
Abstract
Frontotemporal dementia (FTD) is a heterogeneous clinical, genetic, and neuropathological disorder. Clinical diagnosis and prediction of neuropathological substrates are hampered by heterogeneous pictures. Diagnostic markers are key in clinical trials to differentiate FTD from other neurodegenerative dementias. In the same view, identifying the neuropathological hallmarks of the disease is key in light of future disease-modifying treatments. The aim of the present review is to unravel the progress in biomarker discovery, discussing the potential applications of available biological, imaging, and neurophysiological markers.
Keywords
INTRODUCTION
Frontotemporal dementia (FTD) is a genetically and pathologically heterogeneous clinical syndrome characterized by progressive deficits in behavior, executive functions, and language, associated with frontal and temporal lobe degeneration [1–4]. FTD is the second most common cause of dementia in the presenile age group (<65 years of age), and accounts for 5–15% of all cases of dementia, with a prevalence of 3–26 per 100,000 subjects in the age group of 45–65 years [5, 6].
Current redefined clinical criteria identify distinct phenotypes on the basis of presenting clinical symptoms; these include the behavioral variant of FTD, the agrammatic variant of primary progressive aphasia and the semantic variant of primary progressive aphasia [7–9]. In addition, some patients have an associated parkinsonism, as in progressive supranuclear palsy (PSP) and corticobasal syndrome, or motor neuron disease (FTD-MND) [10–12].
Clinical FTD is associated with different types of underlying neuropathology, and the term frontotemporal lobar degeneration (FTLD), characterized by the relative selective degeneration of the frontal and temporal lobes, is used to describe the pathological hallmarks of the disease. Abnormal intracellular inclusions containing Tau, TDP-43, or FUS protein have been identified in the majority of cases; however, the correlation between clinical syndrome and underlying neuropathology remains still largely unsatisfactory. Except for nearly 30% of familial cases, accounted predominantly by the microtubule-associated protein tau (MAPT), granulin (GRN), and the hexanucleotide repeat expansion of the chromosome 9 open-reading-frame 72 (C9orf72), it is still challenging to predict the underlying pathological process in vivo [13]. Furthermore, because of the possible overlap of FTD with psychiatric disorders or other neurodegenerative diseases, such as early onset Alzheimer’s disease or atypical parkinsonisms, diagnosis is often challenging [14, 15].
Taking into account these drawbacks, in recent years numerous studies have made significant progress in understanding the pathophysiology and the progression of FTD from a multidimensional approach. Indeed, biological, imaging, and neurophysiological markers have shed light on the very first alterations in the presymptomatic phases of disease [16–21], or to predict disease prognosis [22–24].
With the development of candidate therapies for FTD likely to occur in the coming years, it is of great importance to develop and validate reliable diagnostic and prognostic biomarkers, with numerous implications regarding stratification for disease-modifying clinical trials and for monitoring disease progression.
The objective of the present work is to review and evaluate available literature data in order to highlight recent advances in biological, imaging, and neurophysiological, markers for the diagnosis and prognosis of FTD.
BIOLOGICAL MARKERS: LOOKING AT PROTEIN-OPATHIES
Looking at biomarkers able to differentiate FTD from other neurodegenerative dementias is one of the hot issues in the current literature, concerning specific therapeutic strategies targeting either TDP-43 or tau accumulations. If neuropathology is clearly predictable in monogenic FTD, in the other cases there is no correspondence between clinical features and neuropathological findings [25, 26].
One of the most comprehensively validated series of biomarkers, which reflect the pathological hallmarks of Alzheimer’ disease (AD), comprises cerebrospinal fluid (CSF) total-Tau (tTau), phosho-Tau181 (pTau), and amyloid-β1-42 (Aβ1-42). An increase in CSF tTau and pTau, and a decrease in Aβ1-42 (and thus an increased tTau or pTau/Aβ1-42 ratio), have been shown to identify AD pathology with extremely high accuracy [27], excluding AD in the diagnostic work-up of FTD. These findings have been confirmed both in clinical and pathological cohorts [28–30] and may be particularly useful for excluding focal variants of AD, which may be clinically indistinguishable from FTLD [31–34].
