
Abstract
Large-scale visualization and analysis of HPIs involved in microbial CVDs can provide crucial insights into the mechanisms of pathogenicity. The comparison of CVD associated HPIs with the entire set of HPIs can identify the pathways specific to CVDs. Therefore, topological properties of HPI networks in CVDs and all pathogens was studied using Cytoscape3.5.1. Ontology and pathway analysis were done using KOBAS 3.0. HPIs of Papilloma, Herpes, Influenza A virus as well as Yersinia pestis and Bacillus anthracis among bacteria were predominant in the whole (wHPI) and the CVD specific (cHPI) network. The central viral and secretory bacterial proteins were predicted virulent. The central viral proteins had higher number of interactions with host proteins in comparison with bacteria. Major fraction of central and essential host proteins interacts with central viral proteins. Alpha-synuclein, Ubiquitin ribosomal proteins, TATA-box-binding protein, and Polyubiquitin-C &B proteins were the top interacting proteins specific to CVDs. Signaling by NGF, Fc epsilon receptor, EGFR and ubiquitin mediated proteolysis were among the top enriched CVD specific pathways. DEXDc and HELICc were enriched host mimicry domains that may help in hijacking of cellular machinery by pathogens. This study provides a system level understanding of cardiac damage in microbe induced CVDs.
Introduction
Host-pathogen interactions (HPIs) between host and pathogen proteins play a crucial role in invasion, infection, and induction of immune response of the host [1, 2]. The study of HPIs is significant for better understanding of infectious disease mechanisms and for developing therapeutic measures [3]. Cardiovascular diseases (CVDs), which are the major cause of deaths worldwide, are known to have microorganisms as one of their etiological agent [4]. The involvement of microbes in CVDs was highlighted in the past by identification of viruses and bacteria in atherosclerotic plaques [5], sero-epidemiological data [6], and a strong association between viral infections with transplant atherosclerosis [7]. While conventional CVDs have extensively been studied [8], the contribution of microorganisms and their protein-protein interactions with human host in the development of CVDs is yet to be explored [9].
Several studies have been conducted on microorganisms and their associations with CVD. Chronic Hepatitis C virus replicates within carotid plaques and promotes a local environment of pro-atherogenic factors leading to the development of atherosclerosis leading to CVD [10]. Evidence also shows HIV infection as an independent risk factor for coronary artery disease and heart failure due to low CD4+ T cell counts related to HIV-associated central nervous system disorders rather than traditional mechanisms of stroke [11]. Acute cardiac conditions including injury, arrhythmia, hypotension, tachycardia, as well as a high proportion of co-morbid CVD have been reported in individuals infected with Severe Acute Respiratory Syndrome-Corona Virus 2 (SARS-CoV-2), particularly those requiring intensive care [12]. The interaction between the viral spike (S) protein and Angiotensin converting enzyme 2 (ACE2), which triggers entry of the virus into host cells, is likely to be involved in the cardiovascular manifestations of COVID-19. ACE2 is a part of Renin-Angiotensin-Aldosterone System that opposes the vasoconstrictive angiotensin (Ang) II functions by converting Ang II to Ang (1–7), that has vasodilatory effects [13]. Apart from this, SARS-CoV-2 infection disturbs the vascular endothelium that has an intricate role in immune regulation and inflammation. The infection leads activates the immune system leading to a hyper-inflammatory state in the vascular system, causing deleterious effects such as endothelial cells dysfunction [14]. The direct interaction between the activated viral glycoprotein Spike 1 with myocardial tissue also leads to direct tissue damage and downregulation of ACE2 receptors, thus resulting in myocardial damage [15]. High-risk human papilloma virus infection is significantly associated with an increased risk of developing CVD but the mechanism has not been fully understood yet [14].
For a better understanding of the role of microorganisms in CVDs it could be beneficial to study the molecular level interactions between host and pathogens. These molecular interactions control important biological processes within a cell and between organisms. In particular, at the cellular and molecular level, interactions between a pathogen and its host play a vital role in initiating infection and successful pathogenesis [15]. The overall mapping of host-pathogen protein-protein interactions (HP-PPIs) can ideally be represented with the help of a large network. Such a mapping can highlight the microorganisms involved in maximum number of interactions and the biological characteristics of the highly interacting pathogen proteins [16] and host proteins [17]. Visualizing the rewiring of host cell functioning by pathogens at multiple signaling pathways and cellular functions using network theory has been used for identifying the potential drug target proteins and common disease mechanisms [18, 19]. Biological characteristics like essentiality and pathogen fitness have been shown to correlate with network topological features like high number of interactions in HPI networks [20, 21]. Network positions of power have also been used to prioritize potential antiviral drug candidates [22]. New proposed measures of centrality coined for HPIs to aid in the design of multipurpose drugs include Connectivity of human proteins targeted by same virus protein, propagation speed, diversity of predators, decreased shortest path, component index, crown centrality and vulnerable centrality [23]. The development of novel drugs, vaccines and other therapeutics for CVDs caused by microorganisms is highly dependent on the knowledge gained from investigating HPIs [24].
