
Abstract
Iron is essential for cellular development and maintenance of multiple physiological processes in the central nervous system. The disturbance of its homeostasis leads to abnormal iron deposition in the brain and causes neurotoxicity via generation of free radicals and oxidative stress. Iron toxicity has been established in the pathogenesis of Parkinson’s disease; however, its contribution to multiple system atrophy (MSA) remains elusive. MSA is characterized by cytoplasmic inclusions of misfolded α-synuclein (α-SYN) in oligodendrocytes referred to as glial cytoplasmic inclusions (GCIs). Remarkably, the oligodendrocytes possess high amounts of iron, which together with GCI pathology make a contribution toward MSA pathogenesis likely. Consistent with this observation, the GCI density is associated with neurodegeneration in central autonomic networks as well as olivopontocerebellar and striatonigral pathways. Iron converts native α-SYN into a β-sheet conformation and promotes its aggregation either directly or via increasing levels of oxidative stress. Interestingly, α-SYN possesses ferrireductase activity and α-SYN expression underlies iron mediated translational control via RNA stem loop structures. Despite a correlation between progressive putaminal atrophy and iron accumulation as well as clinical decline, it remains unclear whether pathologic iron accumulation in MSA is a secondary event in the cascade of neuronal degeneration rather than a primary cause. This review summarizes the current knowledge of iron in MSA and gives evidence for perturbed iron homeostasis as a potential pathogenic factor in MSA-associated neurodegeneration.
INTRODUCTION
Multiple system atrophy (MSA) is a severe, fast progressing neurodegenerative disease characterized by parkinsonism, cerebellar ataxia, and autonomic failure. According to the predominant motor presentation, patients are categorized as MSA-Parkinson variant (MSA-P) or MSA-cerebellar variant (MSA-C). Neuronal loss consistent with pronounced striatonigral degeneration or olivopontocerebellar atrophy reflects the predominant motor phenotype [1]. Additionally, affected brain areas responsible for autonomic or non-motor features are found in the brain stem and spinal cord [2]. Neuroinflammation, oxidative stress (OS), and iron accumulation are increasingly recognized as pathological features of MSA inducing neuronal loss [3, 4].
The diagnostic cellular hallmark of MSA are oligodendroglial cytoplasmic protein aggregates mainly composed of misfolded, hyperphosphorylated, and nitrated α-synuclein (α-SYN) referred to as glial cytoplasmic inclusions (GCIs) [5]. MSA, together with idiopathic Parkinson’s disease (IPD) and dementia with Lewy bodies (DLB), belongs to the group of α-synucleinopathies, which are all characterized by intracellular aggregations of α-SYN. However, the distribution of α-SYN deposits is different among this group of pathologically linked diseases; in IPD and DLB α-SYN deposits are found in neurons referred to as Lewy bodies and Lewy neurites, whereas in MSA oligodendrocytes are the main cells of attack. The density of GCIs is correlated with neuronal loss in affected brain regions and with disease duration [1, 6]. Thus, GCIs are believed to play a major role in the pathogenesis of MSA. However, the underlying etiology and pathogenesis of α-SYN aggregates and neurodegeneration are still elusive.
Dyshomeostasis in transitional metals has been proposed to be involved in neurodegenerative disorders, whereupon iron has received special attention: Increased iron levels and consecutive high levels of OS were proposed to contribute to neuronal loss in Parkinson’s disease, multiple sclerosis and AD [7–11]. Interestingly, a meta-analysis on transitional metals in AD brought up controversies as to which extend iron levels are elevated and that a bias toward reporting increased iron content in AD in review articles might play a role [12].
High iron content in degenerating brain regions as well as regulatory interactions between iron and α-SYN biology, via ferrireductase activity of α-SYN and iron mediated translational regulation of α-SYN involving iron responsive elements (IREs) within the 5’ untranslated region of α-SYN mRNA, have resulted in the idea that iron may contribute to the pathophysiology of MSA [9, 13–19]. However, the relationship between iron accumulation and α-SYN aggregation has poorly been investigated and the mechanisms of iron dyshomeostasis in respective brain regions remain elusive. Additionally, it is unclear whether increased iron levels are the cause of neurodegeneration or rather an epiphenomenon of such. In this review, we have collected data on iron accumulation/dysmetabolism in the brain and its association with OS and neuroinflammation found in MSA. Moreover, the putative functional interactions of α-SYN and iron are highlighted. Before discussing brain iron metabolism in healthy people and MSA patients we highlight excellent reviews that provide a comprehensive understanding on the topic of iron metabolism and toxicity [20–30].