A number of studies have focused on either CSF Tau or TDP-43 metabolism to identify diagnostic markers in FTD patients. Indeed, CSF tTau, pTau, or TDP-43 dosages did not yield convincing results in predicting neuropathological hallmarks [35, 36].
A more recent study by Hu and colleagues has identified in the CSF pTau/tTau ratio a viable biomarker to identify FTLD with TDP-43 pathology as compared to FTLD-Tau [19]. In this view, pTau/tTau ratio has been proposed as useful in detecting patients with amyotrophic lateral sclerosis, which is associated with TDP neuropathology [20].
Later, this result was further corroborated in patients with known Tau or TDP-43 pathology, with high accuracy values in identifying TDP-43 cases [20, 21] (Fig. 1) and with prognostic significance [37].
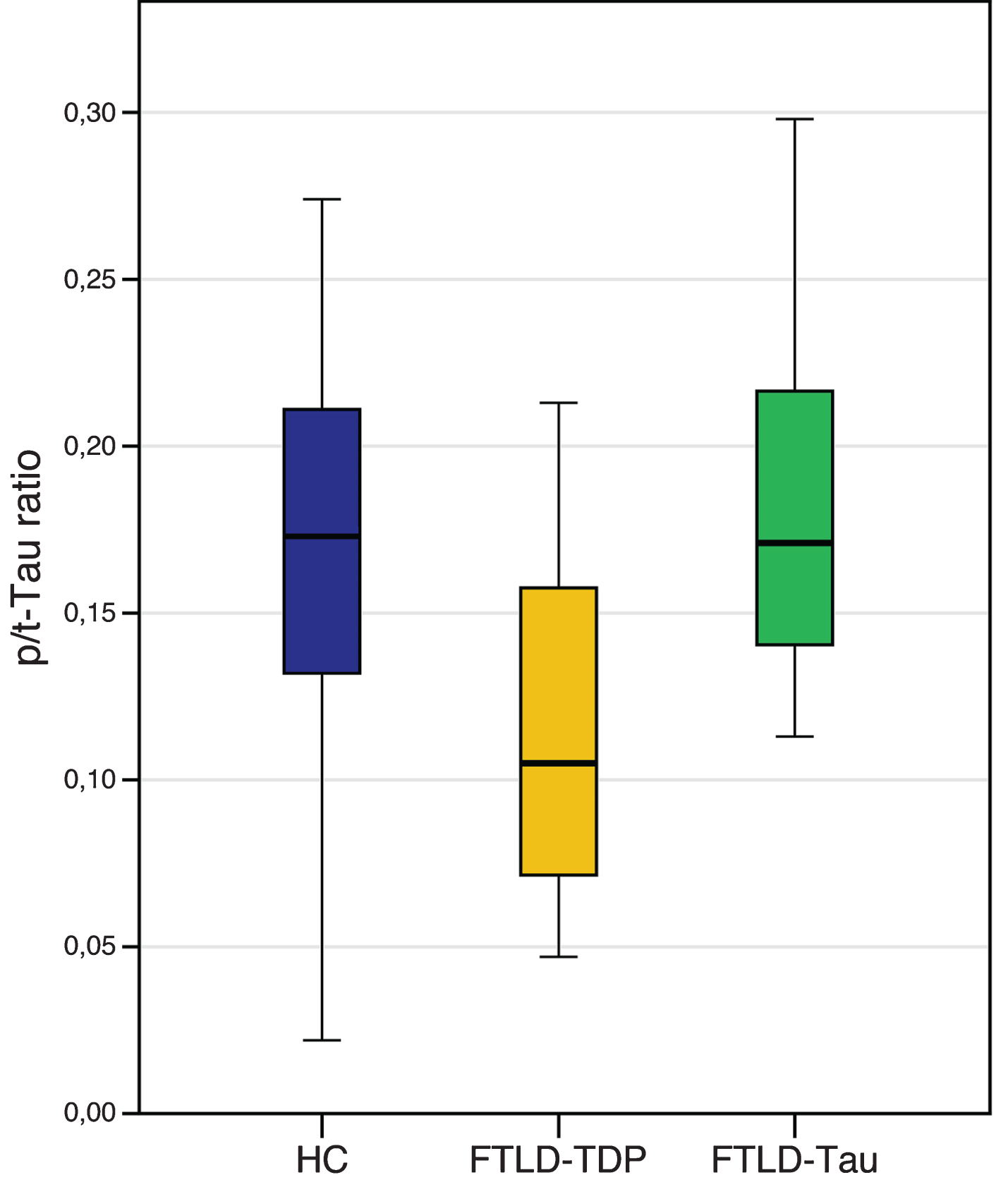
Levels of CSF phospho Tau/total Tau (p/t) ratio in FTLD-TDP and FTLD-Tau, compared to healthy controls (HC). Horizontal thick lines illustrate median cerebrospinal fluid (CSF) values, notches correspond to interquartile range, error bars depict 25th to 75th percentile range of data. HC: age-matched healthy controls; FTLD-TPD: including patients carrying a GRN mutation, TARDBP mutations, C9orf72 mutations and patients with FTD-motor neuron disease; FTLD-Tau: including clinically diagnosed progressive supranuclear palsy (PSP) and patients carrying a MAPT mutation.
The pathological mechanism leading to reduced CSF pTau/tTau ratio in TDP-43 cases is currently unknown but could possibly be explained by a more extensive neuronal damage leading to increased tTau levels, due to the concomitant inclusions of patients with FTD-MND [19, 37]. This hypothesis could also be supported by the association between a reduced pTau/tTau ratio and survival in patients with FTD [37].
