
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the presence in the brain of extracellular amyloid-β protein (Aβ) and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein. The N-Methyl-D-aspartate receptors (NMDAR), ionotropic glutamate receptor, are essential for processes like learning and memory. An excessive activation of NMDARs has been associated with neuronal loss. The discovery of extrasynaptic NMDARs provided a rational and physiological explanation between physiological and excitotoxic actions of glutamate. Memantine (MEM), an antagonist of extrasynaptic NMDAR, is currently used for the treatment of AD jointly with acetylcholinesterase inhibitors. It has been demonstrated that MEM preferentially prevents the excessive continuous extrasynaptic NMDAR disease activation and therefore prevents neuronal cell death induced by excitotoxicity without disrupting physiological synaptic activity. The problem is that MEM has shown no clear positive effects in clinical applications while, in preclinical stages, had very promising results. The data in preclinical studies suggests that MEM has a positive impact on improving AD brain neuropathology, as well as in preventing Aβ production, aggregation, or downstream neurotoxic consequences, in part through the blockade of extrasynaptic NMDAR. Thus, the focus of this review is primarily to discuss the efficacy of MEM in preclinical models of AD, consider possible combinations of this drug with others, and then evaluate possible reasons for its lack of efficacy in clinical trials. Finally, applications in other pathologies are also considered.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative pathology of severe prognosis, currently incurable, and highly prevalent in aged people [1]. The first description of the disease was carried out in 1906 by a German psychiatrist and neurologist, Alois Alzheimer, when exploring the brain tissue of a patient, Auguste Deter, who had died from a mental illness unknown at that time [2, 3]. Today, it is the cause of 50–70% of dementia cases and, according to the Alzheimer’s Association, 13% of people over 65 in developed countries suffer from AD, where it is the fifth cause of death in patients of this age group [1, 4]. If unchecked, global incidence will quadruple to 114 million by 2050 according to population growth projections by the World Health Organization (WHO) [1]. This would imply unsustainable economic burdens for all health systems, in addition to its social impact.
AD can be classified according to the age at which it begins to manifest, that is, if it has an early onset, developing before the age of 65 (3–5% prevalence), or if it occurs after the 65 years of age (95–97% prevalence) [1]. Additionally, the pathology may be of hereditary or sporadic origin, the latter being the most common (98% prevalence). Pathogenesis of late onset AD is very complex. Other than aging, which is the main factor involved in the development of the disease, it may be associated with dietary and environmental factors, including long term consumption of high caloric diets and sedentary lifestyles [1, 4]. Overall, it is the interaction of multiple genetic and environmental risk factors that lead to AD, together with the disruption of epigenetic mechanisms controlling gene dynamics and expression[4–10].
The main symptom of AD is memory loss, which is correlated with a decline of neuron population in the hippocampus, a brain area critical for learning and memory [9, 10]. In addition, the entorhinal cortex, area of interface between the hippocampus and the neocortex for the formation of spatial memory, becomes affected by this pathology. It has been reported that the hippocampus is especially vulnerable to damage at early stages in AD and approximately 25–70% of neurons are lost in this area [9, 10].
In the familial form of the disease, Aβ peptide formation, in particular its longer form Aβ42, is triggered by mutations in the amyloid-β protein precursor (AβPP, chromosome 21), presenilin 1 (PS1, chromosome 14), and presenilin 2 (PS2, chromosome 1)genes. Also, the APOE gene, another risk factor for the development of AD, has been characterized (ɛ2, ɛ3, and ɛ4) [1, 10–12]. It has been described that the ɛ2 allele has a protective role in AD, while ɛ4 has the opposite effect, since it favors the appearance of AD. Although late onset AD appears with higher incidence when the ɛ4 isoform is affected, the exact mechanism of how APOE triggers the formation of amyloid deposits is unknown [11, 12]. The lack of knowledge on the actual mechanisms that trigger the appearance of this multifactorial disease is the exact cause that leads us to having no cure for its treatment [11–13].
Memantine (MEM), a low-affinity voltage-dependent uncompetitive antagonist of NMDA receptors (NMDAR), is currently being prescribedfor the treatment of AD jointly with acetylcholines-terase inhibitors such as galantamine, donepezil, and rivastigmine [11, 14–17]. Since it is a low-affinity antagonist, it blocks the NMDAR but it is rapidly displaced from it, avoiding prolonged receptor blockade and the associated negative side effects on learning and memory that have been observed in high affinity NMDAR antagonists (dissociative anesthetics, ketamine, and MK-801). MEM also has a suitable safety and tolerability limits showing a good therapeutic margin. Another advantage of MEM is that it only interacts with the channel when it is pathologically activated under an excessive glutamate concentration in the synaptic cleft, as is the case with AD. In addition, preclinical data has reported that MEM could block other receptors, such as nicotinic, acetylcholine, serotonin, and sigma-1 receptors [17–19].
Historically, MEM was first synthesized by the Eli Lilly laboratories as a compound for the treatment of diabetes, but it was ineffective [14, 20–24]. In 1972, Merz Pharma applied for a German patent presenting MEM as a potential treatment for various neurological diseases [14, 15]. In the early 1990s, the company initiated the research of efficacy and safety of MEM in nursing patients with severe dementia. In 1999, Forest Laboratories acquired the license for using MEM and initiated a series of clinical studies in both moderate to severe AD. MEM was approved for the treatment of moderately severe to severe AD in 2002 by the European Agency for the Evaluation of Medical Products (EMEA). Complementarily, the efficacy of MEM was evaluated as monotherapy or combined with acetylcholinesterase inhibitors [24]. The dosing regimen of 10 mg bid was based onprevious clinical trial experience.
So far though, the biggest problem that is encountered with MEM and any other approved treatment for AD, is that they do not slow down the progression of the disease, but only have symptomatic effect [26]. That is why current efforts in AD therapy are focusing on the research and development of new molecules that can modify the course of AD progression [27, 28]. Among the major drugs that are in development, we observe some already in Phase III: inhibitors of the β-site amyloid precursor protein cleaving enzyme (BACE) (CNP520, LY3314814, and verubecestat) and antibodies against the Aβ peptide (aducanumab, solanezumab, crenezumab, gantenerumab, and BAN2401) [22, 27].
Because MEM has already been approved and used for AD and data points out possible combinations with other drugs in the future, in this review we will evaluate the different reported results on MEM at the preclinical levels on different stages of AD progression. Also, the different mechanisms involved in the beneficial effects of MEM in the preclinical levels will be discussed. Finally, the lack of MEM efficacy on clinical AD and the potential use of MEM in neurological diseases will be discussed.