BRAIN IRON IN HEALTHY PEOPLE
In the central nervous system (CNS), iron is essential for synthesis of neurotransmitters including dopamine, serotonin, and gamma-aminobutyric acid, and it is involved in the differentiation of oligodendrocytes and myelination [31–33]. Transit of iron to the CNS via the blood-brain barrier and the blood-cerebrospinal fluid (CSF) barrier is coordinated and mostly accomplished by transferrin receptor (TfR)-mediated endocytosis [34]. However, additional uptake mechanisms have been suggested including cellular uptake of ferritin favoring H-Ferritin over L-Ferritin [35] and by the divalent metal transporter (DMT1) [36]. Regarding iron export from the intracellular endosome and the capillary endothelial cells controversies exist to whether DMT1 and ferroportin (FPN) are involved [7, 37–41]. In the interstitial fluid of the brain, iron is bound to transferrin (Tf) which is produced by oligodendrocytes and circulates to target cells within the CNS [8, 20].
Inside the brain there seems to exist cell specific expression profiles determining iron uptake, utilization and storage mechanisms [20, 43]. Neurons possess TfR, DMT1, ferritin, FPN, ceruloplasmin, and hephaestin. Astrocytes express DMT1, ferritin, FPN, and ceruloplasmin. Much less is known about transport mechanisms of microglia cells, although they have capacities to acquire, store and release iron [44, 45].
Ferritin serves as the dominant iron storage protein in the CNS and highest levels are detected in oligodendrocytes, followed by microglia and neurons. The smallest amount is found within astrocytes [44]. Hence, under normal conditions oligodendrocytes are the iron richest cells among brain cells and are therefore regarded the iron regulatory cells in the CNS [44, 46–48]. Nevertheless, the function of the high iron content held within ferritin in oligodendrocytes is insufficiently understood so far. Both isoforms of ferritin are found in oligodendrocytes, albeit H-Ferritin seems to be the major source of iron in oligodendrocytes [49]. Oligodendrocytes acquire iron by TfR uptake and H-Ferritin by receptor induced endocytosis during their maturing process (progenitor cells). Since mature oligodendrocytes do not express TfR alternative iron uptake mechanisms must exist and the more recently reported ferritin (Tim-2) receptor pathway (uptake of H-Ferritin) appears to be a likely candidate [48–52]. Among neurons and microglia, a unique distribution of H- and L-ferritin is observed in association with utilization requirements where H-Ferritin is predominantly found in neurons and L-Ferritin in microglial cells [33]. Further, CNS distinct storage arrangements are found in neuromelanin (NM) containing cells of the substantia nigra (SN), and the locus coeruleus (LC) [53]. Similar to systemic iron homeostasis, brain cellular iron metabolism is also operated by IREs and iron responsive proteins (IRPs) and mutations/deletions of IRP2 have been demonstrated to result in neuronal damage [28, 54].
It is important to note, that in the CNS iron distribution is distinct among different brain regions and cell populations and that iron accumulation in the CNS is a phenomenon of the aging brain [28, 55]. Not only the levels of iron but also the degree of iron accumulation vary in different brain regions. Physiological distribution of iron has been extensively studied by different histological, chemical and spectro-analytical procedures using Prussian blue/Perls’ stain, atomic absorption spectroscopy, inductively coupled plasma spectroscopy, instrumental neutron activation, and colorimetry as well as magnetic resonance imaging (MRI) based techniques. There are multiple contributors to MRI signals, particularly at iron concentrations in the range of white matter and cortical grey matter. In the areas of the brain with higher iron concentration, like the basal ganglia, the bulk of the signal on these iron-sensitive MR sequences is thought to originate from iron, although there certainly remains the possibility of other elements contributing. Highest levels of iron are found in the extrapyramidal system: globus pallidus (GP) (lateral > medial part) > SN > putamen > caudate nucleus (NC) > hippocampus > amygdala > grey > white matter of cerebral cortex = cerebellum [56–63]. Advanced MRI techniques offer in vivo brain iron estimations and confirmed postmortem biochemical studies [64–69]. Additional changes of cellular storage capacities among neurons and glial cells and their different storage modalities including ferritin, NM, and hemosiderin are not fully understood [44]. Unlike microglia, astrocytes, and neurons, oligodendrocytes do not physiologically accumulate iron upon aging [44]. The cause of cell specific iron accumulation upon aging is unclear. However, the fact that extrapyramidal brain regions tend to contain more iron than non-motor linked regions [70] has led to the association of iron accumulation and movement disorders. In the context of IPD in which severe neuronal loss is found in SN, detailed studies on iron, storage proteins ferritin and NM have been performed and indicated that physiologically the SN is more prone to iron induced damage due to accumulation of iron, ferritin and NM in contrast to LC [44, 71–74].
BRAIN IRON IN MSA
MSA has been proposed to be a primary oligodendrogliopathy due to the pathological hallmark of cellular inclusion bodies mainly composed of α-SYN predominating in oligodendroglial cells [75]. Oligodendrocytes are the iron richest cell population in the CNS and there is compelling evidence that iron plays a decisive role in the pathogenesis of MSA being associated with regions of neurodegeneration and α-SYN aggregation in the context of OS and neuroinflammation.