Another approach to identify TDP-43 pathology was to directly measure levels of phosphorylated TDP-43 (pTDP-43) aggregates in blood or CSF. Significantly increased levels of CSF and plasma pTDP-43 have been found in small cohorts of patients with C9orf72 or GRN mutations [38]. However, in a pathology-proven cohort, pTDP-43 levels did not differ between FTLD-tau or FTLD-TDP [31]. The quantification of CSF pTDP-43 still needs further refinement to overcome technical issues, as the relatively low concentrations of pTDP-43 in CSF with possibly different isoforms, and the presence of various antibodies that recognize different epitopes and thus vary in diagnostic accuracy [31, 32].
Intriguing results have been obtained by qualitative analysis of CSF Tau protein instead of quantitative evaluation. If CSF total Tau or CSF phospho-Tau181 dosages were not able to identify different neuropathological signatures, the assessment of post-translational modifications of Tau protein in CSF yielded more convincing findings.
Post-translational modifications of Tau protein have been demonstrated to be crucial in FTLD-Tau pathogenesis [39]. However, while the phosphorylation mechanism has been studied extensively, proteolytic processing has received less attention. Literature data reported that different Tau fragments, produced by endogenous proteases, may modulate Tau aggregation itself [40, 41]. Accordingly, the measure of CSF Tau proteolytic forms has been demonstrated to be specifically altered in patients with Tau pathology, namely in PSP cases. CSF Tau isoforms ratio in PSP was 50% lower than in patients with either neurodegenerative parkinsonism [42, 43] or other Tau-related dementias such as Alzheimer’s disease [44].
Accordingly, a previous autopsy study demonstrated that Tau-related pathologies undergo a different Tau proteolytic processing, generating distinguishable deposits of cleaved Tau fragments in PSP brains [45], and explaining biological differences among Tauopathies in CSF. More recently, neurofilament light chain (NfL) has been proposed as diagnostic and prognostic marker in FTD [46]. Neurofilaments are the major components of the neural and axonal cytoskeleton and perform a fundamental role in axonal transport and in synapse functioning, and NfL is one of the most abundant subunits which increases after neuronal death and axonal degeneration [41].