SYNAPTIC AND EXTRASYNAPTIC METHYL-D-ASPARTATE RECEPTORS (NMDAR)
The NMDAR are cationic channels controlled by the excitatory neurotransmitter glutamate, which plays an essential role in the physiological process of learning and memory in the central nervous system (CNS) [14, 20–26]. The NMDAR can be classified in different subunits called NR1 and NR2A–D [14, 21–25]. In AD, an excessive release of glutamate and, an increasing concentration of soluble Aβ, which has been hypothesized to increase the activity of the receptor [23] and lead to an excessive Ca2 + flux within the cell, as well as free radical generation. The intense influx of Ca2 + impairs mitochondrial function activating transition pores in the inner mitochondrial membrane, which causes the release of cytochrome c, the posterior depletion of ATP, and simultaneous formation of reactive oxygen species (ROS) [23]. These cytotoxic events are associated with synaptic dysfunction and tau phosphorylation. [23].
Depending on the localization of the NMDAR in the neuron, they have been classified in two groups [20, 22]: The synaptic NMDARs have both presynaptic and postsynaptic locations on neurons [29]. The presynaptic receptors play a role in synaptic transmission and plasticity of neuronal networks, while postsynaptic only have a role in the control of plasticity [28–31]. Both types are involved in the activation of neuronal protective and survival genes [29, 30]. Extrasynaptic NMDARs are located on dendrites and require high glutamate concentrations in order to be activated [31]. These NMDARs are characterized by favoring the NR2B subunit which, when excessively stimulated, contribute to neurotoxicity and the control of neuronal cell death. Moreover, extrasynaptic NMDARs are involved in the regulation of Aβ production and thus in the neuropathology of AD [29–32]. They are the main target of MEM.
According to this information, MEM is the only approved antagonist against extrasynaptic NMDARs and the treatment of AD. Its selective action without affecting the physiological NMDAR synaptic activity allows neuroprotective effects brain [20–23].
CLINICAL PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
Initial postmortem brain histopathological examination of AD patients led to the characterization of the two classical hallmarks of the disease: senile plaques and neurofibrillary tangles (NFT) [1, 32]. Senile plaques are extracellular deposits,diffuse or compact, that can be found in the brain of patients with AD, and that are composed of insoluble Aβ peptides of 39 to 43 amino acids [21]. Diffuse plaques are Aβ deposits that do not visibly alter the surrounding neuropil, or induce glial response around them, and are considered relatively benign, since they have been found in cerebral cortex samples of elderly subjects that presented no cognitive impairment [4, 10]. In contrast, compact plaques are often surrounded by dystrophic neurites, reactive astrocytes, and activated microglia, rendering them more neurotoxic. In addition, senile plaques may also contain mucopolysaccharides, α1-antiquimiotripsin, immunoglobulins, complement factors, fibrinogen, APOE, cholesterol, presenilins, cytokines (IL-1β,IL-6, TGF-β, TNF-α), intercellular adhesion molecule 1 (ICAM-1), clusterine, α-synuclein, and inorganic elements [10].
The original hypothesis on the origin of AD was focused on alterations on the usual activity of the amyloid cascade [33–35]. This hypothesis is based on the following process: first, AβPP is processed by the amyloidogenic route, a pathway in which β- and γ-secretases cleave AβPP, causing an excessive production of Aβ peptide and/or a defect of its elimination [33–39]. This process causes its oligomerization and accumulation in the form of senile plaques in the brain cortex, hippocampus, and other brain areas. To better understand the mechanism, it will be explained further:
Initially, the Aβ peptide is generated from the catabolism of AβPP, a plasma membrane protein with a single domain found in different cell types, including neurons, astrocytes, oligodendrocytes, and glial cells. It is encoded by a gene located on chromosome 21, which when expressed gives rise to 8 isoforms, of which APP695 is the most abundant in the brain [38]. This protein is cleaved by α-, β, and γ-secretase enzymes and a complex of proteins containing PS1. In a physiological situation, following the non-amyloidogenic pathway, AβPP is catabolized by α-secretases, producing a sAβPPα fragment that remains in the extracellular space, and a carboxyl-terminal fragment of 83 amino acids (C83), that is anchored on the plasma membrane [35–39]. sAβPPα regulates neuronal excitability, improves synaptic plasticity, learning, and memory, and also increases the resistance of neurons to oxidative and metabolic stress. However, in neuropathological situations, AβPP is metabolized by the amyloidogenic pathway in which the β amyloid cleaving enzyme 1 (BACE 1; β-secretase) breaks AβPP by theN-terminal end while γ-secretases cleave theC-terminal end, obtaining fragments Aβ40 and Aβ42 that remain in the extracellular space and cause negative effects.
In addition, it is believed that in non-pathological conditions the Aβ peptide is produced continuously and that its aggregation and deposition in AD is the consequence of its rise on concentration that makes it precipitate [38, 39]. In physiological conditions, the amyloid protein is produced in normal amounts in its soluble form due to cleavage of AβPP by the α-secretase enzyme, and is eliminated by the action of degrading enzymes such as neprilysin, the insulin-degrading enzyme, and the angiotensin converting enzyme I. Yet, when sporadic AD occurs, the balance between the non-amyloidogenic and the amyloidogenic pathway becomes altered and, there is an excessive production of Aβ, resulting from the cleavage of AβPP by the β- and γ-secretase enzymes and a lower degradation of Aβ due to a lower activity of the degrading enzymes previously mentioned, thus causing an imbalance between the production and elimination of Aβ [33–40].
