
Abstract
The field of Alzheimer’s disease (AD) research has grown exponentially over the past few decades, especially since the isolation and identification of amyloid-β from postmortem examination of the brains of AD patients. Recently, the Journal of Alzheimer’s Disease (JAD) put forth approximately 300 research reports which were deemed to be the most influential research reports in the field of AD since 2010. JAD readers were asked to vote on these most influential reports. In this 3-part review, we review the results of the 300 most influential AD research reports to provide JAD readers with a readily accessible, yet comprehensive review of the state of contemporary research. Notably, this multi-part review identifies the “hottest” fields of AD research providing guidance for both senior investigators as well as investigators new to the field on what is the most pressing fields within AD research. Part 1 of this review covers pathogenesis, both on a molecular and macro scale. Part 2 review genetics and epidemiology, and part 3 covers diagnosis and treatment. This part of the review, diagnosis and treatment, reviews the latest diagnostic criteria, biomarkers, imaging, and treatments in AD.
INTRODUCTION
This is the third and final part of our three-part review series covering the most influential advances in Alzheimer’s disease (AD) research since 2010. This part covers the information most pertinent to the clinician: the diagnosis and treatment of AD. In the past, AD dementia was difficult to differentiate from other dementia-causing pathologies; however, the advent of advanced medical imaging techniques, including molecular imaging, have given clinicians and researchers a much greater insight into the neuropathological processes of their patients aiding in diagnosis. Regardless of these advances, the diagnosis of AD remains one primarily based on the clinical history and presentation of the patients as examined by the physician.
This part of this review covers the latest criteria for the diagnosis of AD, fluid biomarkers, imaging, and current treatments. The diagnosis of AD covers preclinical, mild cognitive impairment (MCI), and AD dementia. The classical terms of AD progression have been used even though the International Working Group for New Research Criteria for the Diagnosis of AD has proposed a new lexicon of AD diagnosis [1] because they do appear in more recent authoritative criterion [2–4]. Table 1 outlines the differences in this terminology.
Table approximately comparing the major salient points of the classical and proposed updated lexicon
*MCI in updated lexicon may be any etiology- cognitively impaired but biomarker negative. Neuronal Injury biomarkers may be tau or FDG-PET. Aβ+ may be decreased CSF Aβ or PET+.
Both advances in cerebrospinal fluid (CSF) and in plasma biomarkers are discussed. Because of the invasiveness of CSF collection, fluid biomarkers from more accessible locations in the body are an active area of research, especially fluid biomarkers that provide a predictive measure of impending AD.
Advances in brain imaging technology have allowed researchers to non-invasively probe into the brain to explore phenomena never dreamed possible. For example, diffusion tensor imaging (DTI), an advanced magnetic resonance imaging (MRI) technique, makes it possible to characterize the network of neuronal connections (the connectome) [5, 6]. Meanwhile, positron emission tomography (PET) has allowed for the detection of picomolar concentrations of high affinity molecular probes to detect minute concentrations of amyloid within the brain. These techniques provide a secondary source of information to clinicians diagnosing AD.
This part concludes with a review of the greatest advances in the treatment of AD. While the standard of care for AD has remained unchanged for several years, consisting of only a handful of effective symptom managing pharmaceuticals, recent advances in genetic research and immunotherapy provide hope that AD may one day be delayed to longer than the six to eighteen months afforded by current pharmaceutical therapies [7]. Of particular interest are therapies that may prevent the onset of AD early in the disease progression. It is estimated that a treatment that slows AD progression by 50% would reduce the risk of an AD dementia diagnosis by nearly half [8]. While advances in the AD treatment field lag behind those of other subfields of AD research, it is not for lack of effort. Only once greater knowledge about the pathology of AD and its molecular relationship to other parts of the brain is obtained, will further progress on the treatment front be made.
DIAGNOSIS
AD is the most common cause of dementia symptoms, followed by vascular dementia and a myriad of other neurodegenerative pathologies. Despite over a century of research progress and modern brain imaging techniques, differentiating AD from the numerous other dementia-causing neuropathologies remains difficult. Furthermore, the overlap of symptoms of the neuropathological diseases and the insidious onset of AD makes accurate early early clinical differentiation and diagnosis nearly impossible. An increasing body of literature published within the last several years has attempted to differentiate the symptom-based term, “Alzheimer’s disease”, which is a diagnosis, from the pathological features traditionally associated with AD [9]. The reason for this separation of clinical and pathological concepts is that extensive data have demonstrated that many individuals exhibit AD-related neuropathological changes postmortem without any significant symptoms of dementia antemortem [9]. While current imaging modalities using PET provide clear evidence of amyloid burden in vivo, many histopathological features of AD, such as synaptic loss, gliosis, Lewy bodies, neuron loss, granulovacuolar degeneration, and cerebral amyloid angiopathy are difficult to quantify antemortem.
AD commonly exists with other co-morbidities, such as Lewy body disease and vascular insults, which may exacerbate cognitive impairment [10]. Lewy body disease is a subset of neurological pathologies that involve the aggregation and accumulation of the α-synuclein protein, a protein which is known to form fibrils similar to amyloid-β (Aβ). Both Parkinson’s disease and dementia with Lewy bodies are included in the Lewy body disease subset. Additionally, postmortem examination of the brains of AD patients reveal cerebral vascular disease and vascular brain injury [11, 12].
According to the latest diagnostic criteria, the clinical onset of AD can be divided into three broad periods: preclinical, MCI, and AD dementia [2–4]. The lengths of time elapsed during these different periods of disease is on the order of decades [13].
Preclinical
According to the amyloid cascade hypothesis, it is now believed that the preclinical stage of AD-related pathology typically begins many years, or even decades, before the onset of AD symptoms during the preclinical phase of AD [13]. It is also well established that many asymptomatic individuals will also accumulate amyloid pathology [14]. The preclinical phase begins with asymptomatic cerebral amyloidosis. Monomeric Aβ begins to aggregate, leaving its reservoir in CSF, and begins to accumulate on the neuronal surface and synapses. During the first stage of the preclinical phase, however, these changes are not yet detectable using current techniques. Cognitive deficits have not yet manifested. During the second stage of the preclinical phase, certain CSF biomarkers, such as increased CSF tau, hypometabolism in the posterior cingulate, and cortical thinning become detectable [4]. The patient, however, still does not display any dementia-type symptoms. In stage 3 of the preclinical phase, the patient begins to detect subtle symptoms while biomarkers continue to increase. The patient may still perform within the normal range on cognitive function tests such as the Montreal Cognitive Assessment (MoCA) and Mini-Mental Status Examination (MMSE), but there is a measurable decrease from the patient’s baseline performance [8]. It is believed that once a person reaches stage 1 of the preclinical phase of AD, they will eventually develop AD if they live long enough [15].
By reviewing the available literature on various biomarkers of AD, Jack et al. proposed a model to indicate how different biomarkers change with AD progression (Fig. 1) [15]. The figure by Jack et al. provides an excellent graphical presentation showing that Aβ deposition precedes neuronal injury and AD clinical symptoms and occurs very early in the disease process.