NfL blood and CSF levels have been shown to be considerably increased in FTD patients compared to healthy controls, being significantly associated with disease severity and survival, and correlating with decreased gray and white matter volume in FTD-associated regions in the frontal and temporal lobes [37, 46–49]. No difference in NfL levels were observed between the different FTD endophenotypes, whereas markedly increased levels have been observed in FTD-MND [37, 46–49]. In presymptomatic carriers, CSF and blood NfL levels have been shown to be within normal range, with a distinct increase after conversion to the symptomatic stage [46]. Whereas some studies showed elevated levels in FTD compared to AD [49], others did not reveal group differences [48]. Furthermore, NfL levels have been shown to be increased in patients with multiple system atrophy, PSP, corticobasal syndrome, and vascular dementia [47, 51].
As other CSF biomarkers reported above, also NfL levels could possibly discriminate between FTLD-TDP and FTLD-Tau, resulting increased in patients with TDP-43 pathology [37, 49]. However, this difference could be accounted for the co-occurrence of FTD-MND in the TDP-43 cohort.
NfL could become a promising, non-invasive biomarker for disease staging, and the strong correlation between CSF and plasma levels [46] makes it even more appealing for monitoring disease progression and treatment response.
NEUROIMAGING MARKERS: LOOKING AT CONNECT-OPATHIES
In the last three decades, neuroimaging techniques have shown a tremendous development, and have been suggested as powerful tools to elucidate disease mechanisms, track disease progression and explore neuroanatomical correlates of clinical and genetic characteristics [52]. Considering FTD, magneticresonance imaging (MRI) has been widely used for in vivo visualization of brain atrophy in the different clinical phenotypes of FTD, clearly reporting abnormalities in frontal and temporal grey matter regions and related white matter bundles [53–55]. As biomarkers of the underlying neurodegenerative process, these structural perturbations have been used to study the pattern of damage in the sporadic and monogenic forms of FTD [56–58] also tracking disease severity and progression [59–62]. The visual assessment of grey matter atrophy on MRI scans through a series of visual rating scales has been described as a fast, clinical reliable and inexpensive approach to increase diagnostic accuracy in the everyday practice [63, 64]. Interestingly, structural damage (in particular grey matter atrophy) has been considered as an effective biomarker for FTD not only when the disease is overt [65], but also in the presymptomatic stage, years before clinical onset [18, 66]. More recently, several neuroimaging approaches for the study of functional connectivity have been applied to FTD, studying coherent patterns of activation among brain regions, in particular at rest [67]. Spontaneous blood-oxygenation level dependent (BOLD) fluctuations, in the context of functional brain networks, have been adopted as sensible and specific biomarkers reflecting connectivity changes early in the disease course, even before structural alterations are detectable with conventional MRI at single-subject level [16, 68–70]. In contrast with AD, which shows a peculiar involvement of the default mode network, FTD is characterized by a predominant breakdown of anterior functional networks, in particular the salience network [71]. Altogether, FTD functional connectivity approaches have shown a progressive functional network disruption [72–74], both at the regional level [69] as well as the whole-brain level [75, 76] using graph-theory measures. In line with these findings, in presymptomatic FTD functional connectivity perturbations (primarily involving salience, frontoparietal, and executive networks) were detectable years before the clinical onset, opening the road for the utilization of these indexes as preclinical biomarkers [16, 68–70]. However, the definition of which markers, i.e., either structural or functional, and which approach are best to track the disease and to assess the response to disease modifying therapies remain challenging. Just recently, Premi et al. adopted a machine-learning approach with multivariate analysis (multi-voxel pattern analysis, MVPA) to different structural and functional MRI metrics in order to study Granulin-relateddisease, from presymptomatic carriers to symptomatic patients with the same mutation GRN mutation) (Fig. 2) [77]. Working as a classifier, MVPA was able to maximize, for each considered MRI measure, the separation between GRN carriers and controls, in both preclinical and clinical phases of disease. Interestingly, structural measures of grey and white matter atrophy represented the best neuroimaging biomarkers in the symptomatic stage, followed by functional connectivity indexes, in particular regional index like fractional amplitude of low frequency fluctuation (fALFF). On the contrary, in the presymptomatic phase, functional connectivity measures (regional indexes like fALFF and degree centrality) showed the best diagnostic accuracy for still asymptomatic GRN mutation carriers, probably capturing the initial disruption of local signal integration [78, 79]. Altogether, considering a global accuracy >80% for either preclinical and clinical phases, fALFF could be considered the best MRI biomarker in the GRN disease continuum [77]. From this point of view, different MRI biomarkers could be defined and applied in the FTD spectrum, in particular with regard to disease-modifying treatments. However, considering future international multicenter clinical trials, a potential MRI biomarker (like fALFF, network and graph-theory indexes) should also prove to be reliable and reproducible, regardless of differences in MRI protocols and acquisition parameters [76].