We here review histopathological and imaging data on the potential implications of iron dyshomeostasis in MSA pathology. Postmortem studies determining tissue iron, mainly used histochemical methods including Prussian blue/Perls’ stain as well as immunohistochemistry targeting iron related proteins (i.e., ferritin). In vivo estimations of brain iron can be obtained by MRI (see below).
Histopathological findings in MSA
Histopathological studies in MSA are scarce, but they all clearly demonstrate that iron deposition in the putamen of MSA patients is a hallmark of the disease [76]. To this end, several postmortem analyses revealed increased iron content and associated neuronal loss particularly in the putamen, while some authors also made these observations in the SN, GP, and NC of MSA patients [59, 77–81]. In MSA, iron accumulation in putamen is accompanied by elevated levels of ferritin [59, 82]. Less pronounced increase of ferritin immunoreactivity is also found in SN in MSA [59].
Regarding the actual amount of iron in basal ganglia of MSA patients, varying data exist possibly due to different methodologies applied to measure tissue iron. Nevertheless, there is evidence that iron content in putamen, GP, and SN is higher in MSA than in IPD, DLB, and controls, being similar to the levels found in progressive supranuclear palsy (PSP) as assessed by qualitative measurements of Jellinger in 2003 [59, 81]. The higher iron levels in SN of PSP and MSA >IPD were suggested to be associated with shorter disease duration and pronounced vulnerability to iron induced cellular damage. Hence, it was proposed that disease duration might be correlated inversely to brain iron accumulation [83]. Aside from basal ganglia, diffuse ferritin deposition was observed in the dentate nucleus of cerebellum of MSA brains [84]. Interestingly, alterations in iron concentrations have not been documented in LC, albeit severe neuronal loss was described in MSA neuropathology [81]. The source of increased ferritin levels in GP and SN of MSA is not known; evaluation of ferritin levels in the CSF of 15 MSA patients revealed no significant change compared to healthy individuals [85] making the CSF as source very unlikely.
In contrast, increased total iron levels have been linked to decreased ferritin and copper content, and increased zinc levels in the SN of IPD suggesting that free bioavailable iron and altered iron handling contribute to neurodegeneration in this area [86]. There are conflicting reports about the stage of disease progression at which nigral iron changes occur, which concurs with the notion that IPD represents different subgroups with different potential pathogenic pathways [83, 87]. Very recently, it has been suggested, that autophagy dysfunction and, less, OS are critical in the iron-induced pathogenesis and iron-induced α-SYN pathology of IPD. Maintaining proper activity of the autophagy pathway is essential for eliminating aberrant protein aggregates like α-SYN [88]. Whether this mechanism of iron induced α-SYN pathology by causing autophagy dysfunction plays a role in MSA remains to be investigated.
Ceruloplasmin acts as a ferroxidase and is essential for the functionality of FPN mediated cellular iron efflux. In two autopsy-proven MSA cases with hypoceruloplasminemia and two MSA controls increased iron deposition in SN and putamen was found, but neither in pons, cerebellum nor in the inferior olive [80, 89]. Although hypoceruloplasminemia and mutant ceruloplasmin have been demonstrated in MSA patients, the role of ceruloplasmin in the pathogenesis of MSA is still elusive [80, 90].
Further analyses of iron and its storage and utilization proteins support the idea of a reduction in bioavailable iron occurring in MSA [91]. Visanji and colleagues performed a detailed postmortem analysis of human brain tissue (MSA, IPD, and control) assessing iron, ferritin, TfR, and FPN distribution in pons, putamen, and SN of MSA tissue and detected region-specific differences: Increased tissue iron, increased ferritin, and decreased FPN were found in pons of MSA tissue and was regarded disease specific. A dysregulation of iron export coupled with an increase in ferritin iron was detected to a lesser extent also in putamen in MSA. A limiting factor of this study may be the small number of only 3 MSA and 3 control subjects [91]. It remains unclear whether neuroinflammation induced elevation of ferritin and consecutively increased intracellular iron levels occur because inflammatory signals and stimuli raise ferritin and decrease FPN expression or whether elevated iron accumulations induce ferritin production and activates microglia via OS [91, 92]. Moreover, it has been speculated that the expression pattern of iron proteins in pons of MSA (with increased tissue iron, increased ferritin and decreased FPN indicating reduction of bioavailable iron) may reflect neuroinflammation with associated induction of hepcidin. Hepcidin originating from the liver acts as a systemic hormone or hepcidin can be produced locally following inflammation-driven generation in macrophage like cells such as microglia then acting in a paracrine fashion resulting in tissue iron retention via FPN downregulation and consequently reduced extracellular iron bioavailability [93, 94].