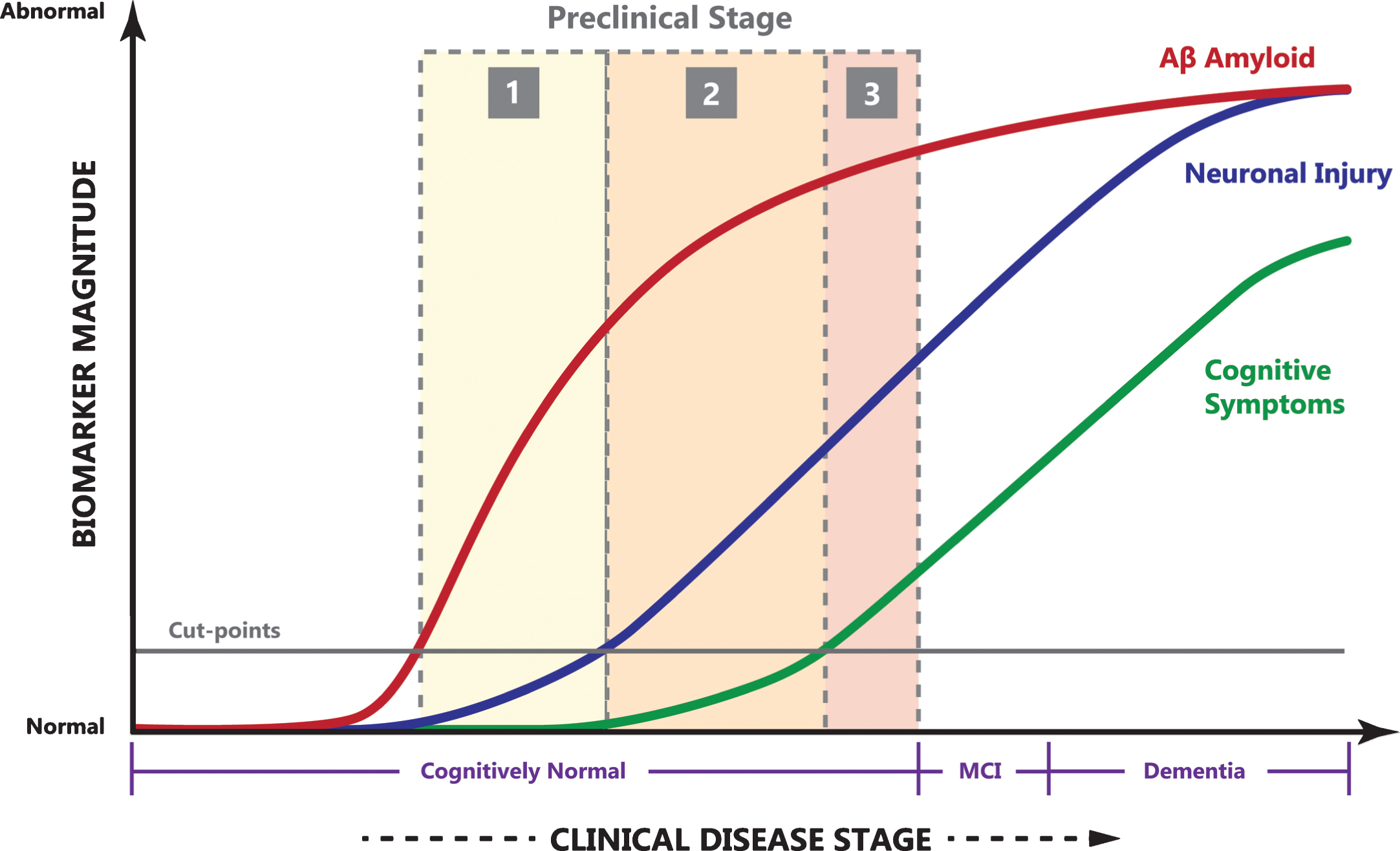
The relationship of clinical disease stage to biomarker magnitude (arbitrary units). Notice the long period of cognitively normal preclinical AD, quickly progressing though MCI to AD dementia. Image adapted with permission from [15].
Mild cognitive impairment
MCI is the phase of AD characterized by a reduction in cognitive function, albeit at a level which still allows the patient to remain an autonomous and functional member of society. It is important to note that despite advances in AD biomarkers and imaging modalities, MCI caused by AD neuropathology still requires a somewhat subjective determination by a clinician through cognitive and functional tests, and cannot yet be diagnosed by a laboratory test [3]. However, PET imaging and CSF analysis are still viable methods of differentiating dementia symptoms. When differentiating dementia, it is important to rule out any other metabolic or structural etiologies responsible for dementia-like symptoms. These etiologies may include deficiencies in vitamin B12, folate, thyroid stimulating hormone, calcium, glucose, complete blood cell count, and renal and liver function abnormalities. Serological tests for syphilis, Borrelia, and HIV should be evaluated. Other etiologies, such as structural lesions and alcohol related dementia, should also be ruled out prior to an AD diagnosis.
A patient with MCI often presents with a complaint of reduced cognitive ability relative to his or her previous level and what would be expected of a person of a similar age and educational background. Whether or not the patient or a family member notice this symptom, the decline in a variety of domains such as memory, attention, language, and executive function will be noticeable over time [3]. This may manifest itself with the patient being less efficient at performing common tasks, such as navigating a common route or shopping. Cognitive tests on the patient with MCI will often reveal scores of between 1 and 1.5 standard deviations below the mean for their age and education level [3]. If CSF or imaging biomarkers are available, a positive Aβ biomarker (decreased CSF Aβ levels and increased Pittsburgh compound B (PiB) PET uptake) and positive biomarkers of neuronal impairment indicate a high probability that the MCI is due to AD pathology. Negative biomarkers strongly suggest that AD pathology is not the cause of MCI symptoms [3].
Alzheimer’s disease dementia
Criteria for an AD diagnosis have demonstrated a reasonably high sensitivity of 81% and a specificity of 70% [2, 17] when confirmed with postmortem neuropathological studies. Unfortunately, the point where AD can be successfully differentiated from other dementia-causing diseases often occurs late in the disease process. Dementia is diagnosed by a gradual onset of cognitive or behavioral symptoms that interfere with the ability to function at work or during daily activities which are unexplained by delirium or psychiatric disorder. A diagnosis of dementia is often made following a thorough history and a cognitive function test such as the MoCA or MMSE. A diagnosis of dementia must include an impairment of at least two of the following domains: impaired ability to remember new information, impaired reasoning, impaired visuospatial abilities, impaired language function, or changes in personality or behavior [2]. Differentiating dementia from MCI is made by determining whether there is a significant interference in the ability for the patient to function at work or in typical household tasks.
Differentiating AD from other causes of dementia can often be difficult, especially at early stages of the disease progression. Probable AD is diagnosed when the patient meets the criteria for dementia, in addition to insidious onset and obvious worsening of symptoms over time. Typically, amnestic symptoms present before non-amnestic presentations such as language and visuospatial deficits. Probable AD should not be diagnosed when the patient has a history of cerebrovascular disease such asstroke [2].
Both MMSE and MoCA tests provide high enough sensitivity and specificity for the diagnosis of dementia compared to non-dementia, to be considered reliable for clinical uses [18, 19]. Beside clinical history and cognitive testing, biomarkers such as CSF Aβ levels and PET scans may yield additional insights into the etiology of the dementia symptoms. Both testing modalities in isolation are inadequate for solely making a diagnosis of AD. The 2007 International Working Group proposed that AD pathology could be recognized without dementia symptoms by low memory recall and imaging or by CSF biomarker evidence of Aβ deposition [20]. Still, reduced memory recall manifests in many diseases other than AD, rendering biomarkers the sole criteria for diagnosis. This places a conflict among different working groups on the role of biomarker evidence for the diagnosis of AD. Additional insights into the role of biomarkers are reviewed below.
BIOMARKERS
Biomarkers for AD are measurable indicators within a patient that can help to evaluate and mark the progress of AD pathology. In fact, research has indicated that the process of AD begins decades prior to the clinical onset of dementia [13], which provides the opportunity of biomarkers to aid in early diagnosis of preclinical AD [21]. This suggests that biomarkers can be integrated into the diagnostic process to pinpoint specific stages of disease progression and help physicians monitor the course of AD. AD biomarkers can be divided into two main classes: direct and indirect. Direct biomarkers are specific key proteins that reflect AD pathology, such as the deposition of Aβ protein and tau. Meanwhile, indirect biomarkers are various indices of neuronal injury and regional patterns of abnormalities that provide less direct or nonspecific evidence of AD pathology [2, 3]. Much refinement still has to be done to ensure that proper attention is paid to various biomarkers depending on the progression of the disease and whether the biomarker is useful in the short-term or long-term. For example, a reduced CSF Aβ concentration alone is insufficient evidence for an AD diagnosis.
Direct Aβ biomarkers of AD pathology include the measurement of reduced Aβ levels in the CSF as well as the evidence of Aβ deposits in the brain in PET images through the use of several specific ligands [2, 3]. Cerebral deposition of Aβ begins in middle-age and increases in frequency with an individual’s age [22]. One study showed that CSF Aβ levels decrease with age, suggesting increased Aβ deposition in the brain as time passed [23]. In this study, 34.2% of individuals aged 70–79 years and 50.0% of individuals from 80–89 years of age experienced lowered CSF Aβ levels, while 25.8% and 30.3% of individuals in these respective groups had elevated mean cortical binding potential (MCBP) [24]. MCBP in PET studies is generally understood to signify the density of “available” binding receptors with reference to a reference tissue [25]. Since these initial Aβ deposits may be upstream of other toxic amyloid species, it has been suggested that preclinical AD can be detected through biomarkers that show evidence of an initial reduction in CSF Aβ levels, followed by elevated MCBP for PiB after the Aβ deposits start to fibrillate [24]. Another study showed that such PiB retention increases as AD progresses, with levels of 31% in healthy controls, 69% in patients with MCI, and 97% in individuals with Alzheimer-type dementia [26]. It has also been shown that evidence of soluble Aβ protein correlates strongly with Alzheimer-type dementia pathology and plays a strong role in the pathogenesis of cognitive decline and eventual failure [27].