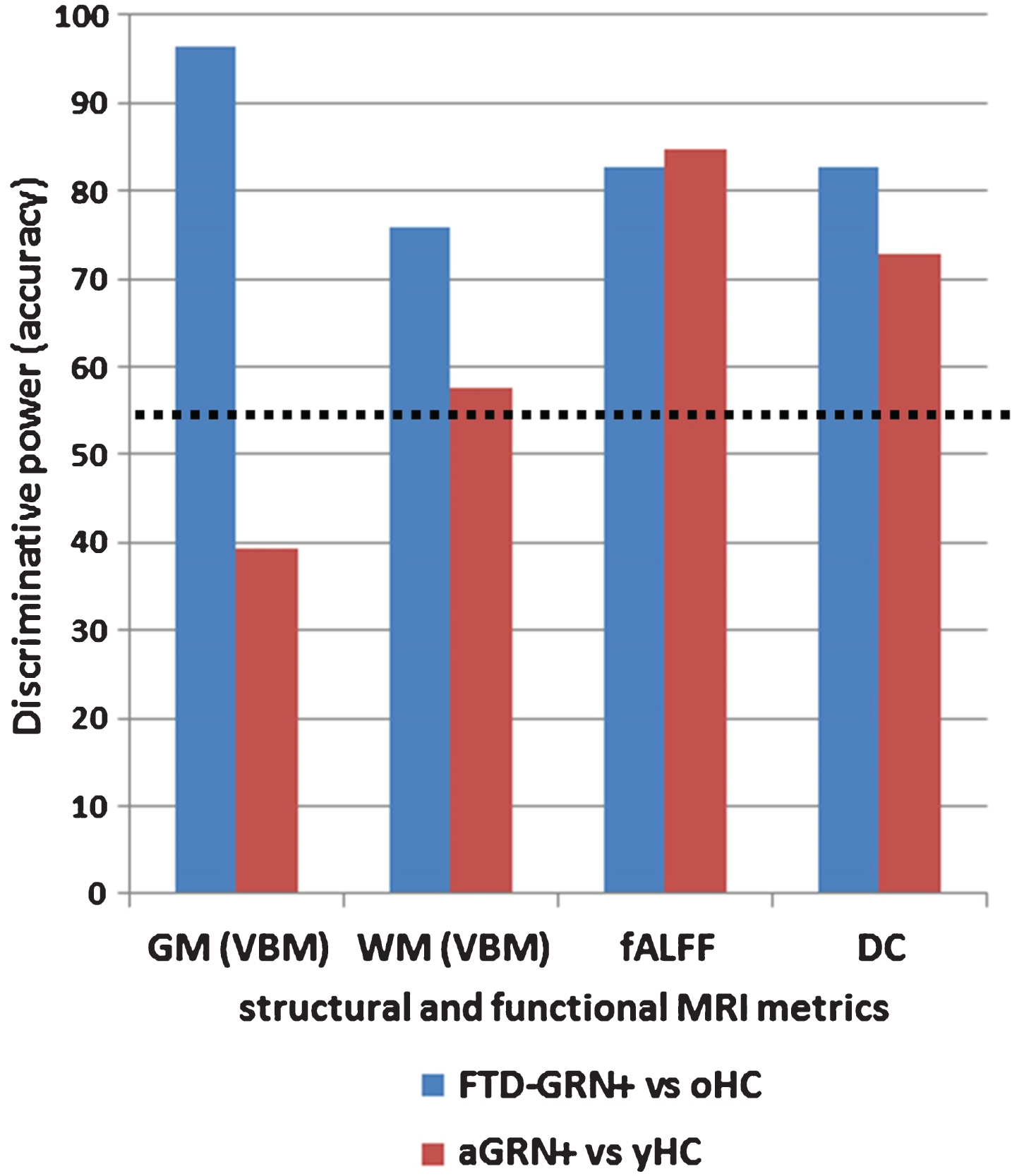
Bar chart showing the classification results (discriminative power) of the different structural and functional measures. The black dotted line represents the significant (>55% of right classification) performance for each measure. Blue is for FTD-GRN+ versus oHC and red is for aGRN+ versus yHC. FTD-GRN+, frontotemporal dementia carrying Granulin mutation; aGRN+, asymptomatic carriers of Granulin mutation; oHC, old healthy controls; yHC, young healthy controls; GM, grey matter; WM, white matter; VBM, voxel based morphometry; fALFF, fractional amplitude of low frequency fluctuations; DC, degree centrality.