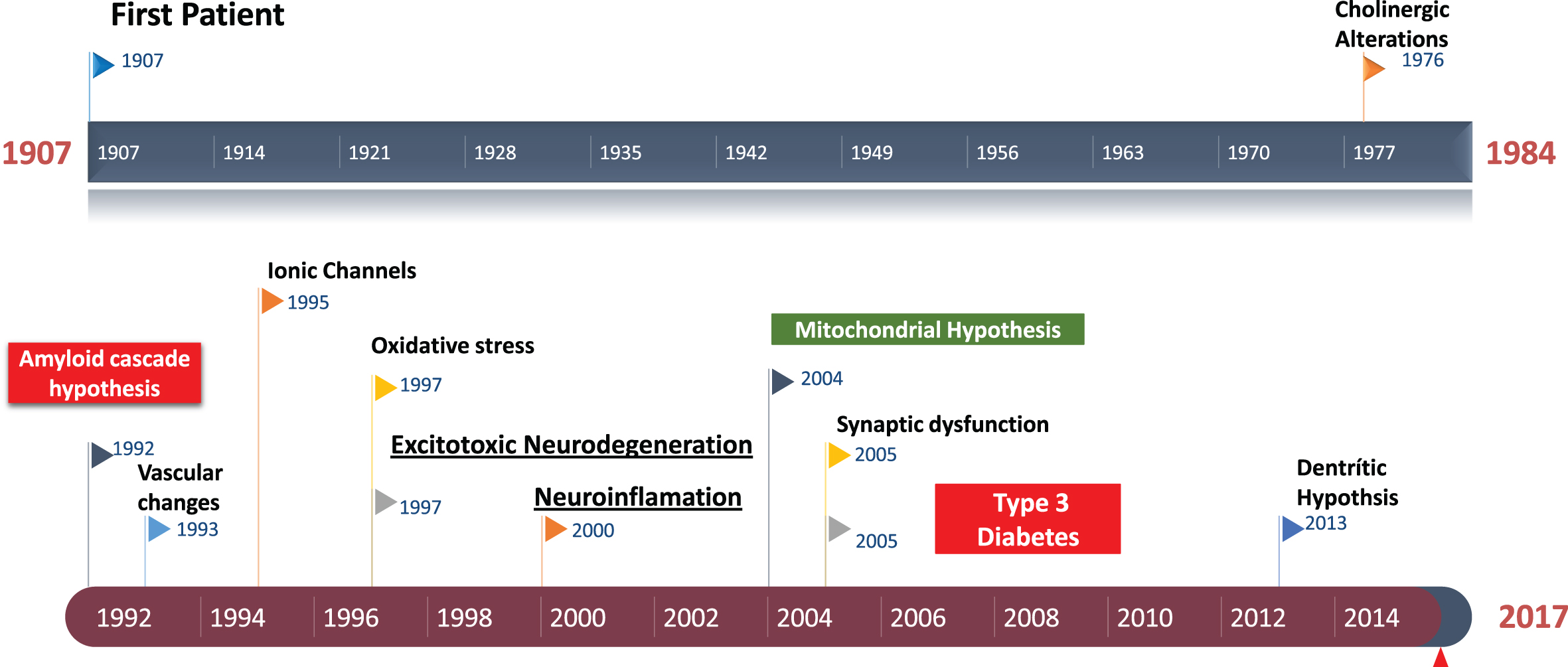
Historical perspective of Alzheimer’s disease. Amyloid cascade hypothesis was suggested in 1992. The role of excitotoxicity in AD was suggested about 1997.
However, even though there is a demonstrated role of senile plaques on this excitotoxicity that leads to AD, these events do not always correlate, and soluble Aβ might the one responsible for synaptic loss and the symptomatology of AD [35–40]. In addition, over the years it has been demonstrated that high Aβ levels in the brain lead to NFT formation, cell loss, vascular damage, and dementia [10, 40]. The presence of NFTs, which are intracellular deposits of helical paired filaments composed of abnormally hyperphosphorylated tau protein, is well characterized in AD brains [36–40]. The physiological function of tau is related to the stabilization of the microtubule polymerization and proper axon functionality and, to a lesser extent, the structuration of dendrites and dendritic spines in neurons when interacting with tubulin [41, 42]. Tau regulates axonal transport of proteins, vesicles, and organelles. In AD, tau hyperphosphorylation causes abnormal associations with tubulin, favoring cytoskeleton alterations [42]. This aberrant association causes for the filaments of tau to become insoluble and precipitate as NFTs in synapses, axonal structures, and neuronal bodies, causing neuronal death by apoptosis [40–42].
In the end, all these cascade mechanisms, along with the previously described alterations issued by the NMDAR interacting with the Aβ, add up to the appearance of dementia due to neuronal activity decline without clear evidence on the real culprit.
EFFECTS OF MEMANTINE ON THE AMYLOID CASCADE
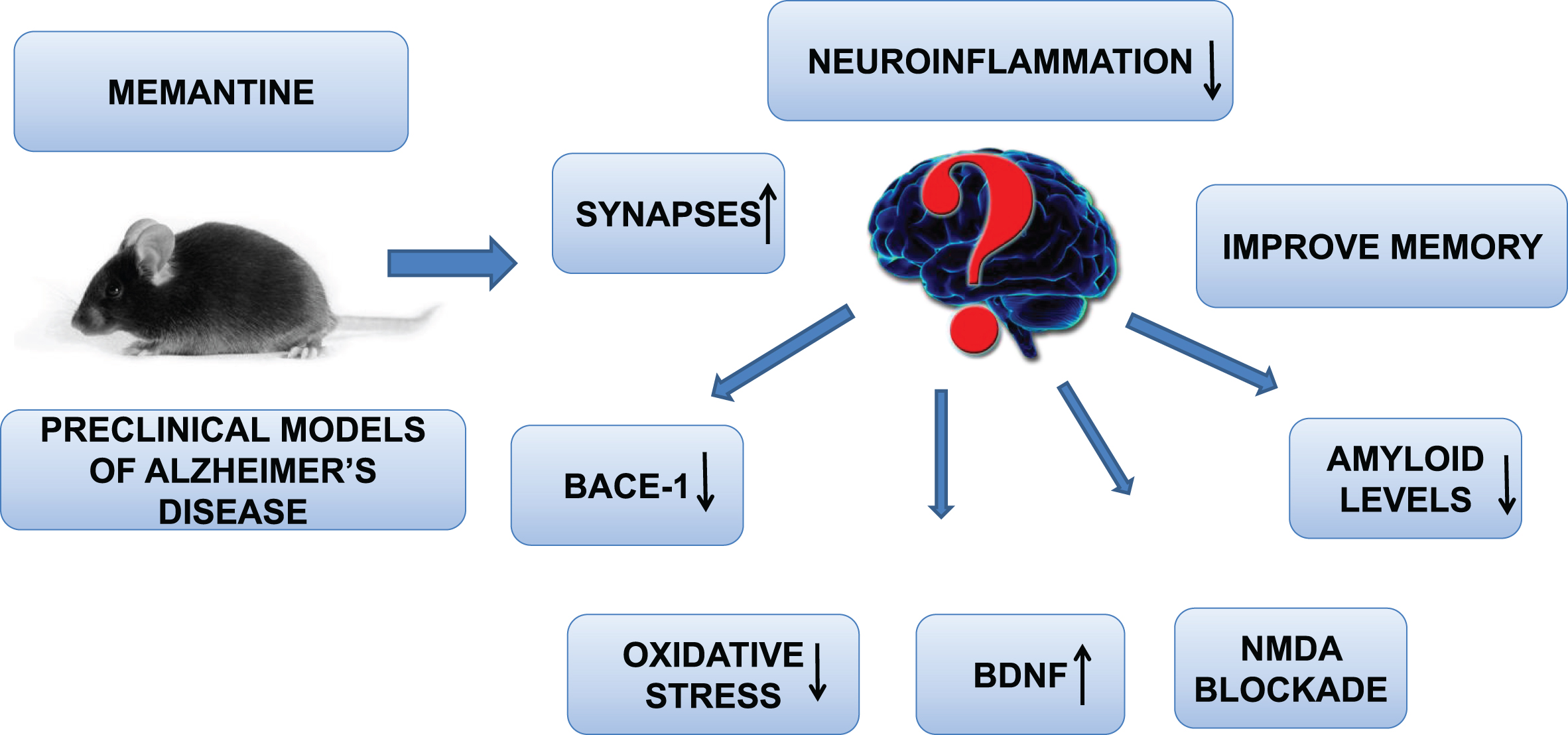
Memantine improve preclinical Alzheimer’s disease neuropathology though increasing BDNF levels, synapses, improving cognition, decreasing neuroinflammation, BACE-1 blockade, and NMDA receptor inhibition.