Direct biomarkers of tau accumulation include total tau or phosphorylated-tau (p-tau) within the CSF [3]. Elevated levels of tau are associated with AD progression, but this phenomenon can also occur in other neurodegenerative disorders such as prion diseases and traumatic brain injury. This can be interpreted to mean that changes to tau can also reflect general neuronal injury. As a result, the direct measurement of tau in the CSF is considered to be a strong marker of damage to neurons and synapses in general, but is also specifically associated with AD [3]. The amount of monomeric tau in the brain’s interstitial fluid can also be measured via microdialysis. A significant decrease of monomeric tau in interstitial fluid has been shown to correlate with the onset of tau aggregation [28].
Apart from tau, other biomarkers that are representative of downstream neuronal degeneration include a decreased uptake of 18-fluorodeoxyglucose (FDG) on PET in the temporo-parietal cortex as well as disproportionate atrophy in the medial, basal, and lateral temporal lobes, and medial parietal cortex, as seen through structural MRI [2, 3]. YKL-40, a CSF biomarker of neuroinflammation, has been shown to predict MCI-AD conversion [23]. Additionally, when compared with CSF Aβ42, YKL-40 has the potential to be a predictive CSF biomarker of AD.
For patients with MCI, a combination of biomarkers that show low CSF Aβ and elevated levels of tau provide extremely informative data that suggests a high likelihood of progression into AD [3, 20]. Two studies showed that such a combination can reach a sensitivity of 90–95% and a specificity of over 90% in AD, outperforming each standalone biochemical analysis [16, 20]. Combined with MRI morphometry to detect hippocampal volume, improvements in AD diagnosis have also been reported [29]. The progression of AD involves an early reduction of CSF Aβ levels, while increased CSF total tau level and adjusted hippocampal volume are biomarkers of downstream pathological processes in the course of AD [30]. Ultimately, the use of multiple biomarkers will be required to allow for the assignment of a likelihood of decline, or progression to dementia in an at-risk individual over a specific time interval of observation [3].
Cerebrospinal fluid
There are three common direct CSF biomarkers currently in use (albeit primarily in research settings): Aβ, total tau (tau), and phosphorylated-tau (p-tau). Evidence has shown that levels of CSF Aβ decrease prior to elevated levels of tau and p-tau, supporting the hypothesis that abnormal Aβ metabolism precedes tau-related pathology and subsequent neuronal degeneration [31]. Due to large variations in biomarker measurements between studies and research facilities, the Alzheimer’s Association launched a global quality control program to monitor the variability of such data [32]. It was shown that between-laboratory variability was larger, and that implementing efforts to standardize laboratory procedures would help to not only lower data variability, but also increase the diagnostic efficiency of AD biomarkers within the CSF [32]. Combining MR imaging morphometry and PET along with the detection of CSF biomarkers can significantly improve the differentiation of AD from otheretiologies [29].
Plasma
Although CSF biomarkers can be an early and ongoing indicator of AD progression and degree of pathology, the procedure required to obtain such data is too invasive for routine clinical use. To overcome these challenges, a strong focus has been placed on the development of blood-based biomarkers that can reduce costs and detect the early signs of AD. Unfortunately, none of these plasma biomarkers have the specificity and sensitivity to detect change in AD pathology. One group, however, was able to discover and validate a set of ten different lipids from peripheral blood and to use their data to predict the patients’ phenoconversion to either MCI or AD within two to three years, with over 90% accuracy [33]. Such plasma markers include serotonin, phenylalanine, proline, lysine, phosphatidylcholine, taurine, and acylcarnitine, all metabolites that decrease in level as AD progresses [33]. Plasma clusterin has also been studied, but it was found to be associated with the severity of AD but not with the incidence of AD [34]. As a result, further research and development will need to be achieved in order to determine which plasma biomarkers can be used in a cost-effective manner and as quick and easy diagnostic tools.
IMAGING
Recent advances in modern imaging techniques have pushed AD imaging to the forefront of AD research and greatly increased our knowledge of the pathogenesis of AD. High resolution MRI has allowed the differentiation of minute structures of the brain, while molecular imaging using PET has allowed clinicians and researchers to stage AD antemortem. The primary imaging modalities for studying AD are MRI to study gross anatomical and connectivity changes and PET as a molecular imaging modality to track the spread of Aβ and the microtubule tau protein. With PET imaging tracking, Aβ deposition has the potential to assist clinicians in differentiating AD from other forms of dementia, however, PET has yet to achieve widespread clinical acceptance for diagnostic purposes. The use of PET has been far more useful for researchers to gain insights into the pathogenesis of AD than MRI.
AD begins in patients with the accumulation of Aβ [35], which can be detected by an increase in fibrillary Aβ (as measured by PET) and a reduction in CSF Aβ42 concentrations, 10–15 years before clinical onset [36]. Additionally, the accumulation of tau fibrils, which form intraneuronal neurofibrillary tangles, has increasingly become a current subject of focus to complement Aβ imaging methods for research purposes [8]. These imaging biomarkers are more predictive of clinical disease progression than CSF analysis. Combining both CSF and imaging biomarkers yields the highest diagnostic accuracy, suggesting a broader application of these imaging modalities for clinical use [29]. It has been suggested that additional biomarkers, such as brain glucose consumption and medial temporal lobe atrophy have the potential to become complementary, or even replace, current biomarker based diagnostic tools [29, 37]. Even if clinicians were armed with these additional tools and a plethora of biomarker data, it is important to access the benefit that would be conferred to the patient. Regardless of its immediate clinical value, it is now beyond dispute that these imaging and fluid biomarkers have greatly increased our understanding of AD pathology.
Amyloid molecular imaging
PET has revolutionized the study of amyloid deposition in situ since its first demonstration by Klunk et al. [38]. Its high sensitivity, capable of detecting picomolar concentrations of amyloid, has allowed researchers and clinicians to quantitatively track the spread of amyloid and has aided clinicians in the staging of AD [39, 40]. Based on the 11C radio isotope [38], the PiB tracer has a nanomolar affinity for fibrillary amyloid species. Newer amyloid PET tracers now use the 18F radioisotope, which has a half-life of more than twice that of its carbon-based cousin [37, 42].
Amyloid load, measured with amyloid PET, is highly correlated with disease progression [43–49]. Aβ deposition begins years to decades prior to the onset of symptoms [50–54]. Even though amyloid deposition is correlated with cognitive decline, some individuals develop MCI and dementia without any amyloid burden. Among these individuals, amyloid positive individuals with MCI convert to dementia much quicker than amyloid negative individuals [55]. Among amyloid positive individuals with MCI, hippocampal atrophy is a better predictor of shorter MCI to AD conversion (Fig. 2) [55]. In gene carriers for early-onset AD, Aβ begins to accumulate approximately 16 years prior to MCI onset and 21 years prior to dementia onset [56].