The role of systems biology approach in predicting potential drugs via understanding the HPIs is advancing rapidly. An atlas of the gene expression signatures of Mycobacterium tuberculosis, their interactions and higher order gene functions in macrophage environment at the time of infection has been prepared using a systems biology approach [25]. Another gene interaction network study made use of systems biology approach to unravel the role of gut bacterial species in cardiovascular diseases [26]. The molecular level interactions between the host cellular components and Francisella tularensis genes were studied to understand the interplay between the host and pathogen and identified the pathways associated with the pathogen offensive strategies in invasion of host defensive systems [27].
Despite the certain involvement of microorganisms in CVDs, a systems level study has not yet been attempted to identify the main pathogens, proteins, domains, and pathways causing CVD effects. Unlike the traditional approach of considering the host or pathogen separately, a systems-level approach, considers the HPI system as a continuum of signaling proteins, and is indispensable to elucidate the mechanisms of infection. This approach is gaining increasing demand as the inference and analysis of HPI regulatory, metabolic, and protein–protein networks shed light on several infection mechanisms simultaneously. The knowledge derived from the HPIs may largely contribute to the identification of new and more efficient therapeutics to prevent or cure infections [28]. Our laboratory recently reported the use of topological parameters from host pathogen interaction networks for development of a highly accurate random forest algorithm for selection of therapeutic targets for treatment of microbial CVDs [29].
In this study, all the unique experimental HP-PPIs were collated from different databases and used to construct a tripartite network composed of three entities, namely: pathogens, pathogen proteins and host proteins as shown by the schematic in Fig. 1a. Next, the HP-PPIs involved in CVDs were also represented as a tripartite network (Fig. 1b). Both the networks were compared so that unique proteins and pathways of HP-PPIs leading to microbe induced CVDs could be discerned. The scale-free nature of the networks was evident at every level, in that a small number of proteins have remarkably high number of interactions in pathogens as well as in the host. The common pathways of CVD and whole pathogens associated HPIs were mainly related to immune system, metabolism, signal transduction, post-translational protein modification and cytokine signaling. However, the pathways specific to microbe induced CVDs were mainly related to signaling by Nuclear Growth Factor (NGF), Fc epsilon receptor and Epidermal Growth Factor Receptor (EGFR), ubiquitin-mediated proteolysis, Epstein Barr virus infection and Human T-cell Leukemia Virus 1 (HTLV-1) infection. The role of specific pathways in mediating CVD was validated by comparison of the pathways to gene expression datasets of myocarditis, endocarditis, and pericarditis in human heart. In an attempt to discern the homologous domains to be involved in molecular mimicry, the DEXDc (DEAD-like helicases superfamily) and HELICc (Helicase superfamily c-terminal domain) were found enriched amongst pathogens. Thus, in this study the proteins and pathways specific to CVDs have been unveiled by of comparison between CVD specific and entire set of HPIs.

A layout of host-pathogen protein-protein interaction networks. a) The schematic of wHPI network with distribution of pathogens shown in blue circular shapes and their respective numbers of proteins in orange pentagonal shapes interacting with total number of host proteins shown in light red ellipse. b) The schematic of cHPI network with distribution of pathogens shown in red circular shapes and their respective numbers of proteins in purple pentagonal shapes interacting with total number of host proteins shown in green ellipse.
A total of 61,218 experimentally determined HPIs were gathered from several HPI databases (listed in the methodology section) between the human host and all the different categories of pathogens and wHPI (whole host-pathogen interactions) network was constructed. Similarly, a comprehensive CVD associated HPI (cHPI) network was constructed for 14,951 experimentally determined HP-PPIs associated with CVDs between human host and pathogens and has been reported previously in the MorCVD database. The statistics for host proteins, pathogens, and their proteins of the resulting wHPI and cHPI networks are shown in Fig. 1a and 1b, respectively.
