
Abstract
The decreased availability of metabolizable energy resources in the central nervous system is hypothesized to be a key factor in the pathogenesis of Alzheimer’s disease. More specifically, the age-related decline in the ability of glucose to cross the blood-brain barrier creates a metabolic stress that shifts the normal, benign processing of amyloid-β protein precursor toward pathways associated with the production of amyloid-β plaques and tau-containing neurofibrillary tangles that are characteristic of the disease. The neuroenergetic hypothesis provides insight into the etiology of Alzheimer’s disease and illuminates new approaches for diagnosis, monitoring, and treatment.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia in developed countries. It is characterized by progressive dysfunction of memory and cognition with a long prodromal period. Pathologic accumulations of amyloid-β (Aβ) plaques and tau-containing neurofibrillary tangles (tau) are found, but an understanding of their cause remains a work in progress. AD represents an ever-growing public health problem that places unique burdens on families, care providers, and the healthcare system in general. The 2017 report from the Alzheimer’s Association notes that more than five million people are living with AD in the United States. Given the absence of effective interventions to slow progress, it is projected to affect 106 million people by 2050 [1]. It is the only disease in the top ten causes of death that cannot be prevented, cured, or even slowed [2].
Hypotheses have attempted to explain aspects of AD, with the most basic being the amyloid cascade hypothesis which associates AD with its characteristic extracellular accumulation of Aβ and tau [3]. The mitochondrial cascade hypothesis expanded the discussion to include effects of inherited and environmental factors on mitochondrial function, correlating these with Aβ and tau formation and the trajectory of AD pathogenesis [4, 5]. Hypotheses have suggested roles of various metals, including copper, iron, zinc aluminum, and calcium [6 –10]. One hypothesis has advanced the relationship between AD and neuroinflammation [11], and there has been a proposal that AD be considered as “type-3 diabetes” [12]. While research has advanced and expanded our knowledge of the pathology of AD, an understanding of its etiology has yet to be fully elucidated, and prevention and control measures remain beyond our grasp.
This paper reviews the evidence and suggests that the status of available metabolizable energy resources in the central nervous system (CNS) is a central determinant of AD pathogenesis. The neuroenergetic hypothesis posits that the chronic, progressive decline in the ability of glucose to cross the blood-brain barrier (BBB)—a normal event of aging—contributes to an energy-deficiency stress in the CNS. In reaction to this glucose/energy-deficiency stress, there is a shift from pathways associated with the normally benign metabolism of amyloid-β protein precursor (AβPP), to a pathway associated with Aβ/tau production and AD progression. Considering AD from this perspective provides insight into its pathophysiology while embracing and clarifying the specific biochemical roles of previously-identified risk factors and correlates, and can thus inform regarding potential approaches for diagnosis, monitoring, treatment, and prevention.
NEUROENERGETICS AND THE ALZHEIMER’S DISEASE PROCESS
Glucose is the brain’s primary source of energy. To enter the CNS from the general circulation, however, glucose must traverse the protective BBB. The primary conduit through the BBB is GLUT1 (SLC2A1), a membrane-bound glucose transporter [13]. Even in the absence of identified pathologies, the level of glucose available to the CNS deteriorates with age [14]. It then follows that available metabolizable energy from glucose would be further reduced by factors affecting its net ability to traverse the BBB and be utilized by the CNS.
To illustrate, the ɛ4 variant of the APOE gene is a known risk factor for AD. Transgenic mice carrying the APOE ɛ4 allele have a 29% reduction in glucose transport through the BBB compared to those having the protective APOE ɛ2 allele [15]. The higher risk and lower age of AD onset in human APOE ɛ4 carriers may, therefore, be the result of their lower baseline of glucose availability in the CNS, this being present for decades before any perceived or reported impacts on cognition [16].
Consistent with the neuroenergetic hypothesis, reduced GLUT1 activity has been reported to correlate with an acceleration of AD symptomatology [17]. Diseases involving glucose dysregulation, such as diabetes mellitus, are associated with an increased AD risk [18]. Reduced availability of glucose in the brain may contribute to obesity, this condition representing a risk factor for AD even if independent of diabetes [19].
Disease processes affecting vascularity and endothelial function, including hypertension, have been linked with increased AD risk [20]. Increased risk of AD has also been associated with viral conditions such as infections with members of the herpes family, including Epstein-Barr virus, human herpes virus 6 and cytomegalovirus; all reported to alter GLUT1 activity through a negative effect on its integral enzyme, protein kinase C (PKC) [21]. Conversely, factors reported to increase PKC activity have been associated with cognitive improvements in memory-impaired individuals [22], such evidence having led to PKCs being referred to as a “cognitive kinase” [23].