Besides MRI, also molecular imaging, like positron emission tomography (PET), has been applied in FTD. The evaluation of metabolic brain alterations by fludeoxyglucose (18F) tracer provided additional clues into the study of FTD, demonstrating a substantial concordance between cortical atrophy and reduced metabolism [80, 81], with different patterns of damage considering the different clinical phenotypes [82, 83]. Moreover, the visual evaluation of FDG-PET images provided greater diagnostic accuracy in differentiating FTD from other dementias [84]; more recently, this single-subject approach has been standardized using an optimized statistical parametric mapping (SPM) [85]. However, the most attractive aspect of molecular imaging relies on the availability of PET radiotracers to identify different proteinopathies in FTD [86]. In the last years, specific tracers for Tau pathology have been developed [86, 87]. At the moment, different Tau tracers are commercially available, i.e., [18F]AV-1451 [88] and [11C]PBB3 [89], with different affinity for neurofibrillary tangles and for Tau isoforms (3R and 4R) [86]. Considering FTD, PET Tau imaging has been recently applied in FTD patients with MAPT mutations (and thus with a known Tau pathology), demonstrating an increased Tau binding in temporal poles and frontal lobes [90, 91]. Interestingly, PET amyloid tracers (like 11C-Pittsburgh compound B or Florbetapir) can aid in the differential diagnosis between FTD and AD, ruling out Aβ pathology [92].
NEUROPHYSIOLOGICAL MARKERS: LOOKING AT NEUROTRANSMITTER-OPATHIES
Several neurophysiological techniques have been implemented for the study of FTD and the historical initial reports on the use of electroencephalography (EEG) did not identify significant changes in patients with FTD, as opposed to patients with AD [93–95]. Subsequent reports using quantitative EEG (qEEG), showed that the typical qEEG pattern for FTD patients was characterized by a decrease in all of the fast activities (α, β1-β3) relative to healthy controls, but did not differ in slow activities (δ and θ rhythms), possibly reflecting the degeneration of frontal regions in FTD patients [96–98]. Compared to AD patients, FTD showed a diffuse higher θ power and a decreased α2 and β1 values in central/temporal regions [99, 100].
Moreover, EEG microstates, which are subsecond (60–120 ms) periods of stable brain state that repeat across time and individuals [101], have been shown to differ significantly in FTD patients compared to AD, schizophrenia, and healthy controls [102].
Just recently, EEG abnormalities have been observed also in a group of genetic FTD patients. In particular, FTD due to GRN mutations showed an increase in high α and decrease in θ oscillations as compared to non-carriers [103].