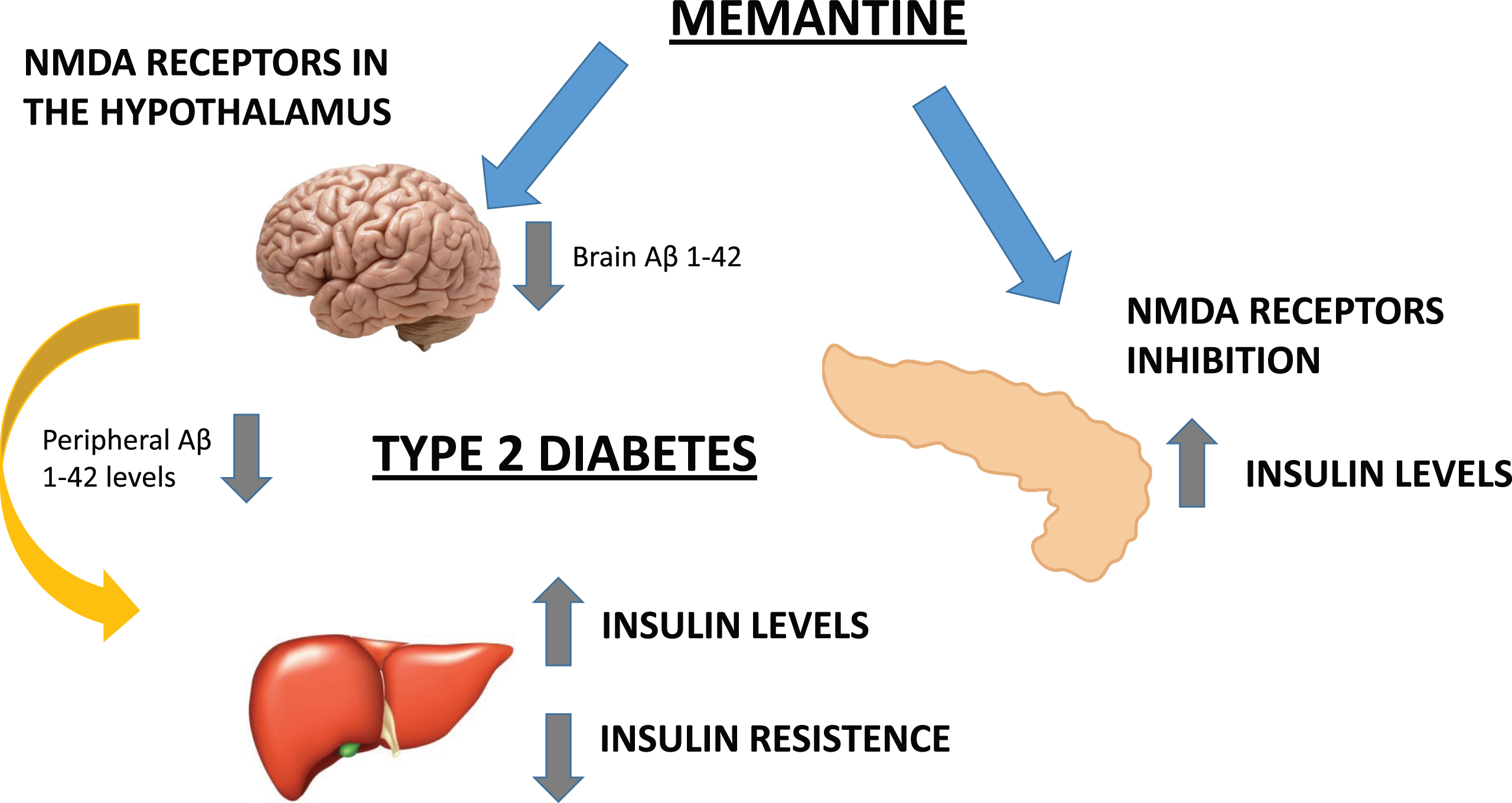
We hypothesized that the potential beneficial metabolic effects of MEM are mediated by the inhibition of peripheral (pancreatic) NMDAR and the blockade of hypothalamic Aβ1 - 42 levels. Therefore, AD could be a cognitive disorder associated to metabolic alterations both in central (insulin receptors) and peripheral areas.
Due to the negative consequences derived of the presence of compact senile plaques in the brain, they have become one of the main targets when trying to design appropriate treatments for AD. Several preclinical studies have reported that MEM leads to a reduction in Aβ brain levels or reduces amyloid plaque burden in preclinical mice models of AD, which contributes to memory improvement [43]. Specifically, 3-month-old APP/PS1 mice treated with MEM (10 mg/kg/day; i.p.) for a period of 4 months showed a significant decrease in amyloid plaque burden in the brain (cortex and hippocampus) [44]. Furthermore, MEM was effective improving short-term memory deficits in APP/PS1 mice, based in exploratory behavior results using the novel-object recognition test [44]. Moreover, Liu and colleagues treated 12-month-old APP/PS1 mice with MEM (5 mg/kg body weight) via intragastric administration and, they demonstrated that MEM treatment improved memory and learning impairment and significantly decreased Aβ deposits in mice brains [45]. Parsons and colleagues demonstrated that after a chronic in vitro treatment, MEM was accumulated in the phospholipid membrane and could have modulatory effects on membrane fluidity [46]. This effect could also modulate activity of membrane-bound enzymes, such as AβPP and BACE-1 and probably explain the anti-Aβ effects of MEM.
Recently, Ito and colleagues reported that MEM reduces the brain levels of Aβ in Tg2576 mice that have plaques and the levels of insoluble endogenous Aβ in aged F344 rat brains. Authors suggest a new mechanism involved through the modulation of AβPP trafficking and stabilization of AβPP on the cell surface, which favors a reduction in Aβ production [47].