Events affecting systemic blood sugar levels can play a role if they contribute to a decreased ability of adequate glucose to cross the BBB. In particular, hyperglycemia has been linked to the creation of advanced glycation end products that can impact BBB functionality and contribute to the risk of AD [24]. Dietary fats, given their role in BBB structure, must also be considered. The Chicago Health and Aging Project (CHAP) reported an increased risk of AD with higher intakes of saturated fats and trans fatty acids, while monounsaturated fats were associated with a decreased risk [25].
Metal toxicity from chronic aluminum exposure is linked with AD [9, 26] and is related to decreases in PKC activity [27]. Ethyl alcohol, which readily crosses the BBB, can affect glycolysis the main glucose utilization pathway, and alcohol, when consumed excessively, is associated with an increased risk of AD [28]. Repeated or severe episodes of systemic hypoglycemia have also been connected with dementia [29]. More directly, cerebral hypoglycemia can induce hyperphosphorylation of the tau protein, possibly contributing to the increased risk of neurofibrillary tangles associated with AD [30].
AMYLOID-β AND ITS PROTEIN PRECURSOR
The initial step in the metabolism of AβPP is critical in AD pathogenesis as it determines whether or not Aβ will be produced as a byproduct [31]. When AβPP is first acted upon by alpha-secretase (ADAM10) [32], the intermediate product is a large ectodomain referred to as sAβPPα, a product in which Aβ is no longer intact. When sAβPPα is subsequently acted upon by γ-secretase, a soluble, non-AD pathogenic p3 peptide is produced that can exit the CNS through the BBB. If, however, AβPP is first acted upon by β-secretase (BACE1) [32], the intermediate product is the ectodomain referred to as sAβPPβ that contains the intact Aβ. When sAβPPβ is subsequently acted upon by γ-secretase, Aβ gets liberated, this being a substance that cannot effectively exit the CNS through the BBB and one associated with AD’s pathology as well as its pathophysiology [33]. Factors, therefore, that promote AβPP metabolism by ADAM10 should be considered as AD benign, while those factors promoting metabolism via BACE1 should be considered as causing pathology characteristic of AD. Neuroenergetic status is hypothesized to be a dynamic determinant of the AβPP degradative pathway; inadequate CNS glucose favoring the AβPP→Aβ pathway (see Fig. 1).
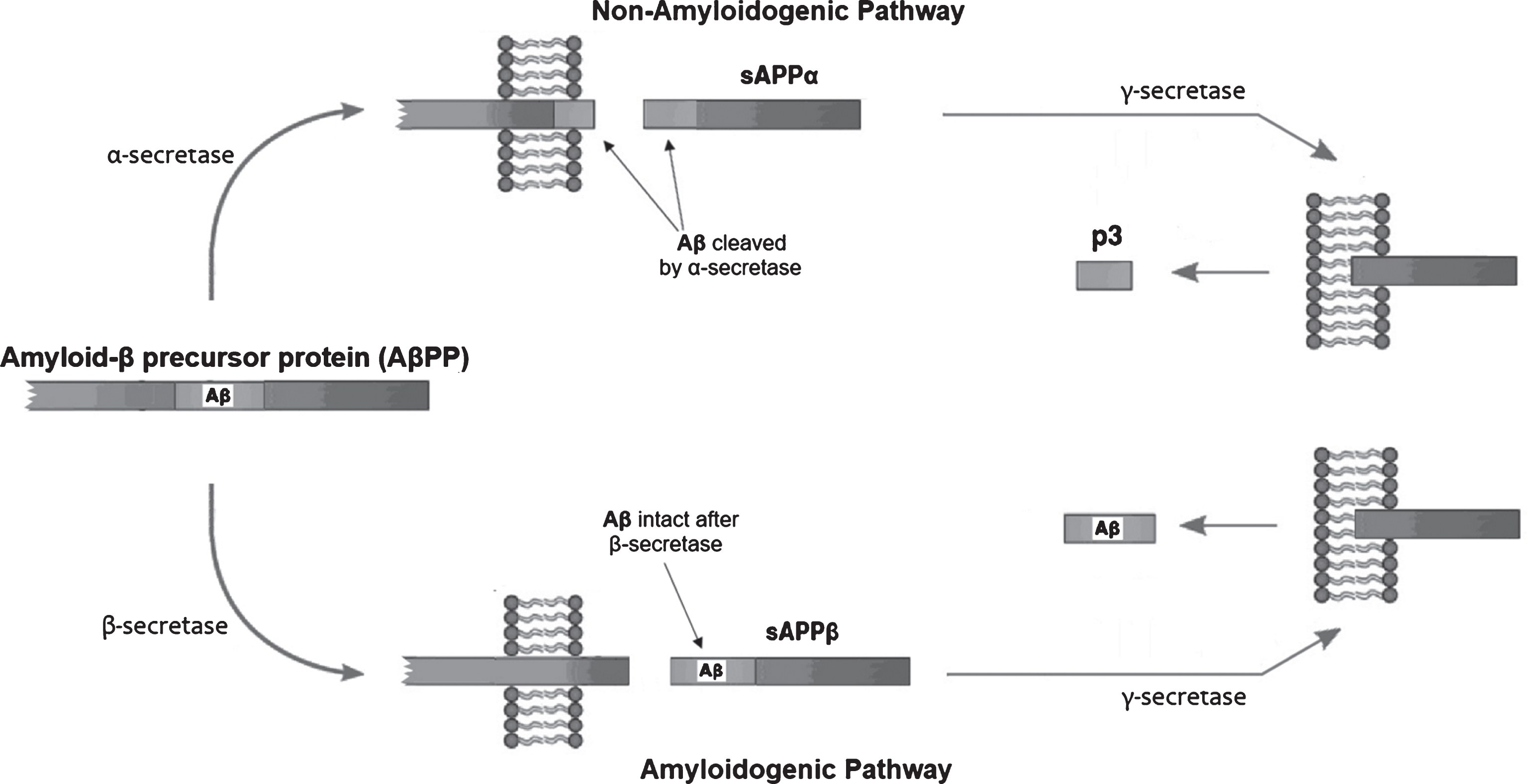
The generation of products from the amyloid-β protein precursor (AβPP). During periods of central nervous system (CNS) energy adequacy, AβPP will get cleaved along a non-amyloidogenic pathway by the action of α-secretase to produce sAβPPα, which, after the action of γ-secretase, produces p3 peptides which have an ability to exit the CNS through the blood-brain barrier (BBB). During periods of CNS energy inadequacy, AβPP gets cleaved along an amyloidogenic pathway by the action of β-secretase to product sAβPPβ, which, after the action of γ-secretase, produces intact amyloid-β (Aβ) peptides that cannot effectively exit the CNS through the BBB, and are associated with the pathogenesis of Alzheimer’s disease. Adapted from [32] (CC BY 4.0).
