive impairment in the elderly, accounting together for more than 70–75% of cases [7]. They also share common risk factors and mechanisms including atherosclerosis, diabetes, and amyloid angiopathy [10–13]. Vascular comorbidity may be present in 30–60% of AD patients [11, 12], while AD pathology may be present in 40–80% of VCI patients [11, 12]. Indeed, the coexistence of the two disorders, termed mixed dementia (MXD), may be more common than “pure” AD or “pure” VCI [14, 15]; moreover, microscopic cortical infarcts, which are not visualized in neuroimaging, may contribute up to 1/5 to 1/3 in the variability of severity of cognitive impairment [16].
Neuropathology usually provides the final and definite diagnosis in clinical neurology. Therefore, there is unanimous accord that the clinical classification of cerebral small-vessel diseases should be based on the distinct neuropathological lesions [13, 18]. Given the fact that VCI may be the result of a number of cerebrovascular pathologies (Fig. 1) resulting from numerous etiopathogenic causes (Table 1), the occurrence of several clinical forms of VCI (Table 2) is therefore understandable. Among the sporadic forms of VCI, particular attention over the last decades has been paid to post-stroke-associated cognitive impairment or post-stroke dementia (PSD); and, more recently to VCI associated with subcortical small-vessel disease (SSVD), a condition that we last reviewed over a decade ago [19]. Although small-vessel disease may cause cortical atrophy (cortical granular degeneration), and secondary localized atrophy from subcortical lacunar strokes, from the clinical viewpoint, however, most cases of cortical atrophy in elderly patients with cognitive impairment are the result of neurodegenerative conditions, not vascular disease. Thus, when approaching such patients with cognitive loss clinicians rely on the presence of “cortical” versus “subcortical” manifestations that allow a first diagnostic impression. These clinical features are confirmed subsequently by neuropsychological tests, brain imaging, and eventually by neuropathology. Therefore, we decided to maintain the time-honored name “subcortical” in the overall approach to this topic.
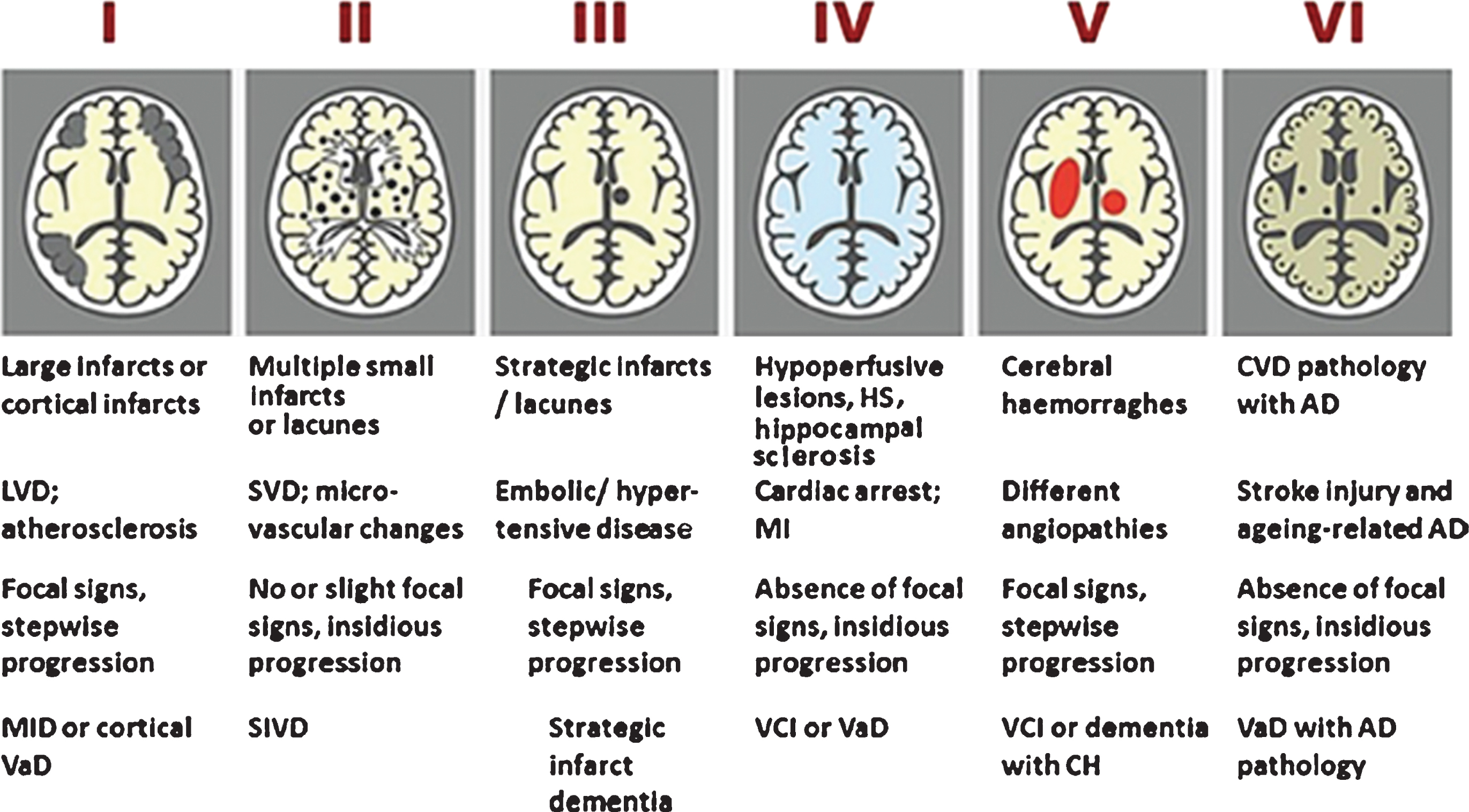
Newcastle categorization in six subtypes of different cerebrovascular pathologies associated with VCI. Post-stroke survivors are included in subtypes I–III. Cases with extensive WM disease in the absence of significant other features are included under SVD. Subtype I may result from large vessel occlusion (athero-thromboembolism), artery-to-artery embolism or cardioembolism. Subtype II usually involves arteriosclerosis, lipohyalinosis and hypertensive, arteriosclerotic, amyloid or collagen angiopathy. Subtypes I, II and V may result from aneurysms, arterial dissections, arteriovenous malformations and various forms of arteritis (vasculitis). AD, Alzheimer’s disease; CH, cerebral haemorrhage; CVD, cerebrovascular disease; MI, myocardial infarction; MID, multi-infarct dementia; LVD, large vessel disease; SIVD, subcortical ischemic vascular dementia; SVD, small vessel disease; VCI, vascular cognitive impairment; VaD, vascular dementia. From: Kalaria RN, 2016 [13].
Pathologic basis of vascular cognitive disorders*
*From Vascog [21].
VICCCS proposed definitions of major VCI subtypes*
*From VICCCS [22].
The numerous diagnostic criteria proposed for the diagnosis of cognitive disorders resulting from vascular disease reflect the difficulties in the field [20]. The most recently updated definitions and criteria have been proposed by the International Society of Vascular Behavioural and Cognitive Disorders (VASCOG) [21] and by the Vascular Impairment of Cognition Classification Consensus Study (VICCCS) group [22, 23].
Based on ample evidence, there is current consensus on the notion that SSVD in the brain is probably the commonest and most prevalent vascular neurological lesion [24], occurring as part of a systemic dysfunction of arteriolar perfusion affecting mainly heavily perfused tissues such as brain, retina and kidneys [25, 26]. SSVD is ubiquitous in elderly patients with cognitive decline. SSVD presents with a more insidious course than PSD and has been associated with prefrontal symptoms affecting cognition, mood, behavior, gait, and bladder control. Moreover, SSVD has been associated with traditional vascular risk factors (VRFs) such as aging, hypertension, hyperlipidemia, diabetes, smoking, peripheral vascular and heart disease, and with common but frequently overlooked VRFs such as obstructive sleep apnea [27] and hyperhomocysteinemia resulting from dietary deficiencies of vitamin B12 and folate [28], as well as from mutations of the methylenetetrahydrofolate reductase (MTHFR) gene [29]. Also, SSVD provided unexpected links as the terrain that precedes and favors the development of neurodegeneration in AD and other cognitive disorders [7–15].
Finally, the pendulum is swinging back and new developments in brain imaging, in particular arterial spin-labeling (ASL) imaging on magnetic resonance imaging (MRI) – a method that allows noninvasive quantification of CBF [30–32]; as well as novel cerebrospinal fluid (CSF) biomarkers [33, 34], have revitalized the studies on hypoperfusion leading to cortical hypometabolism in some forms of cognitive impairment, by mechanisms different from carotid and intracranial large-vessel stenoses.