In summary, histopathological data on iron metabolism in MSA are very limited and number of MSA cases in postmortem studies is rather small. The only truly quantitative assessment of iron concentration in MSA was done by Dexter and colleagues in 1991 and that quantitative iron levels in the putamen in MSA is known from only 8 cases [59]. Additionally, variable histochemical methods make comparisons among studies difficult. More work is needed to corroborate the postulated increase of iron levels in the basal ganglia of MSA patients with putamen possessing highest amounts accompanied by increased ferritin content. Considering that iron bound to ferritin is non-reactive, the findings of increased iron content linked to elevated ferritin levels found in the putamen and SN of MSA patients suggest that a reduction of free – bioavailable iron may occur in these areas. However, the mechanism how iron dyshomeostasis contributes to neurodegeneration and neuroinflammation in MSA remains elusive.
Table 1 gives an overview of histopathological studies investigating iron in MSA.
Summarizes histopathological studies investigating iron metabolism/dysregulation in MSA
AD, Alzheimer’s disease; FPN, ferroportin; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; GP, globus pallidus; HC, healthy controls; HD, Huntington’s disease; LC, locus coeruleus; MSA, multiple system atrophy; NC, caudate nucleus; ns, not significant; IPD, idiopathic Parkinson’s disease; PDD, Parkinson’s disease with dementia; PSP, progressive supranuclear palsy; SN, substantia nigra; SNc, substantia nigra pars compact; TfR, transferrin receptor; IHC, immunohistochemistry; GFAAS, Graphite Furnace Atomic Absorption Spectroscopy; IRM, isothermal remanent magnetization.
Iron imaging findings in MSA
Brain iron can be visualized in vivo using MRI [95]. Iron sensitive imaging sequences include T2/T2*, susceptibility-weighted imaging (SWI), and R2* (= 1/T2) [65, 96]. However, these imaging sequences cannot distinguish between different forms of iron (heme-bound iron; ferritin-bound, Tf-bound, free iron) [65, 97]. Nonetheless, it could be demonstrated that higher magnetic field MRI (3T) is able to more precisely localize iron accumulation within the putamen in MSA [98]. Iron deposits selectively reduce T2 signals (spin echo sequence) appearing as hypointensity in T2 weighted images [79]; whereas gliosis results in hyperintense MRI signal. Superior to T2 imaging in terms of iron detection is T2*-weighted (gradient echo) sequence analysis due to its higher sensitivity for magnetic susceptibility changes [99] similar to R2* and SWI sequences. More recently, development of quantitative susceptibility mapping (QSM) has been introduced in order to quantify brain iron in vivo. However, to our knowledge QSM studies have not been published in MSA so far. Several authors have demonstrated that hypointense T2 signal on MRI in the basal ganglia in MSA correlates with postmortem determined iron accumulation in respective brain regions (see Table 3) [79, 84].
In MSA, the hypointensity of the posterolateral region of putamen received particular attention being associated with highest iron concentrations [100–106]. Therefore, low intensity of putamen of T2/T2*-weighted/SWI modalities of mostly 1.5T MRI has been suggested to be a specific feature of MSA in contrast to IPD and has attracted interest for the differential diagnosis between IPD and MSA-P [79, 106–113]. However, on visual rating of routine MRI images this discrimination between MSA and IPD proved unreliable [101, 114]. Nevertheless, it could be shown that putaminal T2 hypointensity relative to GP is more specific for MSA [101, 104].
The source of iron that causes hypointensity on MRI has been poorly investigated. There are only very few studies combining imaging and histochemistry in MSA [79, 84] and little evidence shows that MRI signal change in putamen might reflect greater levels of ferritin-bound iron, accumulation of hemosiderin and NM in patients with MSA, but not in patients with IPD/healthy individuals [82, 115].
Matsusue and colleagues compared postmortem T2-weighted imaging (1.5T) of MSA-P with histological findings of putamen and found that putaminal hypointensity reflects diffuse ferritin and iron deposition. In MSA-C hypointensity in dentate nuclei was associated with diffuse ferritin depositions [82, 84].
The attempt to quantify brain iron accumulation led to the introduction of a visual grading scale for hypointensity of putamen and subregional examination of putamen by some authors [100, 116–118]. As a result, the inner region of the putamen was proposed to be the most valuable region in differentiating MSA-P from IPD [106]. Grading of putaminal hypointensity among MSA patients revealed that even early stage MSA-P is associated with putaminal hypointensity grade≥2 (like in advanced disease) indicating that iron accumulation might occur early in the disease process [117]. However, no correlation of disease duration and putaminal hypointensity could be demonstrated [110, 117]. Further MRI studies demonstrated that MSA-P could be differentiated from IPD by grade 3 hypointensity of posterior putamen [118]. The results of postmortem analyses revealing higher iron concentration of the posterior putamen in MSA-P compared to IPD complement the MRI based grading of putaminal hypointensity and also allowed differentiation of PSP and MSA/IPD [81, 116].