The interactions computed within the host proteins of the wHPI network resulted in a connected component of 9270(91%) of the total host proteins of wHPI network (Fig. 1a). Similarly, the computation of interactions amongst host proteins of cHPI network showed that 2801(89%) of the total host proteins were connected in a single component as shown in Fig. 1b. In both the networks viral proteins constituted the maximum HPIs followed by bacterial proteins. The distribution of HPIs across different pathogen species having maximum number of pathogen protein interactions in the wHPI and cHPI network is shown in Table 1 that also shows the abundance of interactions of pathogens in CVDs. The overall layout of the tripartite cHPI network for microbial CVDs generated from Cytoscape is shown in Fig. 2.
Distribution of HP-PPIs across top pathogens (Microbes in bold are top pathogens in cHPI network but not in wHPI network)
Distribution of HP-PPIs across top pathogens (Microbes in bold are top pathogens in cHPI network but not in wHPI network)

The layout of the cHPI network. The network shows the host-pathogen interactions between host and pathogen proteins. On the right-hand side there is zoomed version of a small portion of the large network.
Both the wHPI and cHPI network followed the power law similar to other HPI networks [21], with correlation value of 0.845 and 0.728 and R2 value of 0.902 and 0.825, respectively. The high correlation and R value indicate that it is a scale free network with few nodes having large number of interactions and majority of the nodes having a small number of interactions. The average clustering coefficient value of wHPI and cHPI network was 0.453 and 0.521, respectively, much higher as compared to that of 0.018 in the random networks, further validating the node organization. The degree distribution of wHPI and cHPI network is shown in Fig. 3a and 3b, respectively. From the figures, it is apparent that only few pathogens and their proteins are responsible for a remarkably high number of interactions with a subset of host proteins and hence follow the power law.

The degree distribution graphs of the network. The scatter plot of nodes based on their degree values is depicted in the graphs. a) The node vs degree graph of the wHPI network. b) The node vs degree graph of the cHPI network. The red line indicates the fitting of power law in both the networks.
The proteins having a large number of interactions (high degree) and degree exponent <2 were considered central in the network. The comparison of such central proteins in wHPI and cHPI network is as follows:
1) Pathogen proteins
There were 478 central viral proteins in the wHPI network from 44 viruses and 103 central proteins in cHPI network that originated from 28 viruses. Similarly, there were 228 central bacterial proteins of wHPI network from 22 bacteria and 73 central bacterial proteins of the cHPI network from 17 bacteria. The comparison between the mean degree value of central and non-central proteins is given in Supplementary Table 1. After intersecting the central proteins, 113 proteins (52 viral and 61 bacterial) were found to be shared by wHPI and cHPI network. The top 10 central viral and bacterial proteins that were unique to the cHPI network and were not central in the wHPI network are listed in Table 2. These pathogen proteins are likely to have an important role in specific CVD complications due to microbial infection.
Top 10 highly interacting pathogen proteins in the cHPI network specific to CVDs
Top 10 highly interacting pathogen proteins in the cHPI network specific to CVDs
2) Host proteins
There were 287 central host proteins in the wHPI network that had interactions with proteins of 415 pathogens (225 viruses, 168 bacteria and 22 other species) and the 78 central host proteins of the cHPI network interacted with proteins of 124 pathogens, (81 viruses, 28 bacteria and 15 other species). The mean degree value of non-central host proteins was much lower than the mean degree value of central host proteins of the respective networks as shown in Supplementary Table 1.
The central nodes from the intra-species interactions between host proteins are essential for information flow in the network and are more likely to be associated with the disease [30]. In intra-species wHPI network there were 245 and in the intra-species cHPI network there were 81 central host proteins. Comparison of the wHPI and cHPI networks showed that 58 central inter-species and 21 central intra-species proteins were shared by both the networks. However, 20 central proteins from the inter-species interactions and 60 central proteins from intra-species interactions were found unique to the cHPI network. These unique central proteins are likely to be solely associated with CVD effects of microbes rather than the usual host response to pathogenic infections. The high number of central unique proteins among the intra-species interactions of the cHPI network indicates that there is an exclusive subset of host proteins which is specifically involved in the flow of information during CVD condition in the body that is different from the subset of central intra-species proteins of wHPI network. The top 10 central proteins (from both inter-species and intra-species interactions) that are unique to the cHPI network are listed in Table 3. We evaluated the sensitivity of the confidence of interactions on the topological parameters of the network. Upon adding medium and low confidence interactions for the intra-species cHPI and wHPI network proteins, no change was observed in the list of top 10 central proteins reported in this study. However, the degree of a few nodes was observed to change in both the cases.