Other neurophysiological techniques, particularly transcranial magnetic stimulation (TMS), have become promising tools to assess specific cortical circuits in the central nervous system. In the context of dementia, different paired-pulse TMS paradigms have been implemented to assess intracortical inhibitory and excitatory interneuronal activity, namely short interval intracortical inhibition and facilitation (SICI-ICF), dependent on GABAA and glutamatergic transmission [104, 105], long-interval intracortical inhibition, dependent on GABAB transmission [106], and short-latency afferent inhibition (SAI), dependent on central cholinergic activity [107]. Furthermore, specific paradigms of paired associative stimulation [108] or repetitive TMS [109, 110] have shown to increase or decrease the excitability of corticospinal projections of the primary motor cortex (M1), representing a form of long-term potentiation or depression and thus a method of assessing synaptic plasticity.
Neurophysiological studies in FTD have shown central motor circuit abnormalities, even in cases without clinical evidence of motor involvement [10, 111–114]. No significant alterations in motor threshold [10, 112–116], SICI-ICF [112, 114], and SAI [111] have been observed in FTD [117, 118]. However, these studies have been hindered by the small number and by the selection of patients, which has been made exclusively on a clinical basis and not taking into account the significance of CSF proteins (Aβ42, tTau, and pTau) to exclude possible focal variants of AD, or the genetic contribution of known pathogenic mutations.
In this view, Burrell et al. have observed in a large cohort of 40 FTD patients, a significant decrease in SICI and a trend toward a reduced ICF, compared to healthy controls [10]. These findings have been replicated in an even larger study on 64 FTD patients, in which a striking impairment of SICI-ICF was observed compared to 79 AD patients and 32 healthy controls. Furthermore, as expected, an impairment of central cholinergic circuits evaluated with the SAI paradigm, was observed only in AD patients [119] (Fig. 3). Combining both measures of GABAergic and cholinergic transmission (SICI-ICF and SAI respectively), Benussi et al. determined the diagnostic accuracy of TMS to discriminate between AD, FTD, and healthy controls, with levels of sensitivity and specificity >80–85%, even in patients with a high biomarker confidence (CSF Aβ42 and tau determination or amyloid PET imaging) and in the early phases of disease [119].
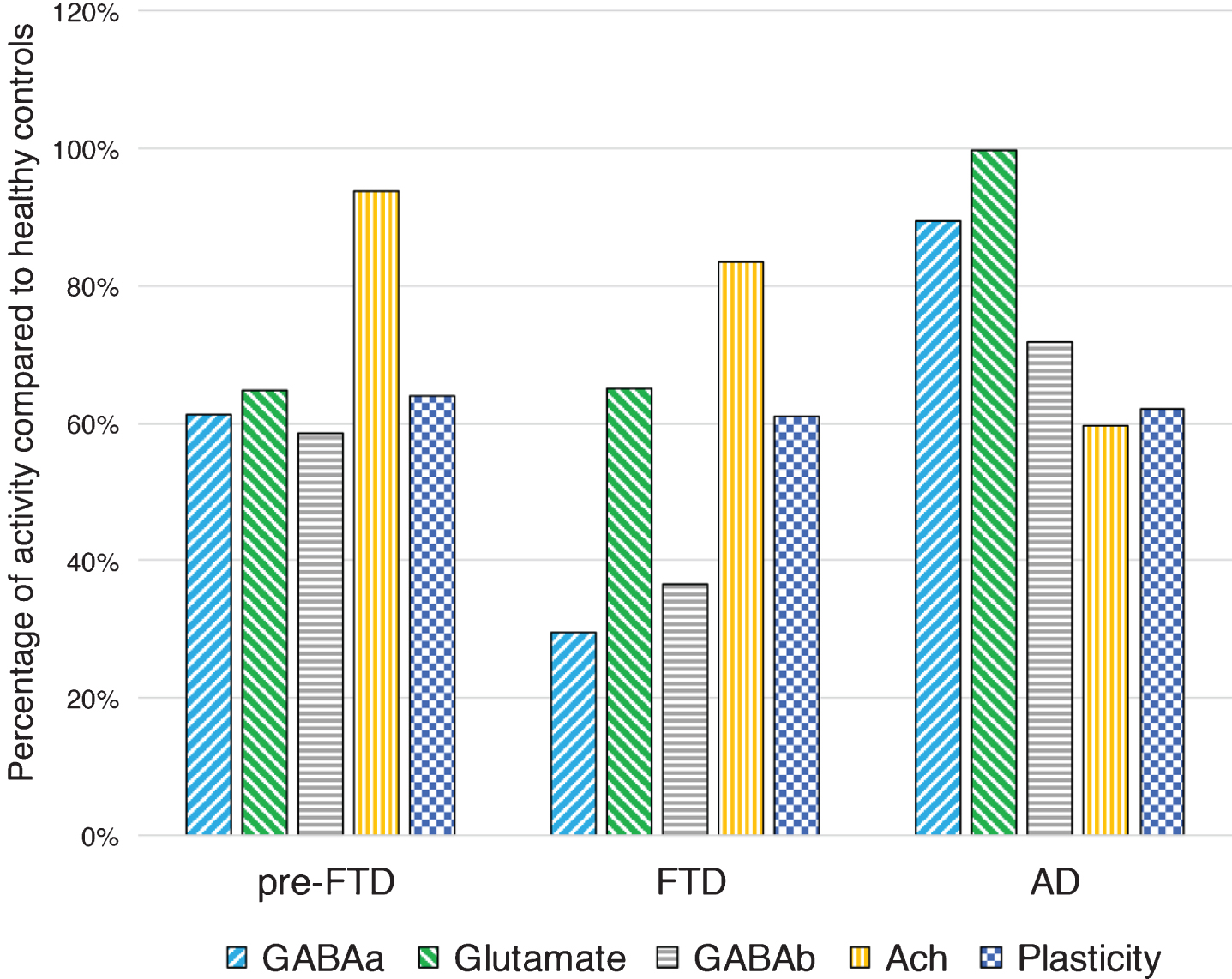