The topography of iron distribution in putamen of MSA was further studied [117, 120]: There is an ascending T2 signal intensity from lateral to medial putamen, even in very early stages of disease without clinical symptoms – and therefore, this pattern was proposed MSA specific [117, 119]. Different iron deposition pattern in MSA-P and PSP were evaluated using R2* values and more severe iron accumulation in posterior/dorsal parts of putamen and GP in MSA-P was shown [120]. MSA-P could be differentiated from PSP by significantly lower R2* values of NC in MSA-P. Furthermore, higher R2* values were determined in the putamen of MSA-P compared to IPD/controls. In a subsequent study, Lee and colleagues evaluated 3T T2*-weighted gradient echo sequences in MSA, PSP, IPD, and controls and found that T2* is superior in detecting iron deposition compared to T2 and demonstrated its usefulness in differentiating patients with MSA from IPD and healthy controls [121].
Interestingly, the highest iron content as assessed by histochemistry and imaging studies in putamen colocalizes with highest GCI density and α-SYN aggregation in MSA brains [79, 122]. In addition to high iron levels in putamen in MSA, hypointensity on MRI is associated with high iron content in NC, SN, the adjacent lateral aspect of GP and in the pulvinar thalami as estimated by T2/T2* imaging analyses [106, 119].
Since contradictory findings regarding hypointensity associated with iron accumulation using different MRI field strengths arose, Watanabe and colleagues investigated the influence of different MRI field strengths (0.35/1.5/3T) on the putaminal signal in patients with MSA. Beside severity of gliosis and iron accumulation determining the MRI signal, the field strengths have profound influence on MRI findings with further implications for the diagnostic value. It has been demonstrated that an increasing field strength was associated with a lower frequency of putaminal hyperintensity (considering putaminal body and margin). In contrast, the occurrence of putaminal hypointensity increased concomitantly with the field strength [98].
Among the predominant motor presentations of MSA it could be demonstrated that putaminal hypointensity is more pronounced in MSA-P compared to MSA-C [79]. Additionally, a longitudinal MRI study by Lee and coworkers revealed a faster progression of putaminal pathology (atrophy and hypointensity) in MSA-P than MSA-C correlating to faster symptom progression in MSA-P [121].
The “putaminal rim sign” is a T2-hyperintense MRI sign at the dorsolateral border of the putamen which is believed to result from gliosis and can help to differentiate MSA-P from IPD [82, 123–126]. This is, however, a non-specific sign and, therefore, not included in the revised consensus criteria [127], while putaminal atrophy shows 92.3% specificity but low sensitivity (44.4%) for distinguishing MSA-P from IPD [128]. It can be speculated that iron deposition at the putaminal rim might be masked by predominant gliosis in this region [129].
Taken together, increased putaminal iron [108, 120] is the most consistent finding in MRI studies and its visualization may be helpful in making the differential diagnosis between IPD and MSA or PSP [106, 116]. Correlating the severity of atrophy and iron accumulation suggests that iron accumulation is a secondary effect of neurodegeneration as significantly increased iron in the putamen is associated with advanced atrophy compared to moderate iron accumulation in GP along with less severe atrophy [120]. Nevertheless, MRI studies need to be interpreted with great caution due to heterogeneous study populations, variation in stage of disease, lacking discrimination of MSA subtypes, different MRI protocols including slice thickness, fast versus conventional spin echo, T2*, R2*, SWI, quantitative and qualitative assessment, and field strengths (see Table 2).
Summarizes MRI studies looking at brain iron content in MSA
GE, gradient echo; GP, globus pallidus; HC, healthy controls; MRI, magnetic resonance imaging; MSA, multiple system atrophy; MSA-P, multiple system atrophy Parkinson variant; MSA-C, multiple system atrophy cerebellar variant; CBD, corticobasal degeneration; NM, neuromelanin; IPD, idiopathic Parkinson’s disease; PSP, progressive supranuclear palsy; PT, pulvinar thalamus; RN, red nucleus; SN, substantia nigra; TH, thalamus; NC, caudate nucleus; FLAIR, fluid attenuated inversion recovery sequences; R2*, tissue relaxation time of MRI (1/T2); SWI, susceptibility weighted imaging; T, Tesla; T2, tissue relaxation of MRI investigation; T2L/T2*L, low-intensity signal within the putamen on T2/T2*-weighted images; ns, not significant; ROI, region of interest; VBA, voxel-based analyses.
An overview about iron on MRI is given in Table 2.
Studies combining MRI with histology are given in Table 3.
Summarizes studies correlating MRI features with postmortem tissue analyses
GE, gradient echo; HC, healthy controls; MRI, magnetic resonance imaging; MSA, multiple system atrophy; MSA-P, multiple system atrophy Parkinson variant; T2, tissue relaxation of MRI investigation; T, Tesla; HE, hematoxylin eosin; GFAP, glial fibrillary acidic protein; IHC, immunohistochemistry.