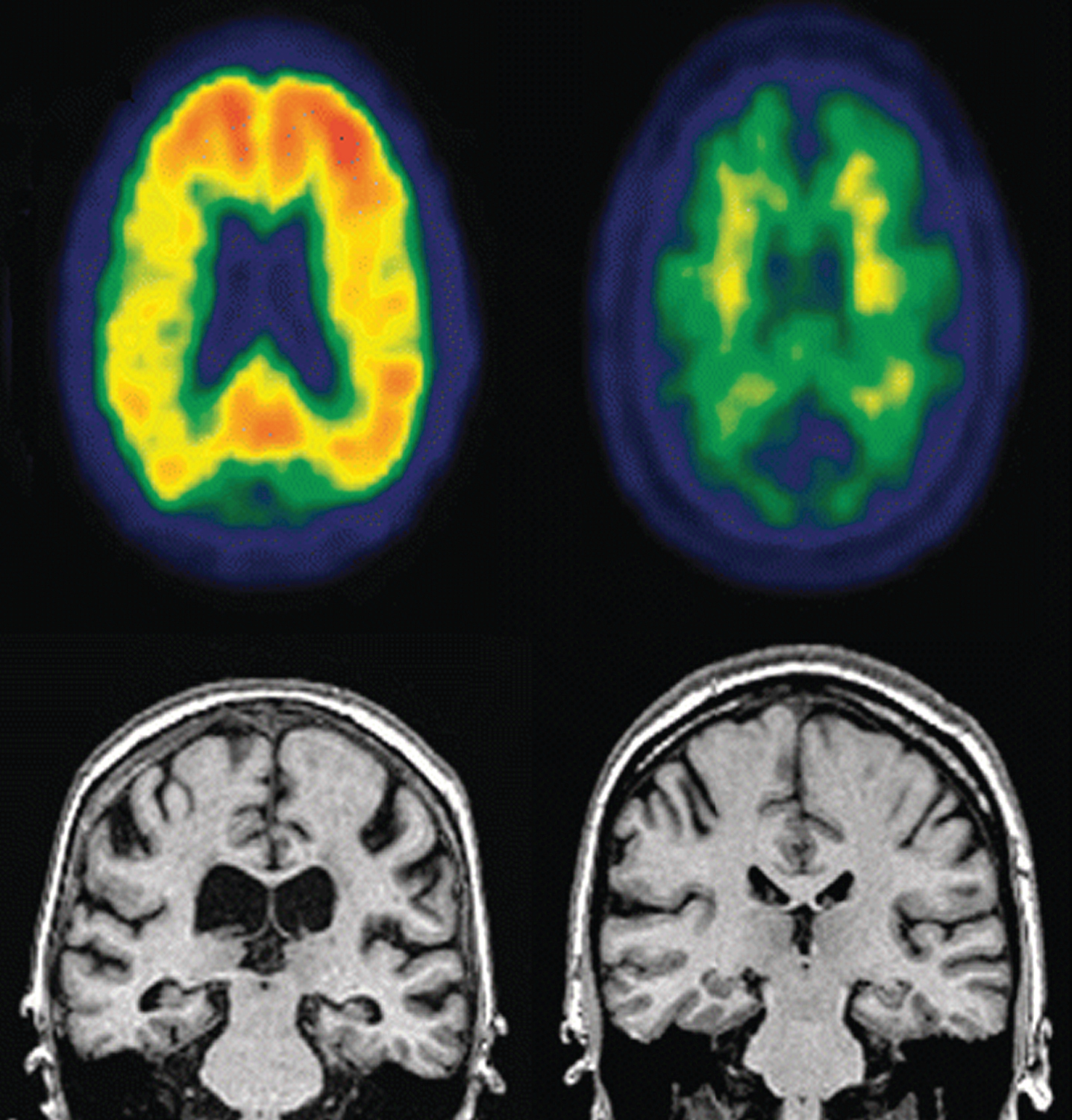
Representative PET (top row) and MRI (bottom row) images of an MCI individual that progressed to AD dementia (left) compared to an MCI individual who did not progress to AD dementia. Notice increased amyloid loading and hippocampal atrophy in the PET images and ventricular enlargement in the MRI images in progressor individual (left). Image reprinted with permissionfrom [55].
Within the last few years, radiotracers based on the 18F radioisotope, such as the FDA-approved Florbetapir, are increasingly replacing the shorter half-life 11C- based radiotracers because of the obviated need for an on-site cyclotron even though 11C- based radiotracers have a higher signal-to-noise ratio. Amyloid load, determined by Florbetapir imaging, has been shown to correlate well with actual amyloid presence and density [37, 57]. There is still some controversy about the lack of correlation between cognitive decline and amyloid load using PET in certain individuals suggesting that more specific imaging biomarkers need to be developed [55, 58]. Other Aβ targeting PET radiotracers have been developed which may provide a higher diagnostic accuracy than other radiotracers [29]. For example, 18F-flutemetamol has demonstrated both a sensitivity and specificity of greater than 93% [59].
With the advent of molecular imaging modalities, primarily based on PET, AD researchers will be able to continue to elucidate increasingly nuanced imaging biomarkers to predict disease onset, MCI-to-dementia conversion, and disease progression.
Tau molecular imaging
While molecular PET imaging detection and tracking of amyloid in the brain has been in use for just over a decade [38], novel radioligands that visualize the presence of pathological tau in vivo have captured the imagination of AD researchers. While PiB binds to amyloid fibrils with high affinity, it does not bind with sufficient affinity to neurofibrillary tangles and is therefore not suitable for tau imaging [39, 60]. This has created a need for tau radioligands to track tau deposition and progression which have been developed within the last few years [58, 61–69]. Since neurodegeneration accelerates following the onset of cognitive impairment, progress in tracking tau deposition is an imperative to gain further insight into the toxicological processes occurring when amyloid and tau interact [70].
In one such early study imaging tau pathology, Maruyama et al. were able to successfully image tau aggregates in the brains of mice [39]. Fodero-Tavolett et al. used 18F-THK523 as a novel imaging radiotracer that showed a 410-fold increase in affinity (KD = 1.7 nM) to tau aggregates as compared to Aβ aggregates (KD = 20.7 nM) [58].
Accumulation of tau, as measured by 11C-PBB3 in the medial temporal region, is well correlated with hippocampal atrophy supporting the hypothesis that tau aggregates are directly neurotoxic [39]. 11C-PBB3 has an additional advantage over 11C-PiB in that it has minimal non-specific interaction with white matter and other myelin containingstructures [39].
Tau load, as measured by PET, is well correlated with CSF tau level and is inversely correlated with CSF Aβ42 levels [70]. There is no evidence of a direct mechanism between increased tau biomarkers (both PET and CSF) and decreased CSF Aβ42. Rather, the relationship is an indirect one. Since the Aβ42 “sink” in the brain uses the “source” of Aβ42 from the CSF, Aβ neurotoxicity triggers tau pathology. An increase in tau production, believed to be signaled by Aβ aggregation in the brain [71], or converted to a more toxic species, especially in the hippocampus [72], manifests itself concurrently in increased levels of tau in the CSF. Work by Wang et al. suggests that Aβ may intensify the spread of tau, leading to neuronal atrophy [72].
Recent work by Brier et al. used tau-based PET to track the progression of tau in a cohort of patients with AD and healthy controls [73]. Brier et al. found that tau deposition in the temporal lobe more closely tracked, and was a better predictor of cognitive decline, than Aβ load (Fig. 3). By comparing various spatial distributions, the authors were able to compare both tau and amyloid loads. Despite a variance between tau and amyloid distribution, the authors were able to reconcile this variance by showing that the tau distributions were associated with disease severity [73].
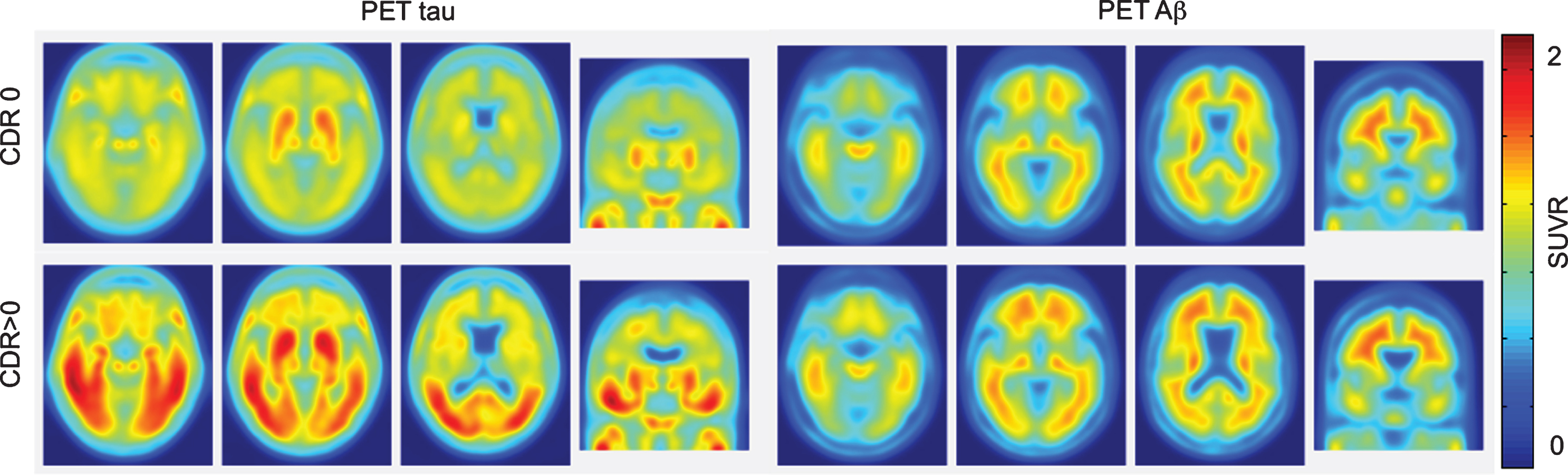
Tau and Aβ PET images in cognitively healthy controls (top-CDR0) and AD patients (bottom-CDR>0). Notice that tau deposition is a better predictor of cognitive decline than Aβ deposition. Figure reprinted with permission from [73]. CDR, Clinical Dementia Rating; SUVR, standardized uptake value ratio
Magnetic resonance imaging
MRI has proven to be an invaluable imaging modality for diagnostic confirmation and research potential given its ubiquity in both research and clinical settings. While molecular MR imaging has been demonstrated over a decade ago [74], molecular based MRI methods have yet to fully develop, largely because of a lack of sensitivity. That being said, modern MR-based molecular imaging techniques may provide for an avenue to detect amyloid or tau aggregates with PET-like sensitivity. Besides its ubiquity in clinical settings, MRI’s strength as a neuroimaging modality lies in its high resolution rendering of anatomical structures and its superior contrast over computed tomography [75].