PATHOLOGY AND MECHANISM
The pathology of SSVD encompasses tissue injury to the cerebral white matter and to the vessels that supply blood to this tissue [15, 17–19]. One of the most important achievements in the past decade was the implementation of the Vascular Cognitive Impairment Neuropathology Guidelines (VCING), a collaborative study performed in the United Kingdom [35]. The study by Barker et al. [36] and some of the other studies referred to here were carried out on brain samples donated after death from elderly donors who were systematically followed during life with regard to their clinical and psychological state at each yearly visit at the Oxford Study to Investigate Memory and Ageing (OPTIMA). From a total brain archive of over 500 donations, this cohort included subjects who were found after death to have only SSVD and no more than Braak and Braak stage 3-tau pathology. We found that cognitive impairment only affected about a third of subjects, the rest had cognition within normal limits [37], using as instruments to assess cognition the Mini-Mental State Examination and the Cambridge Cognition Examination. The findings may have been different using the Montreal Cognitive Assessment test [38, 39] that has been found to be more sensitive to cognitive changes characteristic of SSVD and VCI; or the Brief Memory and Executive Test [40] for detecting cognitive impairment in small vessel disease.
Pathology
Diffuse damage to the white matter (ischemic leukoencephalopathy) is the most common pathology in SSVD [35]. It is usually most severe in frontal and occipital regions [35, 41–44] and the affected white matter exhibits loss of myelin and axons and a chronic inflammatory infiltrate (Fig. 2A, B). There is also evidence of ongoing axon damage in surviving axons, detected with immunocytochemistry for phosphorylated neurofilament precursor protein [36]. Evidence for axonal depletion in white matter lesions also comes from silver-stained preparations [41, 42], correlated with reduced fractional anisotropy in diffusion tensor imaging [43].
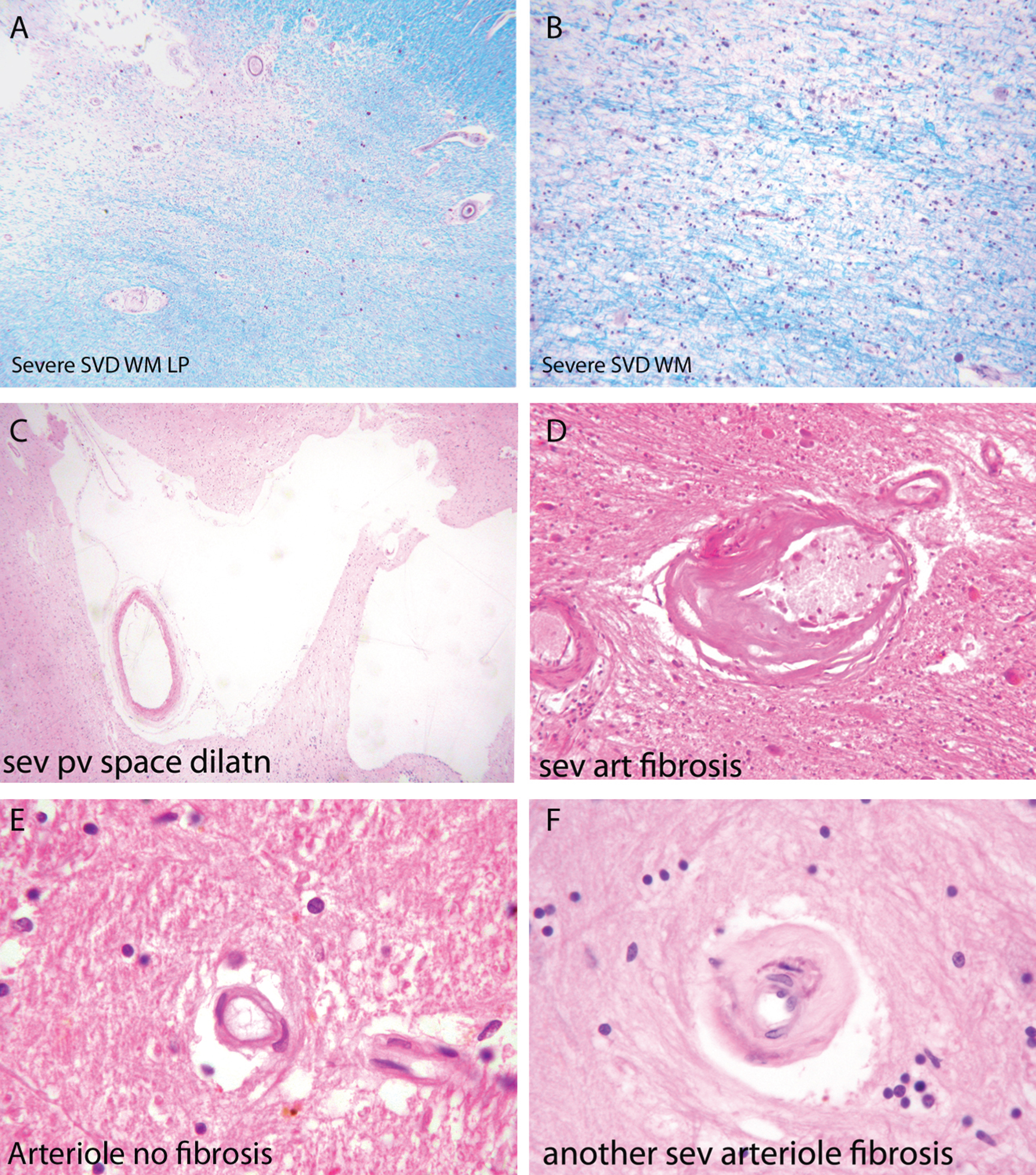
Components of the pathology due to SSVD. A, B) Low power (A) and higher power (B) views of a histological section from a case of SSVD. The section has been stained for myelin (blue) (Luxol fast blue/cresyl violet stain). There is diffuse pallor of staining and, at the top left corner of the section in (A), the tissue is necrotic. B) Damaged white matter at higher power. The nuclei (purple) are chiefly those of infiltrating macrophages. C) Greatly dilated perivascular space (hematoxylin and eosin stain). D) Small artery with a grossly thickened wall in which collagen has replaced smooth muscle (hematoxylin and eosin stain). E) Normal white matter arterioles in which the deeper pink cells are smooth muscle cells. F) Severely fibrotic and stenosed arteriole from a case of SSVD (E and F, hematoxylin and eosin).
Associated with this diffuse white matter damage, Skrobot et al. [35] found six other pathological features predictive of cognitive impairment: 1) arteriolosclerosis, 2) perivascular space dilatation, 3) leptomeningeal cerebral amyloid angiopathy (CAA), 4) microinfarcts, 5) lacunar infarcts; and 6) large infarcts.
Small arteries and arterioles supplying blood to cerebral white matter in SSVD have thickened walls and basement membranes and diminished luminal spaces (Fig. 2D, F) [44–46]. A thrombus within these vessels is rare but arterioles are sometimes tortuous [47–49]. Smooth muscle in the walls of the vessels is characteristically replaced by collagen, and perivascular spaces are commonly enlarged [50] (Fig. 2C). Perivascular macrophages are frequently present and may contain hemosiderin, indicative of likely earlier small bleeds. A marker of endothelial activation, thrombomodulin, was found to be elevated in vessels affected by SSVD, a change that was interpreted as likely to be a protective response to SSVD rather than causative [47]. Similar changes to those in cerebral white matter are often seen in deep grey matter. Veins adjacent to the lateral ventricles often have thickened walls [48], but the significance of venous pathology in SSVD is only beginning to be systematically studied and related to cognitive state. The same is true of capillaries, although imaging techniques such as Dynamic Susceptibility Contrast (DSC)-MRI perfusion has shown alterations of cortical capillary transit time in AD, consistent with low tissue oxygen tension in white matter hyperintensities (WMH) areas [51].
It is evident that the blood supply to cerebral white matter in SSVD is compromised and there is ischemia and oxidative stress in affected regions [44, 52]. From imaging studies the process appears to progress slowly; the rate of WMH in T2 weighted MRI, indicative of damage due to SSVD, is associated with decline in verbal IQ [53] and executive function [54].