Intracortical connectivity and LTP-like plasticity profiles in presymptomatic GRN carriers, in symptomatic FTD and AD patients, compared to healthy controls. Pre-FTD, presymptomatic granulin (GRN) carriers; FTD, frontotemporal dementia patients; AD, Alzheimer’s disease patients; GABAa, GABAAergic activity evaluated with average short interval intracortical inhibition (1, 2, 3 ms); Glutamate, glutamatergic activity evaluated with average intracortical facilitation (7, 10, 15 ms); GABAB, GABABergic activity evaluated with average long interval intracortical inhibition (50, 100, 150 ms); Ach, cholinergic activity evaluated with average short latency afferent inhibition ( +0, +4 ms); Plasticity, LTP-like plasticity evaluated with paired associative stimulation (mean +10, +20, +30 min). Values are expressed as percentage of activity in healthy controls.
The impairment of intracortical inhibitory and excitatory circuits (SICI-ICF) has been observed also in a cohort of presymptomatic carriers and symptomatic patients bearing a pathogenic GRN mutation, with a progressive decline in GABAergic transmission in the symptomatic phases of disease. In addition, long-term potentiation-like cortical plasticity, assessed by the paired associative stimulation protocol, has been shown to be strikingly impaired in the presymptomatic phases of disease, at more than 18 years before expected symptom onset [17] (Fig. 3).
CONCLUSIONS
The field of biomarkers in FTD has made significant progress in the past few years and numerous studies have tried to shed light on the most significant issues in the realm of FTD. In the first place, the study of the earliest physiopathological modifications in FTD has gained much drive from the observations in presymptomatic carriers of known FTD gene mutations. Secondly, biomarkers have shown to increase the diagnostic accuracy of FTD, both between different diseases, as for AD, and between different subtypes of FTD, as for FTLD-TDP and FTLD-Tau. Thirdly, the prognostic capacity has amply improved.
Taken together, these findings have led to major advances in the knowledge of FTD pathophysiology. However, a more profound integration of these biomarkers probably holds the key to unravel the most cunning issues in the realm of FTD, by investigating multiple interconnections between the domain of protein-opathies, connect-opathies, and neurotransmitter-opathies.
With a new emphasis on multi-modal approaches, the next years hold promise for even greater understanding of the physiopathology of FTD, with crucial implications for clinical management, diagnosis and therapeutic trials.
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0584r1).