While Aβ and tau-based PET molecular imaging has generated intense interest in the AD research community, research by Walhovd et al. demonstrated that simple MR anatomical morphometry data measuring hippocampal volume, corrected for cranial volume, was more sensitive to AD diagnosis than both CSF and PET biomarkers. The addition of these biomarkers did result in a significant improvement in the classification of AD [29]. Both hippocampal volume and FDG-PET provided for better prediction of cognitive decline than CSF biomarkers on their own.
Studies of carriers of early-onset AD genes, demonstrated that functional and structural MRI abnormalities occurred more than two decades in advance of cognitive decline, and well in advance of amyloid deposition as measured by PET or CSF biomarkers [36, 76].
Cortical thinning has been demonstrated to be a predictor that is closely correlated with cognitive decline. Dickerson et al. demonstrated that the risk of cognitive decline triples for each standard deviation of cortical thinning [77]. This biomarker is especially interesting because it provides both a predictive measure of cognitive decline, while avoiding the negative effects of CSF collection and the expense and availability of PET equipment.
DTI is an MRI based technique that measures the diffusion of water protons within the body. Different tissues, such as white matter and gray matter, have different diffusion coefficients and therefore show different levels of contrast on a DTI image. Furthermore, water protons diffuse at different rates parallel to, or perpendicular to, neuronal axons. The white matter pathways can be inferred from the eigenvector of the diffusion tensor [78]. This property, referred to as the diffusion anisotropy, can be exploited to probe the network of neuronal connections (connectome) of the brain. Changes in myelin, from edema or atrophy, will also cause a change in the diffusion anisotropy, which is detectable using DTI. Fractional anisotropy is a quantitative measure of diffusional anisotropy and is independent of fiber orientation, which makes it ideal for quantitative comparisons across groups [79]. As shown in Fig. 4, compared with healthy controls, AD is associated with white matter structural changes, demonstrated by widespread reduced anisotropy and increased diffusivity, especially in the frontal and temporal lobes, cingulum bundle and the uncinate fasciculus, corpus callosum, and posterior cingulum [80].
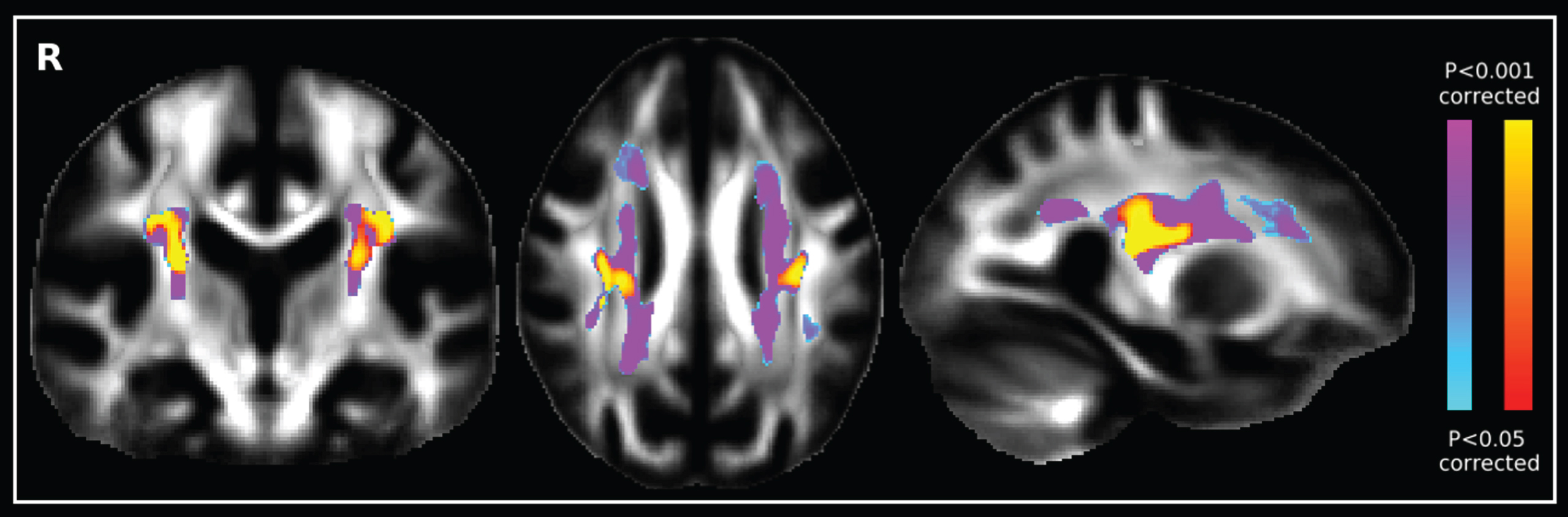
Diffusion tensor image comparing the increase of fractional anisotropy of AD patients (blue-purple scale) with healthy controls (red-yellow scale). Image reprinted with permission from [5].
DTI has routinely demonstrated that the white matter network contains “hub” regions that are highly networked. These hub regions are predominate in the precuneus, posterior cingulate gyrus, and medial prefrontal cortex [6, 82]. AD patients have shown increases in the shortest path lengths of white matter networks, and decreased global efficiency, which indicates a topological disorganization in the AD-associated networks, especially in the frontal lobe [83]. These connectivity abnormalities increase with decreased cognitive abilities.
In addition to anatomical connectivity networks, functional connectivity networks, as measured with function MRI (fMRI) are also affected in AD. Work by Sheline et al. demonstrated that brain activity in the default mode network, which is activated during resting states, is disrupted in AD individuals as compared to healthy controls [84]. Furthermore, individuals with an abnormal APOE4 status had increased levels of abnormal connectivity suggesting that early phenotypic presentations of a specific genotype can be detected with fMRI and may precede the pathological effects of amyloid neurotoxicity [84].
TREATMENT
Currently, there is a lack of, and great need for, effective treatments to prevent and slow the progression of AD. The last 30 years of research into the mechanisms of AD has presented numerous treatment strategies that target the underlying causes of neurodegeneration, with the majority of this work focused on targeting the Aβ cascade which prevents accumulation of toxic amyloid aggregates. Despite many critical discoveries and promising directions, there is a “healthy skepticism” of whether targeting Aβ pathology alone is effective for modifying disease progression in the light of many failures in late-phase clinical trials [85]. Further understanding of disease mechanisms and careful considerations of clinical trial design is providing important insights that will aid in combating AD. This portion of the review pertains to treatment strategies that modify the progression of AD, from molecular treatments to lifestyle therapies, stretching from pre-clinical to clinical trials.