Mechanisms
Epidemiological studies indicate that SSVD is more common with longstanding hypertension in middle age [55–57] and in subjects with diabetes mellitus [58]. Hypertension may damage these small vessels by provoking sheer stresses since these vessels branch out from larger arteries without progressive decrease in size and are therefore liable to experience relatively high pressures. Alternatively, there may be damage inflicted by episodes of hypotension interspersed between episodes of hypertension in people with poorly controlled blood pressure [55]. In the material, we have studied from the OPTIMA cohort we found no relation of SSVD to severity of hypertension or diabetes, but instead we found it to be related to age [59]. Age also had an influence of some SSVD features in the study by Skrobot et al. [35]. Less attention has been paid in the literature of SSVD to the effect of age than of hypertension. This finding emphasizes an interesting similarity between SSVD and sporadic AD for which old age is overwhelmingly the most important risk factor [60, 61]. It is unclear how age may damage small cerebral arteries and arterioles but one change that may be important is stiffening of upstream, larger arteries, reflected in carotid-femoral pulse wave and increased pulse wave velocity which have been linked to WMH [62, 63]. One aspect of vascular smooth muscle that has been shown to increase in expression with age is vascular endothelial growth factor 2 [64]. However, this expression was not related to severity of SSVD in the brains of elderly subjects lacking AD pathology in the OPTIMA cohort [59].
Angiotensin II is a protein capable to damage aging cerebrovascular cells, particularly vascular smooth muscle cells; it shows increased activation with age and is linked to pro-inflammatory effects through activation of leucocytes, cell adhesion molecules, and inflammatory cytokines [65, 66]. It also leads to stimulation of NADPH oxidase, an important source of oxygen species production and downregulation of antioxidant defenses [67, 68].
Though speculative at this stage, it is interesting to note the results of informative parabiosis experiments in animals performed by Wyss-Coray and colleagues. They have found that by engineering a common blood supply between old and young mice they can detect a circulating factor in the blood of young mice that can reduce age-associated cardiac muscle changes in old mice [69]. Clearly, it would be of interest to know if cerebral arterial smooth muscle in elderly mice (and humans) is similarly affected. Another mechanism, which is also worth considering in the pathophysiology of SSVD, is the breakdown of the blood-brain barrier (BBB) [70]. This is common in old age and may at times be severe enough to enable a large molecular weight protein, fibrinogen, to enter the neighboring brain parenchyma from the blood. Bridges et al. [71] examined this feature in material from the OPTIMA cohort. The presence of fibrinogen in cerebral white matter of elderly subjects was readily demonstrable, but its amount was not related to the severity of damage to white matter. This does not exclude a potential role for fibrinogen and its breakdown product fibrin, in the development of white matter damage in SSVD because the damage is likely to have arisen earlier than in the pre-mortem period during which fibrinogen seen in tissue sections is likely to have leaked from blood vessels. It is worth referring to the apparent ability of fibrinogen to damage cerebral cortex in multiple sclerosis [72] and to white matter in EAE, the experimental model of multiple sclerosis [73]. Fibrinogen has also been linked to cortical damage in AD [74]. BBB damage in SSVD may be effected by activation of matrix metalloproteinases [18].
CAA is a factor associated with severity of SSVD and cognitive impairment attributable to SSVD. In a study specifically designed to study the effect of CAA on SSVD [75] we found a complex relationship of CAA to the severity of SSVD-related damage that varied depending on apolipoprotein E (APOE) genotype. There was a modest positive correlation between severity of CAA and severity of SSVD in those who were APOEɛ4-positive, no effect in those who were APOEɛ3-positive and a negative effect in those who were APOEɛ2-positive [75]. CAA has been shown to be associated with microinfarctions in the cerebral cortex. Since the arterioles in the cortex that are affected by CAA also supply subcortical white matter with blood it is plausible that it may promote ischemia in subcortical white matter; Skrobot et al. [35] found that it promoted SSVD in their study. The manner in which APOE genotype may exert this effect is not clear. Nonetheless, Skrobot et al. [35] did not find an effect of the APOE genotype in SSVD.
One further metabolic pathway that would be worth investigating with respect to the effect of age on cerebral arteries and arterioles is the mammalian target of rapamycin (mTOR) pathway [74, 76] which has been shown to have important control over lifespan in animals [77] and to be dysfunctional in AD [78, 79]. The extracellular matrix protein mindin/spondin2 is a protein involved in vascular smooth muscle regulation influenced by the mTOR pathway [80], as is the platelet-derived growth factor-induced control of vascular smooth muscle phenotype [81].
This neuropathology review summarized some of the factors that may contribute to SSVD in the elderly. To be in a position to help those with cognitive impairment due to SSVD, and to help others at risk to avoid it, we need to further clarify mechanisms of damage and find ways to intervene and to combat them.
GENETICS
In 1955, Ludo van Bogaert [82] reported two sisters with a familial form of Binswanger disease and provided the first neuropathological description of the clinical condition that eventually became known as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), the first monogenic form of pure SSVD [83]. Other variants of SSVD with a genetic basis were later reported [84, 85]. However, the mechanisms of contribution of risk genes to the development of sporadic SSVD are not fully known, due to the complexity of the pathogenesis caused by the interaction of genetic and environmental factors. The genetic contributions to the monogenic forms of small-vessel diseases are better known [86, 87] and will not be discussed here; these include CADASIL, Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, Fabry disease, autosomal dominant Retinal Vasculopathy with Cerebral Leukodystrophy, CAA, and collagen type IV alpha 1 chain-related cerebral small-vessel disease. Although some pathogenic mechanisms may be shared between the monogenic and sporadic forms, they are not identical. Therefore, there is still a need to better understand the genetics of SSVD.
Established diagnostic criteria for SSVD have been insufficiently used in genetic studies and the majority of gene-association studies have focused not on clinical diagnoses but on surrogate markers or disease traits, such as WMH, deep intracerebral hemorrhage and cerebral microbleeds, although these markers are not unique for SSVD. Throughout the years, the association of such surrogate markers and diagnoses with gene variants known as single nucleotide polymorphisms or gene loci has been performed in so-called candidate gene studies. A few of these have also aimed at confirming genetic findings with neuropathological studies, where an accurate diagnosis of SSVD is possible [87]. Many of the candidate gene studies have been performed in small patient groups or have given conflicting results when compared. Therefore, important meta-analyses of these previously reported candidate gene studies have been performed. Moreover, a few genome-wide association studies of VCI have been performed [88, 89] but failed to reproduce the findings from the candidate gene studies and meta-analyses. In these studies, rs290227 of the spleen tyrosine kinase (SYK) gene [89] and rs12007229, located on the X chromosome near the androgen receptor gene were associated with risk of VCI [88], but to our knowledge, no follow-up studies of these gene variants have been published to date.
The gene for apolipoprotein E (APOE), a multifunctional protein involved in lipid and cholesterol transport and Aβ clearance, is the best-known risk gene for AD [90] and CAA [91] where the ɛ4 allele increases the disease risk. Meta-analyses of previously published candidate gene studies of APOEɛ4 have shown significant association between ɛ4 allele carriers and increased risk of VCI [92–94]. Meta-analyses [94] have also indicated that the APOEɛ4 allele is associated with an increased risk of cerebral microbleeds [95] but not with WMH [96].
The MTHFR gene codes for an enzyme in the folate cycle, involved in homocysteine metabolism [29]. Although the gene variant C677T (rs1801133) of MTHFR has been shown to be a risk gene for stroke in meta-analyses, it has failed to show associated with WMH and intracerebral hemorrhage in similar studies [96]. In two meta-analyses the T allele of MTHFR was associated with VCI in Asian populations; but this association was not present in Caucasian and Indian populations [93, 94]. A validation study later showed an increased risk of VCI associated with the T allele of rs1801133 in Caucasians [92].
A few genes that have shown associations to VCI have later been confirmed in meta-analyses while some have failed to show associations with disease. The gene variant rs662 for paraoxonase 1 (PON1), a gene involved in homocysteine and lipoprotein metabolism, has failed to be associated with VCI in a published meta-analysis [93] or in a validation study [92]. However, the PON1 L55M (rs854560) variant was found to be associated with VCI, while no association was found with the variant –108C/T (rs705379) of the gene [94]. Variants of two genes related to inflammation, tumor necrosis factor (TNF)-α –850C/T (rs1799724) and transforming growth factor (TGF)-β1 + 29C/T (rs1800470) were found to be associated with risk of VCI in Caucasian and Asians, respectively [94], while the interleukin (IL) gene variants IL-1α –889C/T (rs1800587) and IL-6 –174G/C (rs1800795) showed no significant association in this study. Finally, no significant associations of VCI were found in a meta-analysis [93] and in a validation study [92] of variants of the genes for angiotensin converting enzyme (ACE; rs1799752), alpha-1-antichymotrypsin (ACT/SERPINA3; rs4934) and presenilin (PSEN-1; rs165932). Moreover, ACT A/T (rs4934), APOE promoter–427T/C (rs769446) and PSEN-1 (rs165932) were not associated with VCI in a meta-analysis [94]. The reasons for this could be the reduced number of studies and the limited number of patients included in the meta-analyses, preventing stratification by ethnicity.