An understanding of a physiological basis for significant production of Aβ in normal CNS functionality has been elusive. Evidence now suggests the possibility of an antimicrobial role [34, 35]. It should be considered that one hallmark of viral and microbial success is an ability to outcompete their host for essential resources. These entities commonly rely on glucose for energy, thus potentially contributing to the risk of hypoglycemic stress if affecting glucose adequacy in the CNS. An element of the response to a microbially-induced CNS hypoglycemia could be the shunting of AβPP processing toward BACE1 to produce antimicrobial Aβ, which could ironically be neuroprotective, at least in an acute setting. Consistent with this premise, chronic viral activity, such as that from the herpes simplex virus, has indeed been associated with an upregulation of BACE1 and an increased risk of AD [36]. Inadequate CNS glucose, thus, can act as a cellular stressor contributing to increased BACE1 activity [37].
PKC status plays an indirect role through its association with GLUT1, and the ability of glucose to traverse the BBB. Decreases in PKC activity are linked with a shift from the AD-benign ADAM10 to the amyloidogenic BACE1 [38]. Conversely increased PKC activity could help reduce the risk of hypoglycemic stress in the CNS and help maintain, or even shift, AβPP degradation in favor of ADAM10. This supports a logic behind the present use of PKC activation as a treatment for AD [22]. An existing energy deficiency in the CNS can also drive utilization of myelin lipids for conversion into ketones as an alternate source of metabolizable energy [39], such a breakdown also reportedly associated with the AD process [40]. Factors known or suspected to be associated with the AD process would thus merit study for potential impacts on neuroenergetic status.
It is also noteworthy that a hypoglycemic environment in the CNS has been reported to create oxidative stress with an increased presence of reactive oxygen species [41]. This could help explain key aspects of hypotheses related to metal dyshomeostasis and AD pathogenesis [42], shifting that putative role from casual to opportunistic, that is, the product of events in the developing energy-deficient CNS environment [43, 44].
Consideration of AD as a “type-3” diabetes is of interest, given that bulk delivery of glucose to the CNS through the BBB is not an insulin-dependent phenomenon [45]. Systemic hyperinsulinemia and insulin resistance reportedly correlate with various factors consistent with AD pathogenesis [46, 47], yet improvements in cognition have been reported with the administration of intranasal insulin [48], which bypasses the BBB and gains direct access to the CNS through the olfactory bulb. This phenomenon may be the result of an effect on local neurotransmitter release and synaptic plasticity rather than an increase in net glucose transport through the BBB via the hypothesized impact of neuroenergetic status on the AD process [49, 50].