Amyloid production
Since the Aβ production cascade depends on the cleavage of the amyloid-β protein precursor (AβPP) by subsequent β- and γ-secretase, inhibition or modulation of these secretases is a potential strategy for inhibiting Aβ production and thus prevent cognitive decline. γ-secretase inhibition prevents the formation of Aβ monomers; however, the β-C terminal fragment of AβPP increases as a consequence whereas modulators of γ-secretase reduce Aβ without increasing β-C terminal AβPP. Mitani et al. evaluated the effects of two γ-secretase inhibitors and one γ-secretase modulator on spatial working memory in AβPP transgenic and wild-type mice and assessed their effects on other γ-secretase substrates [86]. They found that subchronic dosing with γ-secretase inhibitors impaired normal cognition at 3 months in the AβPP transgenic (Tg) and in wild-type mice. Immunofluorescence showed accumulation of the β-C terminal AβPP fragment in the pre-synapse of the hippocampal brain regions. The γ-secretase modulator on the other hand was able to correct memory deficits in AβPP Tg mice without affecting spatial memory in wild-type mice suggesting that modulation may be preferable over inhibition [86].
β-secretase (BACE) inhibition may also reduce Aβ, however, difficulties in drug delivery have limited its progress. These difficulties are now being overcome, and orally available small molecule BACE1 inhibitors have been shown to reduce Aβ in animal models and humans. Physiological biomarker studies in AβPP Tg mice, and in non-transgenic beagle dogs with physiological expression of AβPP, show a reduction in CSF Aβ [87]. These results were reproduced in healthy human volunteers, unfortunately, toxicity of the drug has prevented it from progressing to later stage clinical trials; toxicity of this BACE1 inhibitor was not related to BACE1 inhibition as BACE1 knockout mice also exhibited toxic effects [87].
As small molecules have low target specificity, more specific therapeutics, such as antibodies, have been shown to be effective at reducing BACE1 activity and thus amyloid production in AβPP producing human cell lines and cultured primary neurons, as well as reduction in peripheral Aβ concentrations in mice and nonhuman primates. The antibody developed by Atwal et al. has a high-affinity to an allosteric region of BACE1, not the catalytic site as shown by competitive binding assays, and as such, it does not interfere directly with other BACE1 substrates. The efficacy shown here suggests that the antibody has blood-brain barrier (BBB) permeability, however, its therapeutic success will rely on sufficient brain uptake [88].
Direct targeting of Aβ and tau
Proper protein folding is essential for protein function and misfolding can result in amyloidogenic disease. Small molecules which bind amyloid slow the rate of aggregation and thus may aid in regulating protein folding-misfolding homeostasis. Nematode worms (C. elegans) exposed to Thioflavin T resulted in prolonged life expectancy and slowed aging with increases in median lifespan by 60% and maximal lifespan by 43% and 78% at 50 μM and 100 μM, while other amyloid binding and anti-aggregation compounds (curcumin and rifampicin) also increased lifespan by up to 45% [89]. To look at the effects specifically in the context of AD, a C. elegans model (CL4176) which expresses Aβ3–42 in muscle tissue (resulting in paralysis) was treated with Thioflavin T causing a reduction in the proportion of paralysis and toxicity associated with Aβ aggregation [89].
Aβ peptide binding alcohol dehydrogenase (ABAD) is an enzyme expressed in the mitochondria of neurons and its interaction with amyloid is known to decrease mitochondrial and neuron function. To inhibit the interaction between ABAD and Aβ a decoy peptide (DP), chosen from a fragment of the ABAD that binds amyloid was modified with TAT cell penetrating peptide and mitochondrial targeting peptide (Mito) for delivery to the active site in the mitochondria [90]. The DP-TAT-Mito peptide was effective at preventing ABAD-Aβ complex formation, oxidative stress, mitochondrial dysfunction and improved spatial learning and memory in the AβPP Tg mouse model of AD [90].
Immunotherapy
The tau protein is a structural protein that helps stabilize the microtubules of neurons; however, hyperphosphorylation and aggregation may occur, forming filaments within neurons that condense into neurofibrillary tangles that are associated with neurodegeneration. Modulation of the pathological tau protein species may be a viable disease intervention; one such passive immunotherapy has been shown to reduce intracellular tau pathology, resulting in improved results in cognitive assessments. In two well established transgenic mouse models of tau pathology, JNPL3 and the more aggressive P301S, peripheral injections of two different anti-Tau antibodies reduced biochemical markers of tau, and in P301S mice delayed the onset of functional decline [91].
Anti-Aβ antibodies have shown great promise in pre-clinical studies, however, their usefulness in the clinical has not been demonstrate despite the conducting of many clinical trials. Various polymorphisms and assemblies have been discovered, and detrimental immune responses, after active immunization, have been observed [92, 93]. To prevent over-activation of microglia, and to reduce dangerous immune response, Adolfsson et al. have developed a humanized anti-Aβ monoclonal antibody that, not only targets different amyloid assembly states, but is built on an IgG4 backbone that induces a lower effector response than the IgG1 antibody [94]. This IgG4 anti-Aβ antibody, called MABT, was shown to prevent amyloid aggregation, disaggregate preformed protofibrils, and prevent Aβ cytotoxicity in neurons and mixed cortical cell cultures. MABT reduced Aβ toxicity more effectively in mixed primary cortical cell cultures than the same MABT recognition region on an IgG1 backbone, and after assessing microglial response in these cultures, it was found that TNFα pro-inflammatory cytokine production was less suggesting that the reduced effector response of the MABT is superior for increasing neuroprotection. Lastly, MABT was evaluated in a Phase I safety trial, which demonstrated that MABT did not produce vasogenic edema in any patients after single or multidose trials, even in patients with ApoE4 genotype who are at greater risk of amyloid associated vasogenic edema. This passive immunotherapy is now a leading clinical trial candidate that is being funded in phase II/III prevention trials, under the name of Crenezumab, by Genentech and Lilly pharmaceuticals [95].
Therapeutics outside Aβ and tau pathophysiology
MitoQ and SS31 are antioxidants that concentrate in the mitochondria 100-fold more than do typical antioxidants. Resveratrol is a natural anti-aging agent that is known to reduce mitochondrial dysfunction in vitro. Aβ increases expression of fission genes and decreases fusion and peroxiredoxin genes in N2a cells and biochemical assays which showed significant mitochondrial damage, this Aβ damage was mediated by the use of MitoQ, SS31, and resveratrol [96]. In transgenic AD mouse primary hippocampal cells MitoQ, SS31 increased neurite outgrowth, as measured by Drp1 immunostaining, which indicates that MitoQ and SS31 increase synaptic connections in AD model neurons, while resveratrol had no effect. The formation of mitochondrial pores results in oxidative stress and cell death. Decreased CypD, an important protein in the formation of mitochondrial permeability transitions and pores, was observed by immunostaining after treatment with all three antioxidants. This indicates that these three antioxidants inhibit mitochondrial pore formation, and thus, protect the mitochondria in an AD neuronal model; mitochondrial specific antioxidants were more effective at increasing synaptic function after Aβ exposure [96].
Despite evidence suggesting that there is a benefit for the use of antioxidants in animal models of aging and AD to combat oxidative damage in the brain, human trials of antioxidants have had mixed results. To evaluate different antioxidants and their effects on AD, 78 subjects with mild to moderate AD were assigned to three different groups: either 800 IU/day of vitamin E, 500 mg/day of vitamin C, and 900 mg/day of α-lipoic acid (E/C/ALA group); or 400 mg of coenzyme Q 3 times/day; or placebo [97]. This sixteen week study found no change in CSF biomarkers related to AD (Aβ, and tau pathology); however, lower levels of F2-isoprostane found in the E/C/ALA treatment group suggest that there was reduced oxidative stress, but unfortunately this correlated with a faster cognitive decline [97]. The time frame, sample size, and the stage of AD evaluated in this study fail to show whether long term use of antioxidants at earlier stages of AD would be beneficial.
Type 2 diabetes is a risk factor for AD. Glucagon-like peptide-1 (GLP1) is an endogenous 30-unit peptide hormone that is involved in insulin regulation. GLP1 receptor agonists act as a growth factor, increasing neurite outgrowth and reducing oxidative stress on neurons, while the receptors themselves play a role in memory and learning. Liraglutide is a BBB permeable peptide analogue of GLP1 used for treating type 2 diabetes. Liraglutide was able to restore object recognition in Tg mice [98]. Additionally, in the Morris water maze, Tg mice treated with Liraglutide showed improved learning compared with untreated controls. Liraglutide corrected long term potentiation in hippocampal slices of the CA1 region of control AβPP/PS1 Tg mice, and in wild-type controls. After 8 weeks, amyloid plaques in the cortex were reduced by 50%, while plaques stained by Congo Red were reduced by 25%. Moreover, in this study, a 50% reduction in inflammation, and a 65% increase of young neurons in the dentate gyrus, was observed. Interestingly, brain AβPP and Aβ levels were significantly lower following Liraglutide treatment compared to controls [98].