The importance of determining exact genetic mechanisms involved in the pathogenesis of SSVD resides in the possibility of prevention and treatment, as recently illustrated with the use of an agonist Notch3 antibody that prevents mural cell loss and modifies plasma proteins associated with Notch3 signaling in an animal model of CADASIL [97]. Therefore, future genetic analyses of definite SSVD cases in large cohorts and subsequent meta-analyses are warranted.
SYMPTOMS PATTERN AND CLINICAL PRESENTATION
Although cognitive impairment in the elderly may affect the entire range of cognitive functions, there is usually an uneven distribution of deficits, i.e., some cognitive functions are more affected than others. The pattern of cognitive impairment reflects the nature of the disease and the distribution of pathological changes in the brain. From the clinical viewpoint, it is useful to simply consider two major types of cognitive syndromes. The first one is characterized by impaired memory, language problems, difficulty with practical tasks, and loss of the capacity to interpret sensory (mainly visual) impressions including recognition ability. The syndrome originates from dysfunction of posterior cortical association regions (posterior brain syndrome) and is typical of AD. The second syndrome results from executive dysfunction and is characterized by mental slowness and personality changes [98]. The executive control function ensures that mental and physical activities achieve the intended goals. It coordinates cognitive functions such as planning, attention, working memory, abstraction capacity, flexibility, and the ability to take action goals. Consequently, when executive dysfunction is present, various types of activity-related cognitive functions are impaired. Memory disorders also occur as a result of loss-of-set but are less pronounced than in the posterior brain syndrome. Recognition and interpretation capabilities remain relatively intact. Gait pattern is often slower, similar to that of Parkinson’s disease. The syndrome originates from dysfunction of frontal subcortical regions of the brain (anterior brain syndrome) and is typical of SSVD [18, 98–101]. Executive dysfunction may appear also in patients with AD but later in the course of the disease, and it is usually associated with impairment of attentional shifting and working memory [102]. In patients with SSVD, the executive dysfunction manifests itself early in the course of the disease causing impairment in the capacity to use complex information, to formulate strategies, and to exercise emotional and behavioral self-control. In comparison with AD, patients with SSVD show less pronounced episodic memory impairment but more depressive symptomatology and greater variability in the speed of progression of the disease [103, 104]. SSVD specifically contributes to deterioration of psychomotor speed, global cognitive function, and impaired executive control. Executive dysfunction causes the loss of functional capacity required to perform activities of daily living, a requisite criterion for the diagnosis of major cognitive disorder. Patients with AD and prominent impairment of memory may remain relatively independent as long as the executive function remains intact; in contrast, SSVD may lead to early and progressive executive dysfunction with considerable risk of developing major cognitive disorder [104]. Executive function is also a determinant in defining the level of institutional care required.
Recent studies have investigated the cognitive profile of SSVD in more detail. In a comprehensive meta-analysis, Vasquez and Zakzanis [105] compared healthy controls with patients with SSVD and mild cognitive impairment (MCI). Patients showed impairments across all cognitive domains but the greatest decline was in mental speed. SSVD patients exhibited more pronounced impairments in executive function and mental speed compared to the non-vascular MCI group. In contrast, the non-vascular MCI group exhibited more pronounced impairment of delayed memory [105]. Although the differences between vascular and non-vascular MCI are in agreement with the results of previous studies, it is essential to notice that there is overlap in cognitive impairment observed in neuropsychological tests between the groups. Based on the recommendations of the 2006 NINDS-CSN consensus conference [106], a neuropsychological battery for MCI-SSVD was developed using a factor analysis approach [107, 108]. The battery was found to be robust and applicable. Attention-executive dysfunction, memory loss and visuospatial difficulties were the most obvious features. However, the Trail Making Test was poorly applicable to older and cognitively impaired patients.
Age at onset appears to be a crucial factor in determining the cognitive impairment profile [109]. Frontal-executive dysfunction was more pronounced in patients with disease onset before age of 65 in comparison with the late-onset group. The early-onset SSVD group had a more severe small-vessel disease burden, whereas the late-onset group exhibited greater amyloid burden. In comparison with AD, the loss of visual memory is less pronounced in SSVD after one year, whereas the opposite is true for loss of physical independence [110].
A number of simplified approaches to the diagnosis of cognitive impairment in SSVD have been proposed. In a study operationalizing MCI criteria, non-amnestic single-domain MCI was unexpectedly common and the authors concluded that it may represent a previously unrecognized MCI subtype associated with SSVD [108]. The Frontal Assessment Battery is a useful tool to differentiate MCI due to SSVD from AD [111]. The Montreal Cognitive Assessment performed better than the Mini-Mental State Examination to detect MCI-SSVD [112]. The Cognitive Assessment Battery appears to be useful for MCI detection of MCI-SSVD [113]. In an autopsy-defined cohort [114], a combined memory and verbal fluency score was able to differentiate between AD and SSVD with a sensitivity of 85%, specificity of 67% and positive likelihood ratio of 2.5. Amnestic memory impairment and lower categorical fluency characterized AD, while the pattern in SSVD exhibited a tendency toward greater impairment on phonemic fluency and better performance on recognition memory [114]. The CLOX test [116] remains a simple and effective tool to detect executive dysfunction (Fig. 3).
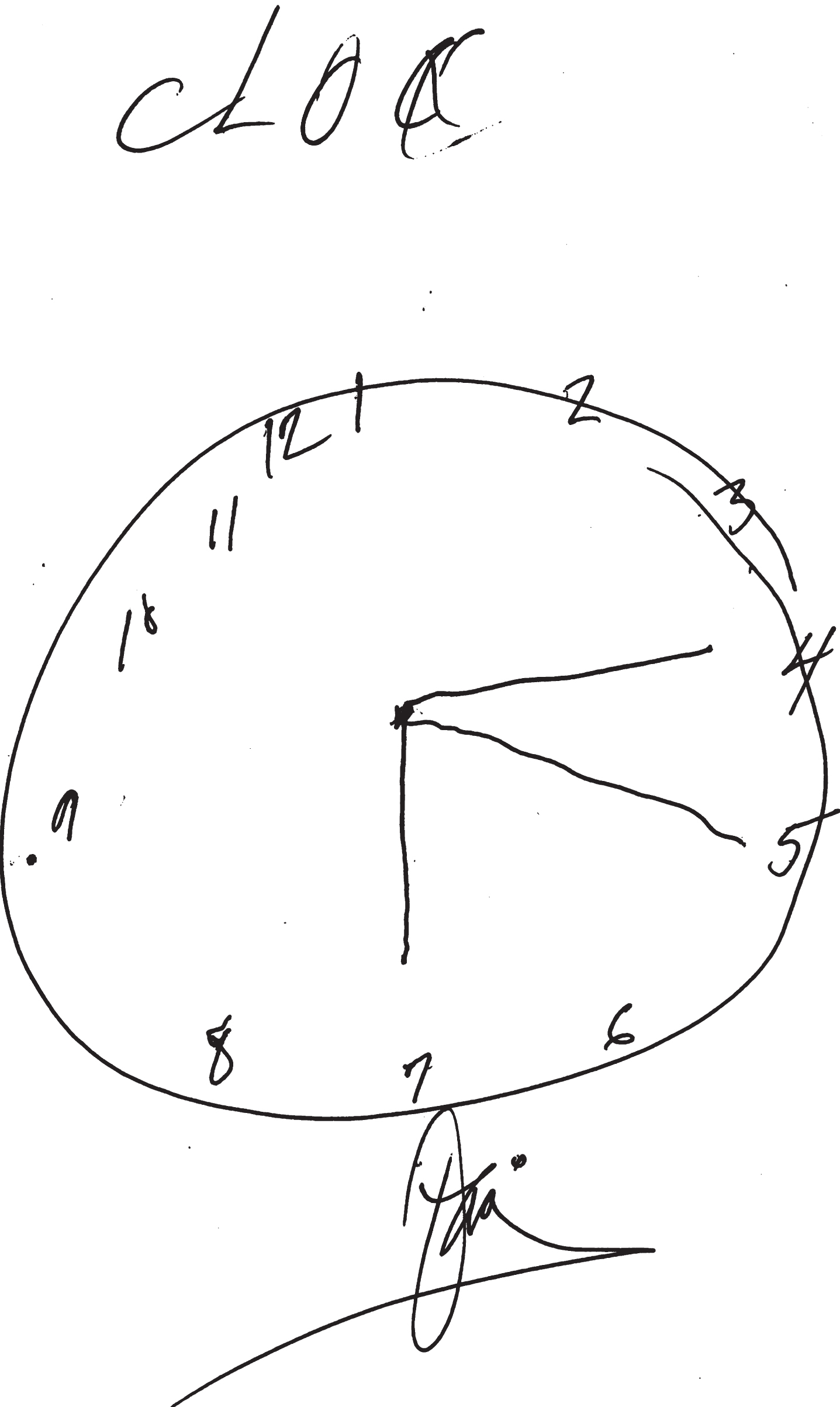
Executive Clock-Drawing Task (CLOX): The patient is instructed to draw a clock on a white sheet of paper. The instructions are as follows: “Draw a clock that says 1:45. Set the hands and numbers on the face of the clock so that even a child could read them.” The instructions can be repeated until they are clearly understood, but once the subject begins to draw no further assistance is allowed. This patient scored 7/15 points. The subject’s performance is scored as follows: Does figure resemble a clock? 1 point; Circular face present? 1 point; Dimensions >1 inch? 1 point; All numbers inside the perimeter? 1 point; No sectoring or tic marks? 1 point; Numbers 12, 6, 3, & 9 placed first? 1 point; Spacing Intact? (Symmetry on either side of 12 and 6 o’clock?) 1 point; Only Arabic numerals? 1 point; Only numbers 1 - 12 among the numerals present? 1 point; Sequence 1-12 intact? No omissions or intrusions. 1 point; Only two hands are present? 1 point; All hands represented as arrows? 1 point; Hour hand between 1 and 2 o’clock? 1 point; Minute hand obviously longer than hour? 1 point; 1 point if none of the following are present: 1) Hand pointing to 4 or 5 o’clock? 2) “1:45” present? 3) Any other notation (e.g., “ 9:00”)? 4) Any arrows point inward? 5) Intrusions from “hand” or “face” present? 6) Any letters, words or pictures? From: Royall et al., 1998 [116].