Iron, α-synuclein, oxidative stress, and neuroinflammation
Collectively, histopathological and imaging data clearly demonstrate that iron levels are elevated in specific MSA brain regions with highest levels in the putamen. However, the mechanisms and consequences of iron accumulation have not been elucidated in MSA so far. In the following part potential interactions and mechanisms of iron dyshomeostasis in the pathogenic cascade are highlighted.
Figure 1 illustrates the vicious circle how iron may contribute to α-SYN pathology: iron accumulation results in high levels of OS/microglial activation leading to iron induced OS/neuroinflammation promoting α-SYN aggregation.
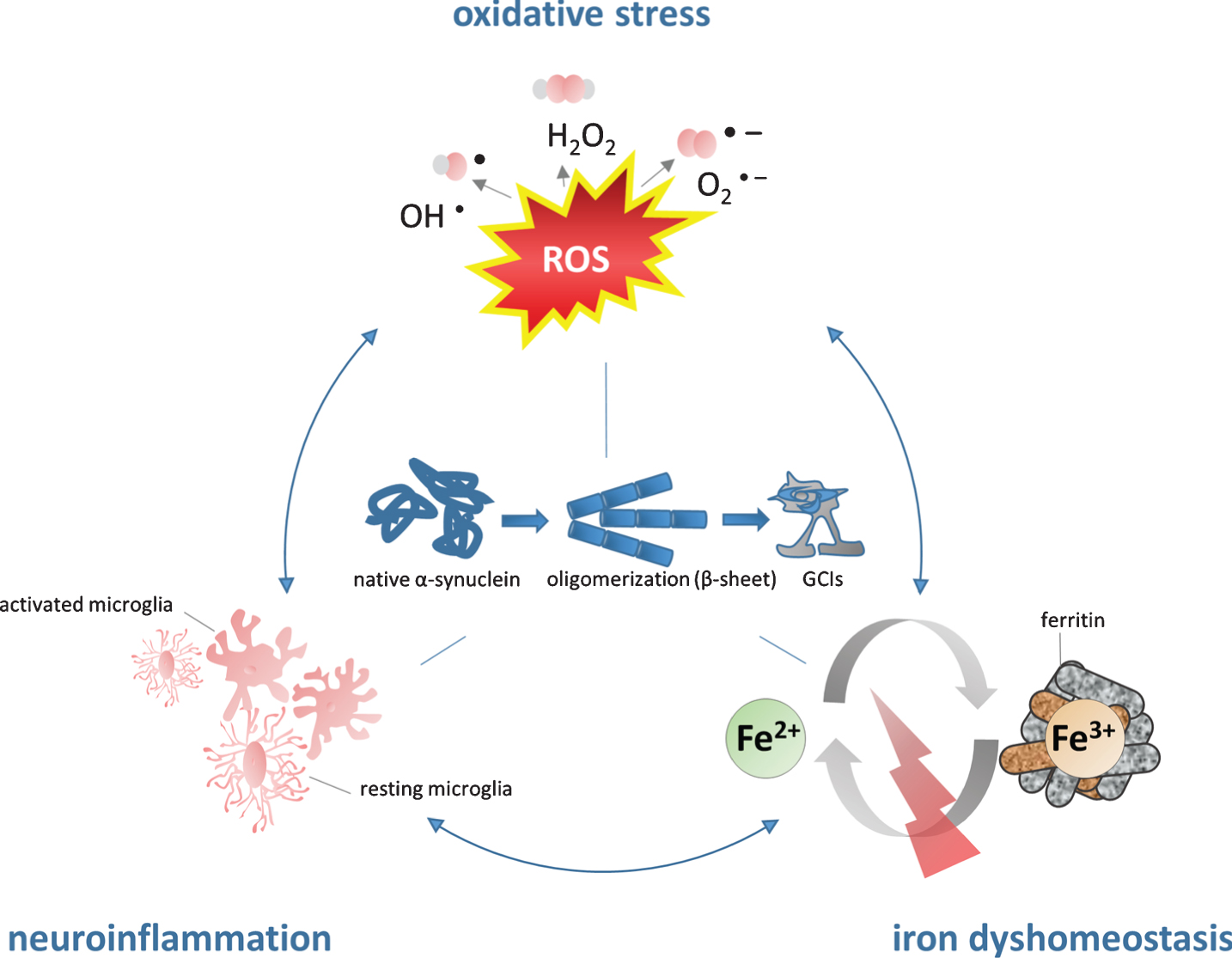
The role of iron in the pathogenesis of MSA. Iron dyshomeostasis is associated with increased levels of oxidative stress and microglial activation. The consecutive formation of a synergistic self-feeding cycle promotes α-SYN aggregation and secondary neurodegeneration. Fe2 +, ferrous form of iron, Fe3 +, ferric form of iron, GCIs, glial cytoplasmic inclusions; H2O2, hydrogen peroxide; O2-, superoxide anion; OH-, hydroxyl radical; ROS, reactive oxygen species.