Metabolism of tryptophan largely occurs via the kynurenine degradation pathway. Kynurenine 3-monooxygenase (KMO) produces metabolites that induce excitotoxicity via NMDA receptor agonism, and as such, high ratios of kynurenine to tryptophan are associated with neurodegeneration. A prodrug KMO inhibitor, JM6, was shown to increase synaptophysin levels in a AβPP Tg AD model, indicating it prevents loss of synapses; however, it had no effect on plaque load and in cognitive assessments, it improved spatial memory, but not spatial learning [99].
Rapamycin is an mTOR inhibitor that has been shown to delay aging and increase lifespan in several eukaryotic models including mice [100]. It has been hypothesized that mTOR inhibition reduces oxidative metabolism, and increases autophagy removing damaged organelles and protein. Long-term administration of rapamycin in AβPP Tg mice reduced high levels of Aβ (most likely by inducing autophagy) and prevented spatial memory loss [100]. In wild-type littermates, rapamycin did not reduce endogenous Aβ, but still improved learning and retention; therefore, it is likely that rapamycin and the TOR signaling pathway are involved in multiple processes that are important in aging and AD [100].
Increasing Aβ clearance from the brain may be beneficial in AD. This can be done by upregulation of endogenous systems for removal of amyloid across the BBB. Apolipoprotein E (ApoE) is an important regulatory protein for maintaining lipid and protein homeostasis across the BBB, including facilitation of Aβ clearance, and as such, various genotypes may predispose individuals for greater risk of AD. Transcription of ApoE is regulated by a variety of receptors, and may be agonized to increase brain clearance of amyloid. One early promising study used bexarotene, a retinoid X receptor agonist [101]. Unfortunately, numerous subsequent studies were unable to replicate those results either in part or entirely [102–106].
Aβ transport out of the brain, across the BBB is accomplished by a transporter protein, termed the lipoprotein receptor-related protein 1 (LRP1). Increasing cannabinoid system activity enhanced clearance of Aβ in vivo by a factor of 2, while parallel studies in vitro showed increases in LRP1 levels by a factor of 1.5, which provides evidence for the role of the cannabinoid system in regulating clearance of Aβ [107]. Cannabinoids have also been shown to reduce the accumulation of intracellular Aβ that precedes the formation of plaques, and is likely an early stage event in AD progression [108]. Central nervous system neuronal cell lines have been developed that produce Aβ in response to removal of tetracycline from culture; these MC65 cells die within 4 days of tetracycline withdrawal due to proteotoxicity of intracellular Aβ accumulation, alongside increases in pro-inflammatory cytokines. When cannabinoid agonists are applied, intracellular Aβ removal increases, and pro-inflammatory cytokine production decreases [108]. These studies show that the cannabinoid system may be an effective target for combating AD by clearing amyloid from neurons, and preventing proteotoxicity and inflammation.
Amnestic MCI (aMCI) is a risk factor AD, and interestingly, hyperactivity of the dentate gyrus within the hippocampus has been observed, which may be attributed to a response to compensate for loss of function, or it may directly contribute to memory impairment. Antiepileptic drugs that target and reduce neural activity in the CA3 region, have been shown to confer benefit in the memory of animals with hippocampal hyperactivity. More recently, FDA approved levetiracetam (an antiepileptic), has been shown to improve cognition in patients with aMCI [109]. A reduction in CA3 over-activity in aMCI patients taking levetiracetam resulted in corrections to their memory, such that their hippocampal activity matched that of controls, which suggests that cognitive deficits caused by hippocampal hyperactivity may not be compensatory, but rather may likely contribute to memory impairment.
Non-pharmaceutical treatment
Physical activity has been associated with cognitive ability in later adulthood [110]. It was observed that, later in life, physically fit individuals have greater grey matter volume than less fit individuals. In a 9-year follow up study, increases in physical activity were correlated with increased grey matter volume, which was shown to reduce the risk of cognitive impairment by a factor of 2, after 13 years from enrolment in the study [111]. It was found that a minimum threshold of activity (walking 72 blocks per week) was required to confer increased grey matter and reduce the risk of cognitive impairment [111].
It is presently unclear what the best forms of physical activity are to delay the effects of cognitive decline associated with aging and AD. Individuals with MCI are at increased risk of developing dementia, and therefore they are at an ideal time for intervention. In a 6-month randomized clinical trial, women who were 70–80 years of age who presented with MCI, responded well to resistance training [112]. Resistance training is thought to result in improvements to associative memory that occurs along with improvements in hemodynamic activity in these brain regions. Compared with controls, in a balance and tone training group, participants performing resistance training had observable functional changes in three regions of the cortex, as indicated by fMRI [112]. This study suggests that twice-weekly resistance training may be a promising strategy to reduce the risk of cognitive decline in seniors with MCI.
With age, functional ability and cognitive decline of older adults is common; cognitive training has been shown to prevent cognitive decline in normal aging. In order to assess its ability to prevent decline in daily functioning, the Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE) study examined the effects of cognitive training in elderly individuals randomly assigned to one of three cognitive training programs: reasoning, speed of processing, and memory training [113]. After the training period, individuals in the ACTIVE study then self-assessed their ability to complete instrumental activities of daily living to assess their functional ability. It was found that reasoning training and speed training improved various cognitive and functional abilities related to the type of training received, even after a 10-year period, while memory training improved memory up to a 5-year period after training [113]. The ACTIVE study has demonstrated that cognitive training, not only improves cognitive ability, but also functional ability.
Since AD is characterized largely by memory impairment, stimulation of neural circuits responsible for memory retention and acquisition may confer benefits. Deep brain stimulation (DBS) of the fornix/hypothalamus was evaluated in a phase I study of 6 mild AD patients, over the course of 12 months [114]. PET imaging showed the reversal of glucose impairment after just 1 month of treatment, while cognitive assessments indicated a possible reduction in the rate of cognitive decline [114]. DBS requires surgical implantation of electrodes deep into the brain and is highly invasive. While these early results show promise, due to the small sample size in this study, however, the only conclusion that can firmly be made is that there were no adverse effects and that the technique was generally well tolerated.
DBS of medial temporal structures such as the hippocampus and the entorhinal cortex may alter memory performance, as they play important roles in transforming short-term memory into long-term memory. Subjects with epilepsy that were non-responsive to drug therapies were given DBS during half of their learning trials in a virtual environment [115]. The task in this trial was to learn locations. Entorhinal, but not hippocampal, DBS resulted in enhanced memory of locations within the virtual environment, as compared to locations learned without stimulation. No adverse events were observed in this trial [115].
Removal and reduction of Aβ burden from the AD brain has yet to be ruled out as a possible disease intervention and prevention strategy. Unfortunately, the leading therapeutics are expensive, as is the case of immunotherapy. A non-pharmaceutical approach to the removal of Aβ involves scanning ultrasound (SUS) treatment to transiently disrupt the tight junctions of the BBB, making it more locally permeable. SUS was found to be safe in mice— no edema, neurodegeneration, or ischemic damaged was observed [116]. In evaluating its efficacy in an AβPP Tg murine model, SUS significantly reduced plaque load (56% less plaque area), and more importantly, reduced extracellular oligomeric (38% reduction) and trimeric (29% reduction) Aβ; working spatial memory function was restored to wild-type levels, while hippocampus-dependent spatial learning and short-term memory were also improved. Mechanistically, SUS treatment induced microglial activation and uptake of Aβ into lysosomes, although the transient opening of the BBB may also be involved in the mechanism [116]. Translation to human trials requires careful consideration due to the much larger brain size, thicker skull, and the potential of damaging the immune response.