Neurological signs have been thoroughly studied in patients with SSVD. A score for focal neurological signs did not correlate with global cognitive performance, but instead with executive dysfunction, as measured by the letter fluency and the Rey-Osterrieth Complex Figure tests [115]. The neurological signs observed more often were the Chaddock sign, stooped posture, decreased arm swing, bradykinesia, and rigidity. Sixty-nine percent exhibited at least one extrapyramidal sign and 58% at least one unilateral lateralizing sign [115]. Motor impersistence and perseveration occurred when cortical areas and underlying white matter tracts associated with the frontoparietal attentional system were affected in patients with SSVD [117]. The LeukoAraiosis and DISability (LADIS) study [118] showed that SSVD was associated with deterioration of gait and balance over time. Also, gait disturbance complaints in non-disabled SSVD patients were, along with age, atrial fibrillation, and the degree of WMH, an independent predictor of disability after 3 years [119]. Interestingly, new studies have found that SSVD plays a role in the development of cognitive impairment in patients with Parkinson’s disease [120, 121].
Neuropsychiatric manifestations such as depressive symptoms, loss of motivation, lack of interest and emotional bluntness are common in patients with SSVD [122]. Small-vessel lesions and amyloid burden independently affect specific neuropsychiatric symptoms [123]. Apathy, depressive symptoms, irritability [123, 124], and sleep disturbances [125] are the most frequent neuropsychiatric symptoms in SSVD. In MCI-associated SSVD both negative and also positive neuropsychiatric symptoms appear; more negative than positive symptoms become more obvious as the disease progresses from MCI to major cognitive disorder [126]. In a pivotal study, both apathy and depressive symptoms were increased in patients with SSVD but typically they did not occur together in the same patient [127]. Diffusion-tensor imaging (DTI) revealed that white matter microstructural changes in SSVD were associated with increased apathy, but not depressive symptoms [127]. These findings have consequences for the formation of treatment strategies for patients with SSVD.
In summary, new studies have confirmed the anterior symptom profile in patients with SSVD [128]. Motor-neurological and neuropsychiatric features have been added to the characteristic frontal-executive pattern of the disorder, which is phenotypically different from that of pure AD [123, 124]. However, the cognitive profile of SSVD is much more complex than typically reported. The results of new studies suggest that overlapping neural networks take part in accomplishing neuropsychological tasks. The field will benefit from further studies on outcome of network pathway disruption [105, 129]. The symptom profiles described here are not sufficiently specific and do not qualify as unique diagnostic markers of SSVD. For diagnostic purposes, other tools such as brain imaging and biochemical markers should be used in combination with the characteristic symptom profile.
NEUROIMAGING
The standard-of-care evaluation in elderly patients with cognitive decline requires at least one brain imaging study [130] to eliminate pathologies different from neurodegeneration and to determine the extent of the vascular lesions [131, 132]. Development of neuroimaging techniques, in particular computerized tomography (CT) and MRI, provided some of the most important tools in the diagnosis of cognitive disorders, allowing accurate demonstration of the location and rate of progression of atrophic changes affecting the brain in AD and other cognitive disorders, as well as the different types of vascular lesions observed in pure forms of vascular cognitive disorders and MXD [133, 134]. Two basic imaging methods are currently used in the clinical evaluation of cognitive disorders: structural brain imaging (CT/MRI) and metabolic imaging (PET/SPECT). CT uses X-rays and the resulting images depend on the electron density and rates of absorption of X-rays by different tissues. MRI uses the changes induced by the magnetic field to alter the rate of spin of the hydrogen atom about its own axis; this change of the electromagnetic properties of protons in water molecules in the tissue added to the small difference in energy produced by the change of spin of the protons is detected and used for the anatomic three-dimensional (3D) reconstruction of the tissue. Positron emission tomography (PET) scans allow measurement of regional cerebral glucose metabolism using 18F-2-fluoro-deoxy-D-glucose (FDG). Cerebral blood flow can be measured with H215O-PET or using single photon emission computerized tomography (SPECT) with technetium hexamethyl-propylene-amine-oxime (99mTc-HMPAO).
Quantification of cortical thickness allows early diagnosis and rate of progression from MCI to major cognitive disorder. White matter involvement can also be quantified with DTI and functional methods including fMRI, functional connectivity, and MR spectroscopy (MRS). Isotope-based techniques include isotope markers for Aβ (11O-PIB, 18F-florbetapir), tau (18FDDNP) and activated microglia (11C-PK11195). Neuroimaging markers are particularly useful at the early symptomatic and preclinical asymptomatic phases of SSVD [131], as well as serving as endpoints in clinical trials. The latter technique allows a significant reduction of the number-needed-to-treat subjects in controlled trials.
According to Thompson and Hakim [24], SSVD is widely recognized as the commonest of all brain lesions. The unusually elevated prevalence of WMH in the elderly was received initially with skepticism, reflected in the name UBOs (unidentified bright objects) used to first describe them [2]. For instance, in 1996, the large population-based brain MRI study on 3301 elderly subjects in the Cardiovascular Health Study [134] found that more than half (64.2%) of all participants had some degree of WMH; only 4.4% had no lesions and 31.4% had minimal lesions. WMH were associated with older age, clinically silent stroke on MRI, hypertension, smoking and lower income indicating limited access to medical services, inadequate diet and living conditions, and poor long-term compliance with antihypertensive treatment [135]; higher grade WMH were accompanied by cognitive impairment and abnormal gait. Correlation studies revealed that the majority of these WMH in the elderly were ischemic in nature [136]. Arne Brun and Elisabet Englund [137] at the University of Lund, Sweden, first called these WMH lesions “incomplete” white matter infarctions. The name “incomplete” indicates that complete ischemic necrosis and cavitation were not present. CT and, more clearly, MRI also revealed the presence of état criblé or dilatation of Virchow-Robin perivascular spaces [138, 139]; lacunes, état lacunaire and Binswanger disease. Joanna Wardlaw and colleagues [140] have extensively reviewed the topic of WMH, including pathogenesis, clinical relevance and imaging features. WMH are not benign lesions since they increase 2-fold the risk of major cognitive disorder and 3-fold the risk of stroke [54, 141]. Recent MRI technical advances [142–144] including manual and automatic volume measurements, DTI, and fMRI should provide further understanding of SSVD lesions. In 2013, Wardlaw and the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) study group [145] completed a major international effort to standardize neuroimaging acquisition, interpretation and reporting of cerebral small-vessel disease. The criteria include the following SSVD detected on conventional MRI: 1) recent small subcortical infarcts, 2) white matter hyperintensities, 3) lacunes, 4) prominent perivascular spaces, 5) cerebral microbleeds, and 6) atrophy (Fig. 4). Criteria for imaging detection and quantification of cortical cerebral microinfarcts on 3 Tesla MRI have been proposed recently [146].