Therefore, after evaluating the results, some authors have suggested that MEM effects on NMDAR do not only associate the neuroprotective effect observed with its administration. Some authors reported that there are additional mechanisms in MEM neuroprotection like nerve growth factor upregulation that activates the tropomyosin receptor kinase A (Trk A) signaling, which eventually inhibits p75 neurotrophin receptor (p75NTR), molecules implicated in neuronal plasticity [48]. In addition, it causes increases on brain levels of the brain-derived neurotrophic factor (BDNF) and Trk B and muscarinic receptors [48]. Furthermore, MEM attenuated Aβ42-induced loss of cholinergic neurons and microglia activation in the neocortex [49]. Interestingly, no beneficial effects were observed after subchronic MEM treatments in 5XFAD mice at 12–15 months of age [50]. Authors suggest that MEM cannot protect from memory loss at advanced disease stages due to high levels of Aβ in the tissue.
Thus, although in preclinical models it has been demonstrated that MEM memory improvement is due to reduction of cerebral Aβ levels and amyloid burden, in this study it was suggested that MEM improves hippocampal memory in young 5XFAD mice by blocking extrasynaptic NMDARs without affecting Aβ levels. In addition, authors suggest that a suitable AD treatment might be the co-administration of an Aβ-lowering therapy (β-secretase inhibitors to reduce Aβ production) together with MEM (which prevents synaptic toxicity of oligomeric Aβ). These data in experimental models of AD with old 5XFAD mice could probably explain why the clinical efficacy of the monotherapeutic MEM approach is lost in moderate to severe memory deficits in advanced AD patients [37]. Likewise, De Felice group reported the importance of Aβ oligomer species in the mechanisms of pathogenesis of AD [51]. Aβ oligomers self-assemble to form multiple types of soluble oligomers (AβOs) including low molecular weight (dimers, trimers, tetramers) and high molecular-weight species ranging in mass from 54 to 150 KDa. AβOs are involved in the process of synaptic and cognitive dysfunction in AD. In this study, authors demonstrate that MEM was effective only on high molecular-weight oligomers and concluded that the limited clinical efficacy of MEM in AD could be explained by this selectivity [51].
MEMANTINE INHIBITS NEUROFIBRILLARY TANGLES FORMATION AT PRECLINICAL LEVEL
It is well known that the process of tau hyperphosphorylation at multiple sites is controlled by several serine/threonine kinases, including protein kinase A, c-Jun N-terminal Kinase (JNK), Fyn kinase, 5’ adenosine monophosphate-activatedprotein kinase, glycogen synthase kinase 3β (GSK3β), cyclin dependent kinase 5 (CDK5), microtubule affinity regulating kinase, extracellular signal regulated kinase 2, and Ca2 + /calmodulin-dependent protein kinase II [52–57]. Thus, these kinases increase the phosphorylation of tau and prevent the ability of tau to promote microtubule assembly and facilitate the polymerization of tau into paired helical filament. The process of tau dephosphorylation in the brain is mainly regulated by the proteinphosphatase 2A.
Kamat and colleagues suggest that NMDAR activation increases tau phosphorylation by a combined process with oxidative stress. It activates the CDK5 signaling pathway and stress-activated protein kinases JNK/SAPK [58]. So, considering the elements we have already reviewed, the interaction of the Aβ peptide with the NMDAR favors an increase on the concentration of intracellular Ca2 +, which activates the CDK5 and SAPK/JNK that, in the end, induce the hyperphosphorylation of tau protein.
Briefly, the mechanisms through which kinases named above phosphorylate tau are described below.
CDK5-mediated tau phosphorylation
CDK5 belongs to the family of cyclin-dependent Ser/Thr protein kinases (CDKs) and carries out the activation and maintenance of numerous physiological functions in the nervous system, including neurite and axon growth [43]. The accumulation of Aβ, as well as oxidative stress and other mechanisms of excitotoxicity, can lead to canonical activation of calpain through significant increases of Ca2 + in the cytoplasm after NMDAR activation [59]. When active, calpain binds to CDK5 and favors its overactivation and phosphorylation of proteins such as tau. Zimmer and colleagues demonstrated that MEM prevented hippocampal activation of CDK5 after brain okadaic acid administration, which induces AD-like phenotype in rats [60].
GSK3β-mediated phosphorylation
Increasingly, AD is being linked to insulin resistance [61]. Insulin molecules bind to their receptors and activates them. The phosphoinositol 3 kinase binds to this receptor by the SH2 domain and converts the phosphatidylinositol-4,5-bisphosphate into phosphatidylinositol-3,4,5-triphosphate, which binds to the protein kinase B (AKT), a Ser/Thr kinase. AKT is then phosphorylated and activated by phosphoinositide-dependent kinase, which in turn phosphorylates GSK3β [62]. The Ser/Thr protein kinase GSK-3β contains two distinct forms: an active form (Ser9 phosphorylated), which is the most abundant in the nucleus, mitochondria, and lipid membrane, and a cytosolic inactive form. AKT inactivates GSK-3β by phosphorylating it, activating anti-apoptotic mechanisms and antioxidant defenses [63–65]. When AD causes insulin resistance, the insulin receptor is not activated, so GSK3β would be on its active form and could phosphorylate the tau and cause neuronal death [61]. De Sarno and colleagues, as well as Martinez-Coria and colleagues, reported that MEM was able to inhibit GSK3β activation in different experimental models [66, 67]. Therefore, the modulation of GSK3β phosphorylation by MEM may contributes to the therapeutic actions of this drug used in the treatment of AD. These results were more significant since MEM improved additional AD hallmarks such as cognitive dysfunction, probably also associated in part, with inhibition of tau hyperphosphorylation, which governs the major synaptic dysfunction by forming tangles.
RESULTS OF ALZHEIMER’S DISEASE APOE E4 CARRIER PATIENTS TREATED WITH MEMANTINE
Although APOE ɛ4 is a well characterized AD risk factor, the role of APOE ɛ4 as a modulator of disease progression, and its pharmacogenetic effects on AD therapies, are a matter of controversy [67].
One the one hand, for example, Han and colleagues compared the efficacy of rivastigmine patch monotherapy or MEM plus rivastigmine patch in patients with mild to moderately severe AD, presenting different polymorphisms of the APOE genotype [68]. The study demonstrated that the association MEM plus the rivastigmine transdermal patch therapy had beneficial effects on the activities of daily living (ADL) in moderately severe AD patients with the APOE ɛ4 allele [68, 69]. On the other hand, Wroolie and colleagues evaluated the effects of MEM on cognition in women with risk of AD and included a control group for the genetic risk factor for AD the APOE ɛ4 in a prospective open-label 6-month long pilot medication trial [70]. The results of this study were negative and authors concluded that this study does not support the potential usefulness of MEM in the prevention of cognitive decline ordementia [70].