Clinical trials
Sertraline and Mirtazapine are commonly prescribed antidepressants for use in dementia associated depression; unfortunately, their effectiveness is presently unclear. A randomized, multicenter, double-blind, placebo controlled trial, assessing the effects of various antidepressants for patients with dementia resulted in an entirely absent benefit, as compared to controls [117]. No statistically different scores on the Cornell scale for depression in dementia between 107 participants with dementia on Sertraline or Mirtazapine, verses 111 in the control group, at 13 or 39 weeks was observed. In addition, fewer controls had adverse reactions and fewer serious events than dementia subjects; this form of treatment for depression in dementia needs to be reconsidered [117].
Insulin signaling and glucose regulation in the AD brain is impaired; as such, correcting this deficiency might be beneficial to cognitive function and cerebral glucose metabolism. Patients with MCI (n = 64), and mild to moderate AD (n = 40), treated with 20 IU of intranasal insulin, had improved delayed memory (40 IU had no effect), while both doses showed no decline in caregiver-rated function; in addition, improved general cognition was observed [118]. At 20 IU, intranasally administered insulin resulted in improvements in primary outcomes, while 40 UI did not. This might be explained by the insulin-dose response curve associated with memory, where optimal levels cause beneficial effects, and doses of insulin outside this range can have negative effects. Although statistically significant results were found, the observed effects were relatively small; further exploration of intranasal insulin therapy and the effects of insulin on AD progression are warranted [118].
Memantine is indicated for use in moderate to severe AD, although it is used off label in patients with mild to moderate AD. Meta-analysis of three clinical trials of a total 431 patients with mild AD, and 697 with moderate AD, found no statistical differences in cognitive function in patients with mild AD between memantine and placebo, either for a within studies analysis or a combined analysis; for the moderate AD group, there were only small differences in some measures of cognition (Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-Cog) and Clinician’s Interview-Based Impression of Change Plus Caregiver Input), but not others (Alzheimer’s Disease Cooperative Study Activities of Daily Living (ADCS-ADL) and Neuropsychiatric Inventory) [119]. This meta-analysis indicates that there is no benefit of using memantine in mild AD, and the evidence is very small for an effect in moderate AD; more trials of memantine alone, and in conjunction with other potential drug candidates, are needed [119].
Evidence suggests cholinesterase inhibitors improve symptom management in mild to moderate AD; whether or not this is true at later stages still needs to be validated. Howard et al. performed a clinical study showing that patients with moderate to severe AD that continued with donepezil over a 52-month period, had a 1.9 point higher score on the MMSE and a 3.0 point lower score in the Bristol activities of daily living scale (BADLS)— this indicates less impairment than patients that had discontinued its use [120]. Patients that continued with memantine over this same period, scored 1.2 points higher on MMSE, and 1.5 points lower on the BADLS [120]. Both donepezil and memantine show efficacy in symptom management in later stage AD, with donepezil slightly outperforming memantine. When used in conjunction, no significant benefits or interference was observed [120]
Both vitamin E and memantine have been shown to be useful in moderate to severe AD; however, vitamin E has been shown to have no benefit in MCI, while memantine has not been shown to be useful in mild AD, and only has minimal benefit in moderate AD. A 4-year long, double blind, placebo controlled, randomized trial of vitamin E with memantine in 613 patients showed that 2000 IU/day of vitamin E slowed cognitive decline as measured by the ADCS-ADL, which translates to a 19% delay per year and a concomitant reduction in caregiver support time; this effect was largely lost in memantine, and memantine with vitamin E groups, which showed no differences between them and only minimal benefit compared to placebo [121].
An early vaccine trial of full-length Aβ was terminated early because of the occurrence of dangerous meningoencephalitis in 6% of the 300 recipients [93]. Nonetheless, those recipients having Aβ antibody responses showed less subsequent decline over a 4.5-year period [122]. Aβ vaccines show potential benefit, provided safety can be addressed. To this end, a novel active Aβ immunotherapy, CAD106, is designed to induce antibodies without T-cell response, which is expected to be the cause of adverse events in earlier vaccine trials. In a phase I clinical trial of CAD106, no adverse immune events were reported during the study or after a 2-year follow-up in 46 participants, although typical injection site side effects of the vaccine were observed, suggesting a favorable safety and tolerability. The antibody response was between 67% and 82% in two separate cohorts, which is promising. Secondary outcomes of CSF biomarkers were statistically insignificant, although this was assessed during the 52-week study, which might have been too early to detectchanges [123].
Passive immunotherapies have been a huge success in many diseases in recent years; this success, however, has not yet been realized when targeting Aβ in AD. Two phase III, double blind, randomized, placebo controlled, clinical trials of bapineuzumab (a monoclonal antibody funded by Janssen) were reported in 2014 and both trials yielded no improvement in cognitive outcomes for patients with AD, despite the fact that one trial enrolled with 1,121 carriers of ApoE ɛ4 alleles showed differences in AD biomarkers compared with controls [124]. Vasogenic edema was observed in participants treated with bapineuzumab, which increased with dose and ApoE ɛ4 allele number; this led to discontinuation of the highest dose regimen during the trial [124].
Another monoclonal antibody— solanezumab, which preferentially binds soluble Aβ— was also evaluated in two phase III, double blind, randomized, placebo controlled trials (EXPEDITION 1 and EXPEDITION 2) in mild to moderate AD patients [125]. Neither trial showed that solanezumab had any effect on ADAS-cog (cognition) or ADCS-ADL (daily function) scores compared to placebo; in terms of safety, incidence of edema was 0.5% higher than controls, but incidence of hemorrhage was 0.7% lower [125]. Interestingly, upon further analyses, a mild subgroup of AD patients, that had begun treatment early, retained some benefit at later time points in the study [126]. This led researchers to assess efficacy in more detail in the mild subgroup, in which they found less cognitive (34%) and functional (18%) decline verses placebo, while various CSF biomarker analysis indicated solanezumab Aβ engagement. While there is no evidence of solanezumab having an effect on patients with moderate AD early in this trial, a third phase III trial, in mild AD patients, is presently ongoing [127]. As this review was in press, Eli Lilly, the developer of solanezumab, announced that this clinical trial failed to produce statistically significant results (p = 0.095) [128]. It is unclear what impact this failure will have on future immunotherapy drugs.
Just recently, Sevigny et al reported the exciting results of Ib clinical trial of a human monoclonal antibody, aducanumab, for slowing the progression of Aβ plaque deposition and cognitive decline [129]. Sevigny et al. demonstrated that a dose of aducanumab of 10 mg/kg reduced plaque load by approximately 50% over a year of treatment. MMSE scores declined by only 0.5 over a year in the treatment group compared to 3 for the control groups. These results are encouraging and provide a glimmer of hope that researchers are making progress in developing efficacious pharmaceutical therapies for the delay of AD symptoms. While we are cautiously optimistic that aducanumab can survive later stage clinical trials, we are reminded of the excitement generated by the successful early clinical trials of other clinical trials, only to fail during later stage trials.
CONCLUSIONS
Advances in fluid and imaging biomarkers have demonstrated that these biomarkers have allowed for a large advancement in AD research. Molecular imaging, primarily using PET, has allowed greater insight into the spread of both Aβ and tau throughout the brains of AD patients. Advanced MRI techniques have allowed investigators to study the effects of AD on brain connectivity.
The last few years have yielded immense insights into the pathogenesis of AD. Macroscopic structural changes are now understood in the context of what is happening pathologically on a molecular level. Modern medical imaging techniques have enabled us to see the “forest”, while nanoscale methods, have let us “see the trees.” Increasingly, the reductionist tendencies of investigators have given way to a more holistic view of AD. While the cumulative sum of the “trees” has been put together to see the “forest”, there is still no effective treatment for AD, let alone a cure, despite hundreds of millions of dollars spent on AD research.
It is becoming clear that at some point in the progression of AD, excessive neurodegeneration is not reversible by simply trying to treat molecular targets such as Aβ; lost neural networks responsible for cognition and memory cannot simply regrow by reducing amyloid burden or oxidative stress, and this is heavily supported by the recent failures of promising monoclonal antibody therapies that have failed in phase III trials. Leading researchers are now shifting their focus to preventative strategies for slowing down AD in the early stages of progression [130]. The recent success of the immunotherapy drug, aducanumab, in prodromal and mild AD hints at the validity of targeting Aβ as early as possible in AD [127, 129]. Many other preventative measures, such as protection of the cell membrane from amyloid toxicity [131], reduction of chronic inflammation and oxidative stress, as well as improvements to overall health and wellbeing through proactive lifestyle choices, may be able to slow the onslaught of AD, provided that intervention begins early enough in the disease process.