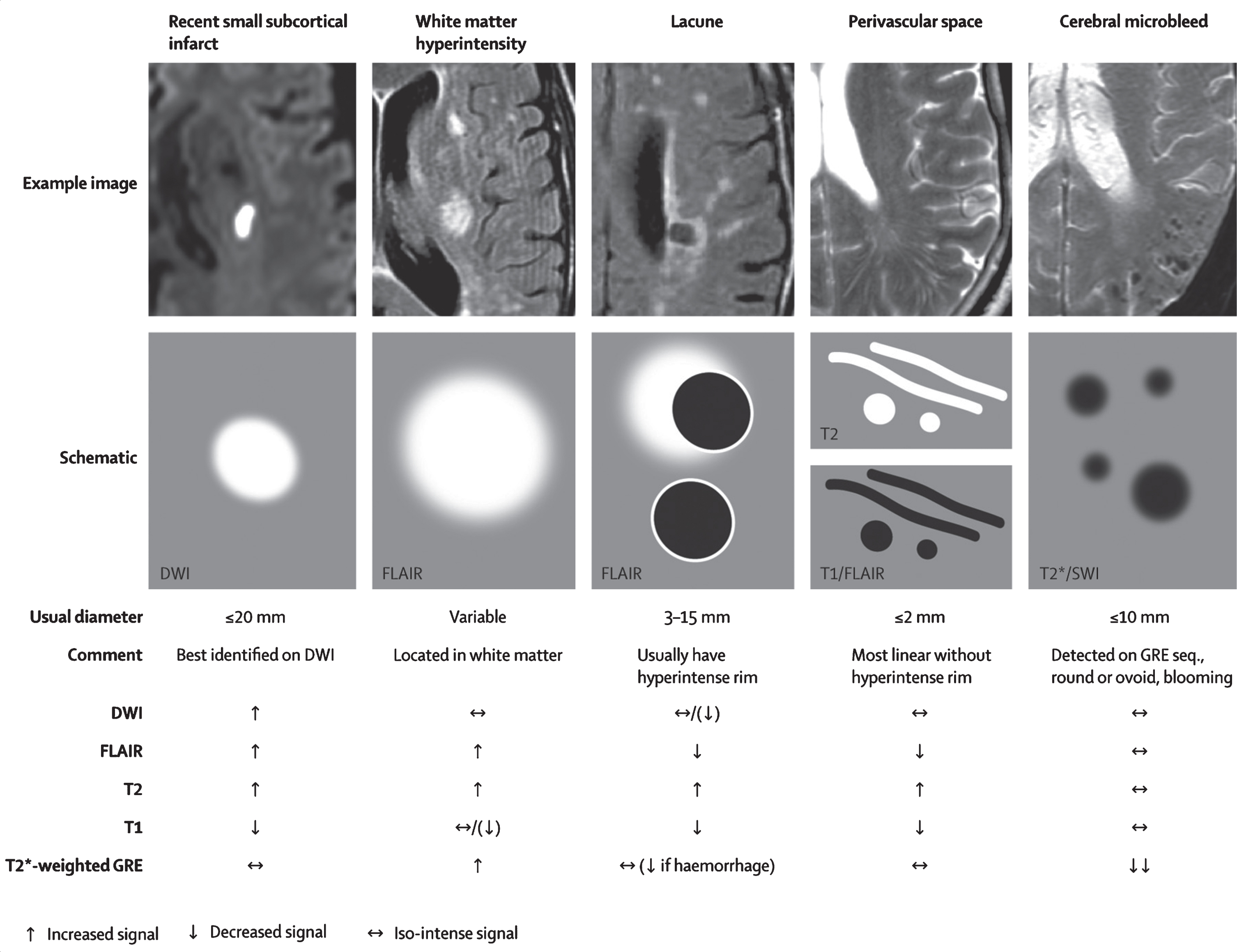
Examples of different features of small vessel disease, including white matter hyperintensities, as published by the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) study group. From: Wardlaw et al., 2013 [145].
Quantification of the total burden of SSVD and the resulting alterations of the BBB leading to decreased CBF has been successfully studied [147, 148]. It has been shown that the severity of the baseline WMH load predates the decrease of CBF after a follow-up of almost 4 years. Cerebrovascular reactivity in SSVD can be measured using blood oxygen level dependent (BOLD) MRI and vasoactive stimulation; preliminary results indicate that cerebrovascular reactivity appears to be decreased in subjects with more severe WMH [147, 148].
Cerebral FDG-PET is widely used in the evaluation of cognitive disorders in the elderly given that cerebral glucose metabolism is an index of neuronal and synaptic function; thus cerebral glucose hypometabolism represents regions of brain destruction such as in stroke, or cortical zones of neurodegeneration in VCI and in most types of degenerative cognitive disorders such as AD [149, 150]. Bloudek et al. [151] performed a meta-analysis to evaluate FDG-PET in AD and found 90% sensitivity (95% CI 84–94%) and 89% specificity (95% CI 81–94%). Kerrouche et al. [152] using voxel-based multivariate analysis of 18FDG-PET separated VCI from AD with 100% accuracy. Hypometabolism in deep gray nuclei, cerebellum, primary cortex, middle temporal gyrus, and anterior cingulate cortex differentiated VCI from AD [152]. In addition to providing a clear diagnostic image of AD and other neurodegenerative cognitive disorders, such as frontotemporal degeneration, FDG-PET is quite useful in the diagnosis of patients with PSD due to multiple strokes and also in determining the contribution of AD to VCI.
The brain is a complex vascular organ; therefore, measurement and understanding of CBF is central to progress in the field of cognition [153]. Neurovascular coupling increases CBF according to local neuronal activity and metabolism [8] serving as the basis for BOLD-MRI and fMRI [150, 151]. A decrease in CBF occurs with normal aging as well as in AD [30–32, 154]. A novel method to measure CBF uses ASL [30, 155], a non-invasive MRI technique that requires neither contrast media nor isotopes [30]. ASL-MRI generates an endogenous contrast by using radiofrequency pulses that “label” water proton spins in blood circulating in carotid and vertebral arteries at the base of the skull. CBF-ASL-MRI images are obtained by subtracting labeled and unlabeled spin exchanges in the brain tissue yielding a quantifiable map of regional CBF (rCBF) [30, 155]. Using 3D ASL-MRI in subjects with SSVD, Sun et al. [156] demonstrated marked decrease in CBF in the cortex of the temporal and frontal lobes, hippocampus, thalamus, and insula that correlated with the degree of cognitive impairment. Also, thinner cortex in frontal, parietal and lateral temporal regions was noted in more severe cases of SSVD.
Excellent correlation of CBF with FDG-PET metabolic rate has been obtained in patients with several forms of cognitive disorders [155, 156], as well as in obstructive sleep apnea [157, 158]. BBB alterations can be detected using a two-stage non-invasive diffusion weighted MRI technique [159] that uses fast and slow diffusion coefficients to differentiate labeled blood in the microvascular compartment (small vessels and capillaries) and in the brain tissue; the ratio of these two signals indicates water exchange rate across the BBB [159]. Large artery integrity can also be determined using flow encoding arterial spin tagging [160].
CBF-ASL in NPH
A promising area of research in SSVD is the use of CBF-ASL is the study of Binswanger disease and normal pressure hydrocephalus (NPH). In 1894, Otto Binswanger described the clinical form of major vascular cognitive disorder that bears his name characterized by extensive subcortical ischemic white matter lesions. Binswanger’s neuropathological report noted the presence of “enormously enlarged ventricles” [4]. Román [2, 161] pointed out the difficulties in clinically separating Binswanger disease from NPH. In fact, both entities are characterized by prominent changes in ambulation, such as small-step gait (marche à petits pas), gait apraxia, frequent falls, and urinary incontinence. A subcortical type of cognitive impairment with changes in mood, behavior, and personality is common in both, as well as pseudobulbar palsy, emotional incontinence, and frontal lobe signs with loss of incentive, drive, and insight. Profound apathy and abulia may be observed in advanced cases of both conditions. Mutism, bradykinesia, rigidity, and dysarthria may lead to confusion with Parkinson disease. Finally, there is frequent occurrence of hypertension and cerebrovascular disease in cases of idiopathic NPH [161], as well as typically noted in Binswanger disease.