CONTROL OF THE DEVELOPMENT OF INFLAMMATORY RESPONSES AND OXIDATIVE STRESS BY MEMANTINE
In AD, it has been demonstrated that neuroinflammatory responses derived of glial activation play a prominent role in the induction of oxidative stress [71]. Thus, several studies have reported a decreased risk of AD in patients taking non-steroidal anti-inflammatory drugs (NSAID) [72]. Furthermore, long-term use of NSAIDs appears to be effective in reducing the incidence of AD [72]. In addition, the vast majority of studies of MEM focus mainly on its effects on neurons, while MEM interacts with other cell types in the CNS [14, 73]. In preclinical models of this disease, neuroinflammatory markers like microglia and astroglia reactivity have been observed, yielding interesting results and showing decreases in neuroinflammatory biomarkers after MEM treatments.
Usually, in the brains of AD patients, microglial cells are highly reactive, mobile, and located close to amyloidal plaques. It has been hypothesized that this cell type could have different roles in the complex mechanisms that lead to the appearance of neuropathological conditions [71]. Firstly, microglia would have a protective role in the initial stages of the disease, phagocyting Aβ peptides. Secondly, when the disease progresses, microglia would be one of the responsible cell types in the development of proinflammatory and neurotoxic responses due to the release of molecules like cytokines [73]. The increase of substances such as TNFα and IL-1β, jointly with mediators such as nitric oxide or ROS, produced by glial cells, favor an exacerbation of AD pathology [71, 74]. In addition, IL-1β appears to induce AβPP production, increasing Aβ brain levels, which in turn would induce further interleukin production by microglia. Thus, a positive feedback cycle would be created. Therefore, although clinical trials do not support the efficacy of NSAIDs, this group of drugs or others with similar properties may have a therapeutic effect on AD [71]. Interestingly, it has been reported that MEM has an anti-inflammatory action on microglial cells through the inhibition of inwardly rectifying K+ (Kir) channels [75].
An excellent study performed by Kaindl and colleagues reported the existence of microglial NMDAR that when overactivated, would cause the development of inflammatory responses and trigger brain damage processes [76]. They suggest a key role of microglial NMDARs in the secretion of neurotoxic factors such as interleukins, TNFα, ROS, and nitric oxide. In addition, these authors hypothesize that in the brain a neuron-glia positive feedback cycle is generated through glutamate release by neurons favoring glial activation [76]. Thus, MEM, through the blockade of glial NMDAR, could exert neuroprotective effects through anti-inflammatory mechanisms. This point was further demonstrated by De Felice and colleagues who reported that neuronal hippocampal cultures treated with Aβ oligomers favor an increase in ROS formation through NMDAR activation [77]. They demonstrated the neuroprotective effects of MEM showing results of protection against synapsis loss.
Wu and colleagues reported that MEM increases the release of glial cell-derived neurotrophic factor (GDNF) from astroglia, which may account for its neuron survival-enhancing effect in primary neuron–glia cultures [78]. Caumont and colleagues also demonstrated on an in vitro study, that MEM administration in a C6 glioma cell line increased GDNF production, suggesting that MEM neuroprotective effects in brain neurons could be also due to the release of neurotrophic factors from glial cells [79]. Interestingly, neurotrophic effects of MEM were significantly inhibited when a neutralizing GDNF antibody was added to the cell cultures. Willard and colleagues reported that MEM also attenuated cytotoxic effects [80]. Specifically, MEM alleviated inflammatory processes mediated by chronical lipopolysaccharide administration into the basal forebrain of mice [81–83]. Consequently, it has been suggested that a chronical combined administration of MEM and NSAIDs could be a suitable strategy to delay the onset or slow the progression of AD [83].
ALTERNATIVE APPLICATIONS OF MEMANTINE IN OTHER PATHOLOGIES RELATED TO THE MODULATION OF BRAIN BDNF LEVELS
Meisner and colleagues were the first group to report new significant pharmacological actions of MEM unrelated with NMDAR blockade, the modification brain protein expression of BDNF [48]. In fact, they demonstrated that when administering MEM to rhesus monkeys (Macaca mulatta) infected with immunodeficiency virus (HIV), brain protein levels of BDNF increase. Likewise, studies with other NMDAR antagonists suggest that they can modulate the levels of brain BDNF, pointing to a potential role of glutamate in the activity of this neurotrophic factor [84]. Interestingly, several preclinical studies have evidenced that in addition to its applications for AD, MEM displays antidepressant-like effects in rodents subjected to experimental models of depression [85]. In addition, the co-administration in rodents with MEM and ketamine or MEM with sertraline, increased BDNF protein levels in the hippocampus of rats [86, 87]. In addition, using other NMDAR antagonists, the association of fluoxetine and amantadine also induced robust significant increases of both BDNF protein and mRNA gene expression in rodent brain [88]. An additional benefit of the use of MEM that is not related to neurodegenerative diseases, is its therapeutic application in the treatment of alcoholism. Specifically, it has been hypothesized that the results would be the consequence of the effects of MEM on BDNF and its role in alcohol consumption [89]. Finally, since BDNF is an inductor of the expression of markers for synaptogenesis, as well as, is a regenerator of on synaptic sites, Zhu and colleagues evaluated the effects of MEM in preclinical models of Parkinson’s disease induced by the administration of 1-methyl-4-phenyl-1.2.3.6-tetrahydropyridine [90]. This models causes for reductions of BDNF levels in the hippocampus, usually being associated with reductions to long-term potentiation. MEM treatment increased BDNF expression and restored long-term potentiation[90].
To summarize, all this data related to the effects of MEM on BDNF opens new possibilities on its possible applications for the treatment of neurological-neurodegenerative diseases [91].
MEMANTINE TREATMENT HAS EFFECTSBOTH IN BRAIN AND PERIPHERAL METABOLIC BIOMARKERS RELATED TO DIABETES AND INSULIN METABOLISM
Some studies have reported a close association between late onset AD and type 2 diabetes, a disorder usually related with obesity, going so far as demonstrating already severe impairments of glucose metabolism in mild cases of AD [8, 92–97]. Thus, lifestyle, particularly in terms of physical activity and diet, has important effects on health and cognitive processes, during aging in humans [13]. However, the interaction between obesity, aging, and AD is complex, with some evidence pointing out to the negative effects of obesity in early adulthood. Specifically, some authors have demonstrated that the development of obesity in those ages substantially increases the risk of developing AD [93–96]. This point is very interesting, since recent data suggests that AD should be considered as an affection of the entire organism and not only the brain, possibly having a partial origin in peripheral metabolic alterations [94, 95].