α-SYN consists of 140 amino acids and physiologically it is a soluble α-helical protein reversibly attached to the cell membrane in the cytoplasm of brain cells. As part of the soluble N-ethylmaleimide-sensitive-factor attachment receptor (SNARE) complex it is believed to interact with the plasma/vesicular membrane in the process of neurotransmission. α-SYN has been implicated in the pathogenesis of IPD, MSA, and DLB [81, 130], which are all characterized by cellular aggregation and fibrillation of the protein. While α-SYN inclusions are mainly found in neurons in IPD and DLB, MSA is characterized by oligodendroglial α-SYN aggregation. As mature oligodendroglial cells do not express α-SYN physiologically, the origin of intracellular α-SYN aggregates as well as the trigger of α-SYN aggregation in MSA remain unknown [131]. It has been speculated that oligodendrocytes actively take up α-SYN that is released by neighboring neurons resulting in accumulation in oligodendrocytes [132]. This hypothesis is further supported by in vitro evidence pointing toward a neuron-to oligodendroglia cell to cell prion-like transmission of α-SYN [133–135]. Furthermore, a link between cell to cell propagation and neuroinflammation has been proposed for IPD [136] which might be translated to MSA. Disease progression and neurodegeneration appear to reflect increased levels of OS and proinflammatory cytokines released by activated microglia in mouse models of MSA and IPD [3, 137–139].
Post-translational modifications have been implicated in the pathogenesis of proteinopathies. In healthy humans phosphorylated α-SYN accounts for only 5% in contrast to synucleinopathies with the majority of α-SYN being phosphorylated [140] characteristically at position serin-129. OS seems to play a major role in these modifications as several in vivo and in vitro experiments demonstrated that increased OS leads to conformational changes of α-SYN structure, phosphorylation, oxidation and nitration and further accelerates oligomer formation, aggregation and increased production of reactive oxygen species (ROS) [19, 140–147].
As we have learned from in vitro experiments excess iron can promote α-SYN aggregation [143, 148] in a dose-dependent manner and dopamine exposure can further stimulate this process [19]. The exact mechanism remains unknown, although increased iron levels as well as iron catalyzed OS have been shown to promote the conversion of the α-helical conformation into β-sheet or spherical oligomer structure resulting in further α-SYN aggregation [17, 143] similar to that emerging upon incubation of (misfolded) α-SYN alone [149]. Whether a direct interaction of iron and α-SYN is involved is not known, although an iron binding site has been identified on α-SYN among other metal binding sites, resulting in conformational and solubility changes of the protein [17, 150–152]. Both ferric and ferrous iron were shown to bind to α-SYN [153], and it has been demonstrated that ferrous iron promotes α-SYN aggregation [154]. Interestingly, phosphorylation of α-SYN, occurring during OS, results in increased affinity to the metal binding site for iron [155].
Where OS arises, the oxidation of iron to its ferric form leads to aggravation of α-SYN pathology. To this end, the in vitro identification of an enzymatic activity, namely ferric-reductase activity within α-SYN by Davies and coworkers may explain the contrasting relative distribution of ferrous and ferric iron in SN: ferrous >ferric iron = 2:1 in healthy controls versus 1:1 in IPD [61, 156–158]. Consequently, an increased abundance of ferric iron and the consecutive propagation of OS via the Fenton reaction [159] may further accelerate α-SYN aggregation [13].
Another putative mechanism of iron induced α-synucleinopathy may be proposed by the discovery of a functional IRE within the 5-untranslated region of human α-SYN mRNA underlying iron mediated stimulation of α-SYN expression via translational regulation [160]. In vitro experiments demonstrated that in the presence of iron α-SYN mRNA is upregulated by IRP-knockdown resulting in pronounced cellular α-SYN aggregation [13, 161]. MSA is associated with disruption of long intervening non-coding RNAs (lincRNAs) along with protein-coding genes related to iron metabolism and immune response [162].
Generally, the crucial role of oligodendrocytes in MSA becomes clear by its intracellular aggregation profile of α-SYN. Their high intracellular iron levels along with low concentrations of anti-oxidative agents (glutathione) in combination with their high metabolic rate make them particularly vulnerable to oxidative damage [163, 164]. Plus, cytokines released by activated microglia in the context of inflammatory response have been shown to induce oligodendroglial cell death [165, 166]. Furthermore, oligodendroglial cytoplasmic aggregation of α-synuclein itself leads to increased susceptibility to OS and tumor necrosis factor alpha (TNF-α) [167, 168] building a self-sustaining damaging system.