Bradley et al. [162] compared the presence and degree of periventricular WMH in patients with treated and untreated NPH and in age-matched control subjects. A highly significant statistical association was found for WMH in NPH when compared with controls. Patients with MRI evidence of WMH but without clinical symptoms of NPH had increased frequency of communicating hydrocephalus. Diffusion of CSF into the periventricular areas and SSVD contribute to periventricular WMH in NPH.
Due to Pascal’s hydrostatic pressure law, the net effect of CSF pressure on the dilated ventricles is an increase of interstitial pressure in the brain parenchyma or transmantle pressure [163]. Perfusion of the periventricular regions proceeds in a centripetal pattern from the surface of the brain toward the ventricles by means of long medullary arteries; therefore, an increase in the intraventricular pressure results in an opposite gradient of centrifugal pressure capable of producing cerebral hypoperfusion [164] and ischemia of the watershed periventricular white matter, particularly in elderly subjects with loss of CBF autoregulation [164], but also in children with hydrocephalus from posterior fossa lesions [165, 166]. As illustrated in Fig. 5, global CBF-ASL perfusion increases post-lumbar puncture (post-LP) in patients with diagnosis of NPH; in contrast, Binswanger disease cases show no change in CBF post-LP.
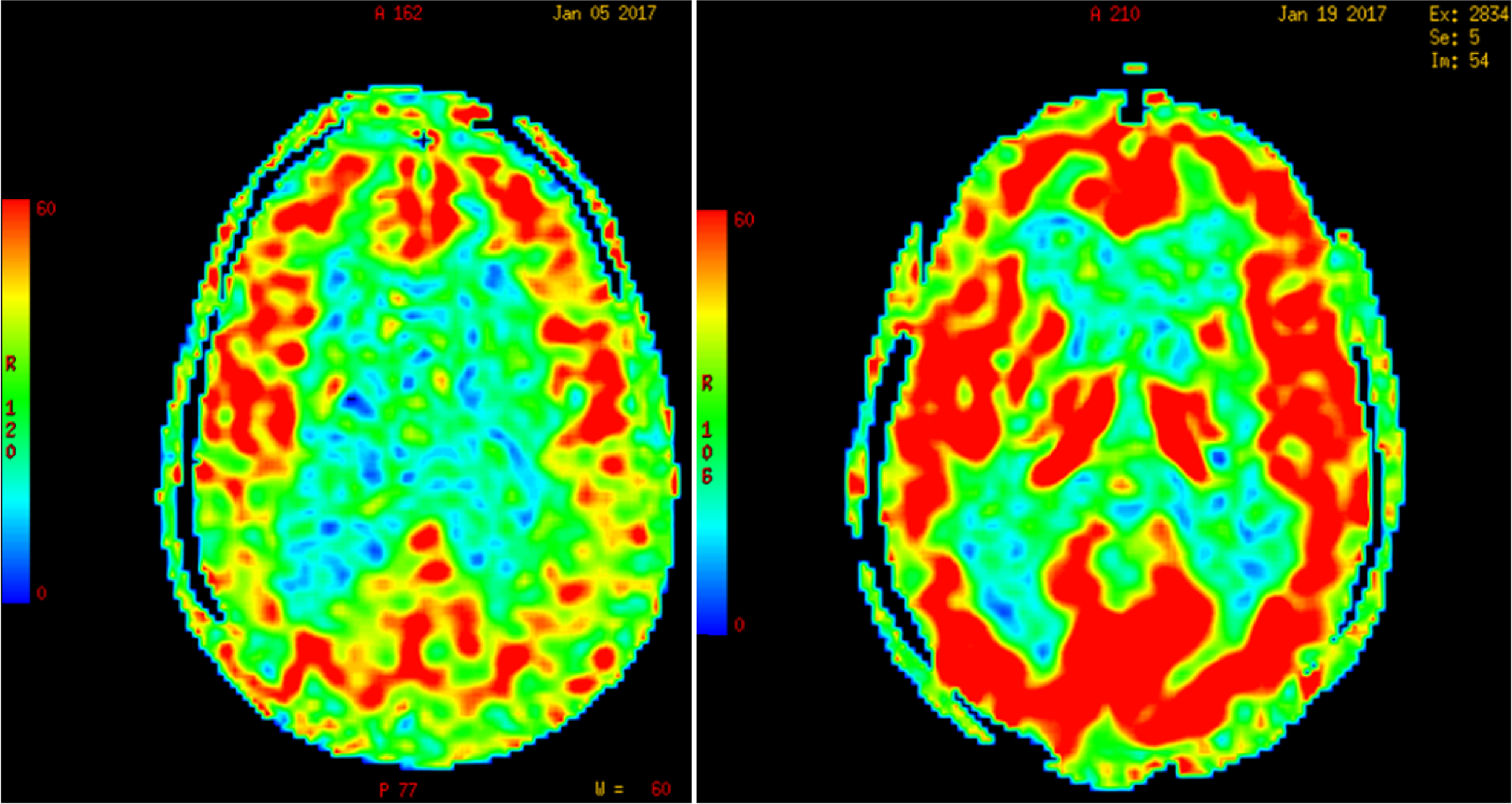
Increase in global cerebral blood flow determined by arterial spin label MR imaging post-lumbar puncture in a patient with normal-pressure hydrocephalus. (Román GC and Fung S, unpublished data).
In summary, most of the advances in the diagnosis, pathogenesis, epidemiology, and overall understanding of SSVD in the last decade have resulted from the clinical and research application of brain imaging, in particular MRI. Further studies on the vascular mechanisms underlying SSVD could be expected from research using MRI-based CBF-ASL studies and newer imaging modalities. Several groups in Europe recently recommended the implementation of ASL-MRI studies for numerous clinical applications including patients with dementia [30]. Brain imaging should continue to open novel therapeutic paths for these patients.
CEREBROSPINAL FLUID BIOMARKERS
As stated above there are biochemical changes in the brain of patients with SSVD, which may be reflected in the CSF since it is in direct contact with the extracellular space. This assumption and strategy has proved fruitful in the field for AD research. Recently, the index between tau (τ) protein in its total (τT) or phosphorylated form (phospho-tau, τP) and Aβ peptide 1–42 (Aβ42), has been incorporated in the research criteria for AD [33, 168]. The differentiation between AD and VCI, and especially between MXD and “pure” VCI, is not always easy in everyday practice. VCI is a heterogeneous entity with SSVD increasingly recognized as the most common pathological substrate. Here we summarize the diagnostic value of these biomarkers in the differential diagnosis of SSVD [33, 169].
Tau (τ) protein is the major component of intracellular neurofibrillary tangles and is present in a hyperphosphorylated form in AD pathology. Both τT and τP tau are increased in the CSF of patients with AD, as compared to controls. τT is better viewed as a marker of neuronal and/or axonal degeneration, while τP is a more specific marker of tau hyperphosphorylation process and tangle formation in AD pathology [170]. Although the diagnostic accuracy may be reduced to some degree when attempting to differentiate AD from some other types of cognitive disorders, when combined with Aβ42 the above biomarkers achieve sensitivities and specificities approaching or exceeding 90% for the discrimination of AD from normal aging.
CSF τT levels in VCI provide conflicting results ranging from normal [171–173] to increased [174–178], or to intermediate between controls and AD cases but much lower when compared with AD [179, 180]. Some patients with VCI may present with high or even very high τT levels [173, 180–183]. When patients with “pure” VCI and MXD or AD pathology with subcortical white matter lesions are clinically separated, the results were again conflicting with τT in VCI reported as comparable to the controls [173], increased [178] or intermediate but much lower as compared to those of AD [180], while patients with MXD presented with increased τT in all studies. However, normal τT levels are found in patients with lacunar infarcts [171], progressive leukoaraiosis [175], or pure SSVD [184–187]. Phospho-tau (τP) levels have been reported as normal in VCI or SSVD [177, 185–187], while in MXD the levels are increased to those of AD cases [180] or intermediate between controls and AD [186]. Aβ with either 1–40 (Aβ40) or especially 1–42 amino acids (Aβ42) is the major component of amyloid plaques; Aβ is present extracellularly in AD and seems to inversely reflect amyloid pathology, i.e., CSF Aβ42 is 50% reduced in AD as compared to normal aged subjects. CSF-Aβ42 has shown high sensitivity and specificity (over 85%) and is recognized as one of the three “core feasible” CSF markers for AD [170].
Reduction of CSF Aβ42 has been reported in VCI [183] or in pure SSVD [185, 187–189], at levels similar to those of AD or intermediate between controls and AD cases. In other studies, Aβ42 levels in VCI have been comparable to those of controls but higher than in AD [172, 176–179]. Some VCI patients present low Aβ42 levels, but some overlap may be present [180].