Following the same line of thought, Marquard and colleagues reported the presence of NMDARs in pancreatic β-cells [93]. These authors demonstrated that dextromethorphan (NMDARs antagonist) regulated insulin secretion in pancreas [93, 94]. Dextromethorphan blocked NMDARs, preventing the activation of K+ channels, reducing its efflux from the cell, and prolonging the time the depolarized state of β-cells [93, 97]. Consequently, intracellular Ca2 + concentration increased leading to insulin release and improvement of the diabetic state. However, even though the results are very interesting, it must be taken into account that strong NMDAR antagonists have considerable cognitive side effects and MEM, being a partial antagonist, might be a better choice for long-term drug usage [97–99].
Considering these results, the use of MEM could not only alleviate the symptomatology in the central nervous system but also in peripheral tissues. In our research group, we have studied the effects of MEM on APPswe/PS1dE9 mice fed with a high-fat diet (unpublished results). The animals were fed with a high-fat diet until 7-8 months of age and they were treated with MEM-supplemented water (30 mg/kg) during the last 12 weeks. In this study, MEM improved peripheral metabolic parameters like insulin resistance. Moreover, MEM reduced the accumulation of Aβ (mechanism already described in this review). This result is especially significant since Arrieta-Cruz reported that rat in vivo hypothalamic Aβ25 - 35 administration altered peripheral regulation of glucose, showing that Aβ plays an important role in regulating glucose homeostasis [100, 101]. Correspondingly, it has been reported that deposits of Aβ peptides can be found in different non-neuronal tissues such as heart, liver, and pancreas as it was reported by Troncone and colleagues [102, 103]. Zhang and colleagues demonstrated in the APP/PS1 mice model of familial AD, that Aβ induces insulin resistance in hepatocytes suggesting an important role of peripheral Aβ in modulating systemic insulin sensitivity and glucose metabolism (Aβ would bind directly to the insulin receptor, affecting its signaling [104, 105]. All these studies are of importance because they reinforce the evidence that late onset AD should be considered as a metabolic disorder denominated type 3 diabetes [8].
Finally, De Felice described that the neuroinflammatory process mediated mainly by the cytokine TNFα could be another mechanism though which insulin signaling would be affected [5]. TNFα would bind to its receptor and induce a signaling cascade that would activate the JNK kinases among others [13]. The JNKs, specifically the JNK1 isoform, would be a direct inhibitor of the insulin receptor substrate 1, the substrate of the insulin receptor, being its activation cause for a reduction of insulin signaling and glucose up-take, an already described symptom of possible appearance of AD [5, 96].
In the end, all the data adds up to the conclusion that there is a real interaction between the central and peripheral systems and that; MEM seems to be an interesting choice to alleviate multiple biomarkers, peripheral and central, at once.
CLINICAL TRIALS WITH MEMANTINE
It is clear that although preclinical MEM treatments for AD are hopeful, the clinical results are ineffective, none of them cure, and at most, they are able to slow the process of cognitive loss [106]. For example, in a clinical study (Clinical Trials identifier: NCT00322153), 28 mg of MEM were administrated daily orally for 24 weeks [107]. The authors used the Severe Impairment Battery, a neuropsychological test for severely demented patients, for the assessment of cognitive function. The authors concluded that high doses of MEM provided behavioral benefits in patients with moderate to severe AD treated in combination with an acetylcholinesterase inhibitor (AChEIs). Howard and colleagues in a clinical study obtained similar results using donepezil (reversible AChEIs) in combination with MEM [108]. Thus, the combined therapy of AChEIs with MEM has the potential to provide benefits that either drug alone cannot produce. Galantamine is also a possible choice for these combined treatments since it inhibits acetylcholinesterase activity in the synapses and can modulate the nicotinic receptor. In a clinical study, the combinatory regimen galantamine plus MEM was evaluated in patients with mild to moderate probable AD. It was a clinical multicenter, double blind, randomized, parallel-group trial of 12 months of duration (Clinical trial: NCT01921972) [109]. The objective was to evaluate the efficacy of a combination of galantamine 24 mg/day plus MEM 10 mg b.i.d. (twice a day) with a dose titration of sixteen weeks. However, results of this study still have not been reported. If the expected results are positive, the drug association should have increased synaptic activity and prevented excitotoxicity. Another reported approach has been the combination masitinib (tyrosine kinase inhibitor; 3–6 mg/kg/day) with MEM or/and cholinesterase inhibitors (donepezil, rivastigmine, or galantamine) (Clinicaltrials.gov NCT00976118) that significantly improved the cognitive response [28]. As a result, the first conclusion we can obtain from these results is that more studies are necessary in order to evaluate potential combinatory treatments in order to delay or prevent AD, which, after all, is a multifactorial disease and might need to be treated through multiple targets.
Annweiler and colleagues reported that the association of MEM plus vitamin D supplements had a statistically and clinically significant effect on the gain of global cognition improvement over AD symptoms [110, 111]. Likewise, the authors demonstrated that the combination was more effective than taking either substance alone. Interestingly, it has been reported that vitamin D shows neuroprotective effects such as antioxidant action, inhibiting the synthesis of inducible nitric oxide synthase, regulating the activity of γ-glutamyl transpeptidase, increasing the synthesis of nerve growth factor, showing anti-inflammatory properties and also attenuating Aβ1 - 42 accumulation [111]. Therefore, the authors suggest that this association could act by a synergic effect increasing the neuroprotective action of MEM and, overall, providing a more complete protection in patients exposed to glutamatergic excitotoxicity. In addition, a new recent clinical trial (NCT01409694) named Alzheimer’s disease Input of Vitamin D with mEmantine Assay (AD-IDEA) is being analyzed; however, no data have been reported from this study about the efficacy of the association improving cognitive function. Recently the same group reported in a clinical trial that MEM cognitive improvement depended on the plasmatic levels of vitamin D [112].
Interestingly MEM has been evaluated in the treatment of frontotemporal dementia (FTD) [113–115]. Lindquist and colleagues reported a familial case of a patient with FTD, an adult neurodegenerative disorder with dementia and behavioral disturbances treated with MEM with a maintenance dosage of 10 mg twice a day. The authors suggest that, in this specific case of a patient with pathogenic mutation (R406W) in the microtubule associated protein tau (MAPT) gene, MEM treatment could stabilize the progression of symptoms of disease [113]. However, a multicenter, randomized, double-blind, placebo controlled clinical trial (NCT00545974) evaluated the efficacy of MEM in mild-to-moderate FTD and the main conclusion of this study was that MEM treatment do not ameliorate these dementia symptoms (Table 1) [114].
Examples of clinical research data reported with memantine
MEM, memantine; MRI, magnetic resonance imaging; AD, Alzheimer’s disease; MRS, magnetic resonance spectroscopy; CSF, cerebrospinal fluid; PD, Parkinson’s disease; MCI, mild cognitive impairment; HD, Huntington’s disease; FTD, frontotemporal dementia; DLB, dementia with Lewy bodies.