Several cell culture investigations, animal models and PET imaging in MSA patients have demonstrated that neuroinflammation parallels α-SYN pathology [169–173]. Hence, neuroinflammation has been accepted as a prominent finding contributing to neurodegeneration and α-SYN mediated toxicity mainly originating from activated microglia cells [3, 174]. Microglial activation in MSA was reported in regions of neuronal loss including putamen, GP, pons, SN, and the dorsolateral prefrontal cortex using [C11]-(R)-PK11195 PET imaging [170] and it has been demonstrated that microglia activation results in morphological and functional changes, plus they release pro- and/or anti-inflammatory cytokines, neurotrophic factors, and cleavage damaged or dead cells [175–177]. However, albeit their indispensable mechanism of host defense and maintenance of neuronal viability, microglial cells can be over-activated leading to chronic inflammation and microgliosis which is found in several neurodegenerative diseases including MSA. As a result, neurotoxic factors including TNF-α and interleukin-1 beta as well as superoxide radicals including ROS and nitric oxide are released, which further provoke neuroinflammation and protein aggregation, creating a positive feedback loop facilitating neuronal damage and cell death [178–184]. Variable forms of α-SYN including native, misfolded α-SYN as well as nitrated α-SYN can trigger neuroinflammation by microglia activation in a dose and conformation dependent manner [3, 185–193]. Although the exact mechanisms of microglia uptake of α-SYN are not fully understood and an α-SYN specific receptor of microglia has not been identified yet, it has been demonstrated that an uptake mechanism must exist [194, 195] that in turn triggers the release of soluble immune modulators. One possible α-SYN uptake strategy includes the toll-like receptor 4 (TLR4) which is a pattern-recognition receptor that has been identified to play an essential role in microglial activation mediating phagocytic activity, release of proinflammatory cytokine and production of ROS after binding to α-SYN. Following, a modulatory role of TLR4 in neuroinflammation was suggested.
In summary, α-SYN aggregation is associated with GCI density in MSA specific areas and activated microglial cells [174]. Thus, the upregulation of inflammatory mediators and microglia-mediated neuroinflammation are associated with the pathogenesis of MSA [179, 197]. Since neuroinflammation and α-SYN are observed in early MSA neuropathology it remains unclear whether α-SYN induces neuroinflammation or neuroinflammation incudes protein aggregation.
Overall, the role of iron should no longer be questioned in the process of α-SYN related neurodegeneration as iron accumulates in degenerating brain regions, under inflammatory conditions and can aggravate cytokine induced oxidative and nitrosative stress responses along with distinct effects of iron on immune effector pathways [198]. Even though it is still unknown whether neuroinflammation is an initial or downstream effect [122] of synuclein and/or iron accumulation, it has been demonstrated that on the one hand, nitrated α-SYN resulting from OS activates microglia [138, 199] and elevates proinflammatory mediators [200, 201] and on the other hand that iron accumulation activates microglial cells leading to release of proinflammatory cytokines [202–205]. Consequently, concomitant iron accumulation, OS, and neuroinflammation can be particularly damaging in the brain by sustaining a self-promoting vicious circle.
CONCLUSION AND OUTLOOK
This review summarizes the currently available evidence of the putative functional association of iron dysregulation in MSA and points out possible targets for disease modification. Although iron is essential to maintain physiological functions, iron overload is linked to cellular damage and neurodegeneration. An elevation of brain tissue iron levels in MSA has been determined by histopathological analyses and conventional imaging studies using T2/T2* and SWI techniques of MRI. In MSA, highest levels of iron are detected in putamen. Histopathological investigations complement imaging findings in determining increased iron levels in basal ganglia, but implicate perturbation of iron homeostasis even leading to reduction of bioavailable iron in pons and putamen in MSA. Mechanistic analyses of iron and α-SYN point to several potential regulatory interactions including iron induced OS promoting neuroinflammation and α-SYN aggregation. Whether dysregulation of brain iron homeostasis acts as an inductor of neurodegeneration or is an epiphenomenon of cellular death remains to be elucidated. Imaging technologies and consecutive examinations can address this question and, indeed, increased iron levels in putamen have been shown in early disease stages in MSA. Unfortunately, MRI features and corresponding histopathological measurements are scarce and postmortem tissues are usually obtained from patients with advanced disease. Further limitations constitute the inability of MRI to detect low molecular forms of iron, the varying biochemical/histopathological methods used to determine iron in MSA and of course the small sample sizes.
In conclusion, iron seems to play a yet poorly understood role in the dynamic pathogenesis of MSA in the context of neuroinflammation and OS, and clearly has been an undervalued component in the past. Considering no currently available treatment options in MSA, the goal is to identify treatable targets to establish disease-modifying therapies. The complexity and multifactorial nature of MSA requires multiple strategies. Iron chelators which may either reduce brain tissue iron content or modify pathological iron accumulation in specific cells [206] may be promising in this respect for MSA treatment [207]. Indeed, treatment with the oral iron chelator Deferiprone in IPD resulted in significant decrease of iron deposition in SN using MRI-based measurements and motor scales [208, 209]. To access this question systematically, we need good animal models to study MSA and to understand the contributing role of iron in this process. This may also help to evaluate the potential of iron targeting strategies by iron chelators or other iron trafficking modifiers such as calcium antagonists or anti-hepcidin treatments [210–212].