In summary, most of the above studies agree that Aβ42 levels in VCI are reduced to a degree comparable to AD. The ratio of Aβ42/Aβ40, which is decreased in AD, may be comparable to the controls in pure VCI [190]. Thus the above index, if normal, may strengthen the diagnosis of SSVD, when used along with clinical imaging and other biomarker data.
Taken together, most of the above studies agree that τP is usually normal in pure SSVD, while some patients may present with low Aβ42 and/or high τT levels. A recent study [191], using NINDS-AIREN criteria [192], showed that only 29% of VCI patients presented with increased τP levels; however, reduced Aβ42 was found in 53% and increased τT levels in 41% of cases [191]. Such deviations from the usual pattern may indicate clinical overlap with AD or MXD, thus reducing sensitivity and specificity of individual biomarkers when used as tools in every day practice; therefore, combination of several biomarkers is required [190].
Overall, for the diagnosis of SSVD and separation from AD—the most common clinical scenario—the use of the above biomarkers as diagnostic tools is of value, but further studies are needed to evaluate sensitivities and specificities in various clinical situations. Combinations of these biomarkers with neurofilament light, metalloproteinase-9, TIMP metallopeptidase inhibitor 1, albumin index and metalloproteinase-2 index may further increase the diagnostic accuracy [178–180].
METABOLIC FACTORS
Risk factors for SSVD include systemic hypertension [193] and conditions causing hypoxia such as smoking [194], chronic obstructive pulmonary disease [195], and sleep apnea syndrome [196]. Hypercoagulation [193], chronic kidney disease [197], and homocysteinemia [198] could worsen the consequences of hypertension and hypoxia. Moreover, the metabolic syndrome due to an adverse lifestyle and aging could also influence the risk of SSVD [199].
Overnutrition and sedentary lifestyle result in excessive adiposity and metabolic disturbances. The risk profile is dependent on the distribution of body fat and the presence of metabolic aberrations [199]. The metabolic syndrome (METS) includes the clustering of abdominal obesity, insulin resistance, dyslipidemia, and elevated blood pressure [199]. In the French Three-City (3C) cohort [200], the presence of METS increased the risk of incident major VCI over 4 years. In 308 healthy older French persons from the longitudinal ESPRIT study [201], those with METS increased two-fold (Odds Ratio [OR] = 2.74) the chance of presenting with high levels of WMH volume, compared with subjects without METS. Furthermore, in 1151 healthy old Japanese people [202], METS was associated with silent brain infarction and WMH, whereas less strong associations were found with increased body mass index alone [202].
In the Japanese study by Bokura et al. [202], dyslipidemia was associated with WMH. Although there have been variable results in terms of the role of hyperlipidemia in WMH/SSVD progression [203], several studies have shown a positive association with hypertriglyceridemia [204]. ApoE is the major transport protein for cholesterol in the central nervous system [205]; ApoE levels in the CSF are significantly lower among patients with AD compared with controls [206], as well as in other forms of cognitive disorders including VCI [207]. ApoE transports cholesterol to neurons via ApoE receptors [208], thereby being involved in the mobilization of lipids for repair, growth and maintenance of myelin and axonal membranes [208, 209]. Thus, the deficiency of ApoE could possibly worsen the consequences of WMH by reducing repair mechanisms. ApoA-I levels in the CSF were reduced in AD but levels were not associated with ApoE ɛ4 allele distribution [207].
Insulin resistance is part of the clustering of risk factors seen in METS [210]. Using transcranial Doppler and MRI in subjects with diabetes mellitus type 2, the estimated WMH volume correlated negatively with CBF velocity [211], which could indicate a possible mechanism underlying WMH development. The results of several studies have suggested the association of impaired glucose homeostasis or diabetes mellitus with VCI or with increased WMH volume [212, 213]; but not all reports are concordant [214]. Moreover, postmortem studies have revealed that in the AD brain, there is resistance to signaling through the insulin receptor and the closely related insulin-like growth factor-I (IGF-I) receptor [215–217]. Neurons resistant to insulin and IGF-I action could lack trophic signals and therefore degenerate [218]; also, insulin and IGF-I are associated with the quantity of oligodendrocytes and myelin production [219–221]. However, it is unclear whether brain insulin resistance is a primary or secondary event in AD and it is unknown if there is resistance to insulin or IGF-I activity in the brains of patients with SSVD.
Subclinical inflammation is associated with both METS and SSVD. Several studies have shown that the inflammatory markers C-reactive protein, IL-6, TNF-α, TGF-β, and vascular endothelial growth factor levels are elevated in SSVD patients [222–225]. Furthermore, some studies have reported stronger associations between inflammatory markers and risk of SSVD/VCI compared to the corresponding associations seen in AD [226, 227]. However, data are not fully conclusive given that unchanged or even decreased levels of inflammatory markers have also been observed [227–229]. An additional hypothesis posits that immunosenescence could be of importance [229]. This means that an aged and defective immune system could be a risk factor for cognitive decline VCI. In conclusion, although inflammation is likely to occur in SSVD development, the exact nature of this involvement is still unclear.
Exposure to chronic stress may result in cognitive decline by activation of the hypothalamic–pituitary–adrenal (HPA) axis [230]. Although glucocorticoids display both neuroprotective and neurodegenerative effects [231], the results of most experimental studies suggest that prolonged or excessive increases in cortisol levels result in neuronal injury and reduced cognitive function [232, 233]. Increased circulating and CSF levels of cortisol have been observed in AD [234, 235], and there are reports of disturbed cortisol secretion also in VCI [236]. In the Lothian Birth Cohort 1936, higher cortisol at the start and the end of a mild cognitive stressor was associated with higher WMH [237]. Chronic stress with activated HPA axis could therefore be a risk factor for the development of SSVD. These concepts will need to be explored in the future.
In summary, sedentary lifestyle combined with overeating, chronic stress, and aging increases the risk of SSVD. However, the underlying mechanisms have not been elucidated in detail, and little is known concerning metabolic changes in the brain. Therefore, further research is needed to explore the role of metabolic factors in WMH development. Lifestyle intervention and medical treatment of metabolic alterations must be studied in clinical trials to determine the potential effect in reducing the risk of SSVD.
CONCLUSIONS
The most important research advance in the field of VCI in the last decade has been the scientific demonstration that age-related white matter involvement is a sign of subcortical small-vessel disease that leads to cognitive failure and impaired functional capacity, i.e., SSVD. Although this is the most homogeneous and common type of VCI and clinical criteria for the disease exist, it is often underdiagnosed both in incipient and manifest stages. Likewise, given the magnitude of the problem, a surprisingly limited number of pharmacological studies on SSVD have been conducted to-date. Interestingly, in a subgroup analysis of the Scandinavian Multi-Infarct Dementia Trial, nimodipine was found to be effective in patients with SSVD [238]; a more recent controlled clinical trial of DL-3-n-butylphthalide (a nutriceutical extracted from celery, Apium graveolens) showed positive results in patients with mild VCI caused by disease of the subcortical small vessels [239]. As mentioned earlier, a therapeutic experiment in an animal model of CADASIL demonstrated that the use of an antibody targeting Notch3 signaling effectively prevents mural cell loss [97]. A similar method could eventually be developed in animal models of SSVD.
One of the most important steps toward providing a greater knowledge about SSVD, and subsequently better care options for the disease, is to accept it as a distinct entity, as has been the case with AD. Indirectly, the lack of success in more than 100 AD trials has facilitated this approach [240, 241]. Recent advances towards that goal have emerged including: 1) increased knowledge of the symptom profile of SSVD, in addition to the characteristic frontal-dysexecutive cognitive profile; 2) new MRI studies have found that white matter involvement is associated with reduced cerebral blood flow; 3) a characteristic non-AD profile of CSF markers reflecting subcortical degeneration, inflammation and extracellular matrix breakdown has been identified; 4) the clinical and pathological criteria for SSVD have been updated; 5) the potential importance of deep vein collagenosis has been studied [242]; and 6) interesting associations between SSVD and NPH have been found. Although several of the new findings need to be replicated, they already now imply that focusing on SSVD using various methods is a promising way of addressing the great challenge of preventing and treating age-related cognitive impairment. Future studies investigating the relationships between sporadic SSVD and genetic factors are needed as well as research on the various mechanisms of causation presented above. These goals aim primarily at finding targets for intervention in patients with SSVD early in the course of the disease but may also have positive consequences for knowledge and treatment of AD [243].
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0803r1).