The inflammatory hypothesis suggests that CNS inflammation helps cause, or at least promote, AD. The concept is supported, in part, by reports of AD-sparing effects observed in individuals consuming anti-inflammatory drugs [51]. Inflammation is an integral part of the body’s first line of defense against invasive pathogens. mRNA levels for peroxisome proliferator-activator receptor gamma (PPARγ), a hormone-regulating receptor that plays a role in the body’s anti-inflammatory response, tend to be elevated in AD [52]. The chronic, progressive decline in the ability of glucose to cross the BBB could provoke an inflammatory response if responded to as suspected pathogenic activity in the CNS, given the sufficiency of energy elsewhere in the body. Although a local phenomenon, neuroinflammation must also be acknowledged as a glucose-consuming event with the potential to further drain this energy resource in the CNS [53]. The reduction of energy-consuming local inflammation could, in theory, represent a mechanism behind AD-protective effects from anti-inflammatory activity, but it is unclear whether, or to what degree, it could function as a block to overall neuroenergetic inadequacy and AD progression.
ALTERNATE SOURCES OF METABOLIZABLE ENERGY IN THE CNS
Another source of metabolizable energy in the CNS are ketone bodies that include D-β-hydroxybutyrate (β-HB) and acetoacetate, these substances being able to traverse the BBB independent of GLUT1 functionality. β-HB and acetoacetate are made and utilized in the human body during periods of starvation, diabetic ketoacidosis, or when there is a prolonged lack of digestible carbohydrates in the diet [54]. β-HB is also produced when unabsorbed food components, often polysaccharide in structure, are metabolized by microorganisms in the human microbiome to produce butyric acid, a short-chain fatty acid that appears in the blood as β-HB after passage through the liver [55]. By way of its contribution to available β-HB, a high-fiber diet has been suggested as an asset to brain health [56, 57]. Butyric acid is also present in milk fat, representing about 3% of the lipids in butter [58]. Nutrients or agents contributing to increased circulating levels of β-HB could, in effect, serve as alternative substrates for CNS energy metabolism, helping to explain reports of cognitive improvements in AD populations given β-HB directly from external sources [59 –61]. Caprylic acid, an eight-carbon fatty acid comprising approximately 8% of the lipids in coconut and 4% of the lipids in palm kernel, also gets metabolized to β-HB in the liver and has been reported to produce ketonemia in a case of AD, with improvements in cognition tracking with the plasma ketone level [62]. Triheptanoin, already in use clinically to treat inherited metabolic disorders, is another source of metabolic energy for the CNS not reliant on GLUT1 that would merit study for use as an AD intervention [62]. In animal models, the induction of ketosis in dogs reportedly reduced the accumulation of Aβ/tau [63]. Using the mouse model of AD, ketones were reported to reduce amyloid in the brain and improve cognition [64].
EPIDEMIOLOGICAL CONSIDERATIONS
A theme emerges when epidemiological studies examining lifestyle factors are viewed through the lens of the neuroenergetic hypothesis. Lower risks of AD have been reported in cultures with a traditional high-fiber cuisine, such as those in Sub-Saharan Africa, as well individuals adhering to the Mediterranean, or Mediterranean-DASH Diet Intervention for Neurodegenerative Delay (MIND) diets [65 –68]. Exercise contributes to vascular health, which has a positive impact on BBB in aging [69]. Lactate, created during exercise, is another fuel that can traverse the BBB via monocarboxylate transporters that are independent of GLUT1 and the BBB’s age-related changes [70, 71]. Consistent with this premise, pathologies associated with negative effects on vascular health and BBB performance are associated with an increased AD risk, including obesity [72], oxidative stress [73], hypertension [74], hyperinsulinemia [75], metabolic syndrome [73], and insulin resistance and glycemic disorders [76].
CONCLUSION
The neuroenergetic hypothesis seeks to explain the process of AD as resulting from the chronic decline in available metabolizable energy resources, mainly glucose, in the CNS. The range of metabolic consequences of this age-related, progressive decline is consistent with existing hypotheses and epidemiological observations. Viewing AD from this perspective can provide new insights and approaches to diagnosis, treatment, and risk mitigation. In order to fully test this hypothesis, CNS energy resources consistent with normal health will need to be assessed at various ages and developmental stages. There will also need to be a better understanding of the role of GLUT1, PKC, BBB, and any other functional element known to affect metabolizable energy resources in the CNS, and AD progression. Assessments can then be made of the impact of genetic factors, and volitional elements such as diet, disease, co-existing drug and dietary supplement regimens, and lifestyle issues. The goal would be to grasp the range of AD’s neuroenergetic footings to develop an effective method of integrated, personalized AD risk assessments. Interventions could then be investigated for their potential effects on the net availability of energy resources in the CNS. Additionally, non-GLUT1-dependent energy resources can be studied for their ability to affect AD’s characteristic Aβ/tau proclivity directly, or indirectly through increasing available glucose or alternate energy resources in the CNS. Research efforts could then focus on examining various methods to maintain adequate CNS energy substrates. The key will be an identification and mainstream application of factors associated with a non-amyloidogenic CNS energy profile.