Since Parkinson’s disease (PD) dementia and dementia with Lewy bodies have some pathological and biochemical similarities to AD, MEM have been proposed that could provide an improvement in these brain disorders (Table 1). Thus, the clinical trial NCT00855686 evaluated the efficacy and safety of MEM (20 mg per day over 6 months) in patients with mild to moderate PD dementia or dementia with Lewy bodies (DLB). In this study, patients were stratified according to diagnosis and randomly assigned to MEM or placebo in a double blind, parallel-group study. The main conclusion of this study was that the incidence of adverse events of patients treated with MEM was low and similar to the placebo group. The authors suggest that MEM could be a suitable strategy for mild to moderate DLB [115].
As a note of interest, and as it can be seen in Table 1, MEM has been evaluated as a potential therapy for other diseases such as PD. This is because MEM is an organic molecule of an aminoadamantane class similar to amantadine, which produces moderate symptomatic benefits in PD, related to tremor, tardive dyskinesia, chorea, and dementia [116]. However, the potential benefits of this drug are not mediated through the potentiation of dopamine effects, so further studies are necessary. Serafini and colleagues evidenced that the glutamatergic system could be involved in the pathology and treatment of depression via the modulation of post-synaptic ionotropic receptors including the NMDAR [86]. Thus, MEM has been reported that in association with sertraline, a selective inhibitor of serotonin reuptake, it shows a suitable efficacy profile in patients with major depression [88, 117–119]. Likewise, previous preclinical reported data demonstrated that the association of MEM with antidepressant drugs, such as fluoxetine and venlafaxine enhances the antidepressant effect of classical therapies. MEM associated with galantamine was investigated in the treatment of cognitive deficits in with schizophrenia [120, 121]. Others clinical trial evaluated, a part of MEM, additional non-pharmacological strategies for example the Environmental Ecological Therapy in AD [122, 123].
In addition, MEM has been evaluated for cognitive impairment associated with multiple sclerosis patients, and the objective is that MEM could improve the ability to work in severe cases of this devastating disease [124]. The results reported in Huntington’s disease from a randomized, placebo-controlled study, which examines both efficacy and safety of MEM during long-term administration (52 weeks) in non-demented multiple sclerosis patients, were negative for cognitive impairment improvement. Ondo and colleagues reported that the administration of 20 mg of MEM daily significantly improved motor symptoms, but failed to improve patient’s cognition [125].
Cankurtaran and colleagues reported a case study of a 32-year-old woman with Huntington’s disease where the drug association of risperidone with MEM was effective in the alleviation of cognitive deficits in this patient [126].
CONCLUSION
Although MEM shows an adequate profile in preclinical AD models, clinical data have not reported satisfactory results for curing the disease [127, 128]. Both cholinergic and glutamatergic dysfunctions are currently key neurochemical targets for the symptomatology of AD but drugs that alter these two pathways seem to have no real applications in the clinical cure of this disease, including MEM. Thus, the design of combined treatments seems to be the method to improve the current situation [129, 130]. For example, therapy with AChEI and an MEM is an appropriate treatment strategy as the disease progresses, since there is a lack of other treatment options in moderate to severe AD [129, 131].
The neuroprotective mechanisms of MEM in AD against neuronal loss (apoptosis decrease and mitochondrial alteration) and reduction of Aβ production could be attributed, at least in part, to its selective action blocking extrasynaptic NMDAR. Furthermore, in addition to extrasynaptic NMDAR blockade, recent preclinical data provides promising results about the brain decrease of Aβ accumulation by MEM in transgenic animals and age normal animals. The new mechanism proposed by MEM would be the reduction in Aβ production through the regulation of endocytosis of AβPP, which affects Aβ production in neurons. The prevention of Aβ excessive generation is important, because the vast majority of the preclinical studies evaluated reported that MEM attenuates or reduces soluble AβO levels and fibrils and plaque generation, which is related with the process ofmemory improvement.
Likewise, MEM inhibition of tau phosphorylation could be a process mediated also by the blockade of extrasynaptic NMDAR and the prevention of intracellular calcium increase. The activation of extrasynaptic NMDAR by glutamate and/or Aβ leads to an increase in intracellular Ca2 + into the postsynaptic cells. Intracellular Ca2 + activates calpain and CDK5 pathway involved in tau phosphorylation. Furthermore, this process can lead to excessive neuronal oxidative stress formation that can destroy synaptic connections between neurons (memory loss) and induce activation of kinases involved in tau phosphorylation. It has also been reported that MEM have beneficial effects in preclinical AD models by decreasing the activation of microglia in the rodent brain and by increasing the production of trophic factors.
In addition, recent reports suggest a link between type 2 diabetes and AD, currently denominated as type 3 diabetes where the alteration of hippocampal insulin receptor favors cognitive loss. Interestingly, pancreatic NMDAR are involved in insulin release, which leads to its use in the alleviation of these situations of insulin resistance. Furthermore, we hypothesized that the beneficial metabolic effects of MEM are mediated probably by an inhibition of peripheral (pancreatic) NMDAR and the blockade of hypothalamic Aβ1 - 42 levels. Therefore, in the end it seems that AD is a cognitive disorder associated to metabolic alterations both in central and peripheral areas. Considering this hypothesis, we suggest that a combined treatment with MEM and another drug with antidiabetic effects could be a potential strategy to delay AD progression.
However, the lack of clinical efficacy of MEM in AD may be due to the administration of the drug too late when the disruption of neurons and/or synapses is advanced and impossible to reverse. Other possible explanations might be: 1) MEM needs to be administered with other drugs (antioxidants, anti-inflammatory, GSK3 inhibitors, and others) in order to potentiate its effect on the delaying and prevention of the disease; and 2) MEM is effective in the treatment of neurons but it is hard to determine whether they remain active.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Spanish Ministry of Science and Innovation PI2016/01, CB06/05/0024 (CIBERNED), the European Regional Development Founds and MAT 2014-59134-R project. NIA 1R15AG050292 from GC. Research team from UB and URV belongs to 2014SGR-525 from Generalitat de Catalunya. ESL and MLG belong to 2014SGR-1023 and ESL, acknowledges the PhD scholarship FPI-MICINN (BES-2012-026083). CBZ is supported by grants from CONACyT Mexico (No. 0177594) and RDCT from Postdoctoral fellowship CONACYT No. 298337 and the Doctoral Program in Sciences in Molecular Biology in Medicine, LGAC Molecular Bases of Chronic Diseases-Degenerative and its Applications (000091, PNPC, CONACyT).