Top 10 highly interacting host proteins in the cHPI network specific to CVDs
1) Pathogen proteins
Mapping of biological characteristics of pathogen proteins was carried out based on virulence prediction and ontology analysis to probe the mechanism of action as described in the methodology. In the wHPI network 1452(74.5%) of the total viral proteins and 1027 (35.87%) of the total bacterial proteins were predicted to be virulent. Similarly, in the cHPI network 628 (66%) of the total viral proteins and 522 (26%) of the total bacterial proteins were predicted to be virulent. The fraction of virulent viral proteins dominated in both the networks.
Virus and bacteria use different types of infection strategies. While many bacterial pathogens are intracellular, others use diverse processes and systems to secrete toxins and virulence factors into the extracellular milieu of the host cell. Bacteria also secrete proteins that interact with host proteins to adhere with host proteins or disrupt the immune response mechanisms [31]. In our study, 381 bacterial proteins of the wHPI network and 92 of the cHPI network were mapped as secretory bacterial proteins. Of the secretory bacterial proteins, 266 of the wHPI network and 72 of the cHPI network were predicted as virulent. Thus, in case of bacteria higher fraction of secretory proteins were observed to be virulent than the central ones.
2) Host proteins
The host proteins were biologically characterized based on essentiality, immune-relatedness, host factor role and extracellular location. Among the total host proteins of the wHPI network there were 5462(54%) essential proteins, 4759(47%) host factors, 741(7.35%) were immune related proteins and 3804(37%) proteins were extracellular in nature. The cHPI network contained 1863 (59%) essential proteins, 1777 (56%) host factors, 627 (19%) immune related proteins and 920 (29%) proteins were extracellular in nature. Overall, the cHPI network host proteins had a higher fraction of immune related and host factor proteins in comparison with the wHPI network proteins.
Biological attributes of the central proteins of wHPI and cHPI networks
1) Central viral proteins
Amongst central viral proteins, 391(82%) in the wHPI network, and 80 (78%) in the cHPI network were predicted to be virulent. In contrast, very few of the central bacterial proteins of wHPI and cHPI networks were predicted to be virulent. However, a higher fraction of non-central secretory bacterial proteins of wHPI (82%) and cHPI (78%) networks were predicted to be virulent. This indicates that for a bacterial protein to be virulent, high number of interactions are not necessary, but the protein is likely to be secretory in nature.
2) Central host proteins
The biological features of the central proteins from the inter-species and intra-species interactions were studied for both wHPI and cHPI networks. Mapping of central host proteins of the wHPI and cHPI networks with biological attributes showed that both inter-species and intra-species central host proteins had high fraction of host factors, extracellular and essential proteins. In the cHPI network, the fraction of immune proteins was much higher in the intra-species central host proteins. The fraction of central host proteins mapping to the biological attributes is shown in Supplementary Table 2.
Biological and network topological feature mapping of the host proteins interacting with viral proteins was also carried out as there were a very high number of interactions between the two. It was observed that the central proteins of the virus interacted with a high fraction of essential (88%) and host factor proteins (72%). The host proteins interacting with the central viral proteins also had a high fraction of central human proteins (96%). Our observations are in line with the previous reports showing that virus interacting proteins of the host occupy positions of power in the network [32]. It has also been suggested that viral proteins tend to target more central and highly connected host proteins that help the viral proteins in invading the host cell and hijack host’s machinery for its own use [33].
Functional enrichment analysis
1) Gene ontology analysis
The KOBAS server mapped 92% pathogen proteins and 98% of the host proteins of the wHPI network. 86.5% pathogen proteins and 99.9% of the host proteins of the cHPI network were also mapped by the KOBAS server. Some common enriched biological processes for the host proteins of wHPI and cHPI network were observed which included apoptotic process, positive regulation of transcription (DNA-templated), protein homodimerization activity, signal transduction, protein ubiquitination and phosphorylation, MAPK cascade, and GTPase activity. Similarly, the molecular functions of the host proteins of both the networks were predominantly represented by protein binding, ATP binding, metal-ion binding, DNA binding, transcription regulator activity and catalytic activity. The location of host proteins involved in wHPI and cHPI networks was predominantly intracellular. Similar to the host proteins, there were some common and enriched gene ontology components observed for the pathogen proteins of the wHPI and cHPI networks. Such common biological processes were small molecule metabolic process, cellular nitrogen compound metabolic process, biosynthetic process, oxidation-reduction process and cellular amino acid metabolic process. The common predominant molecular functions were catalytic activity, nucleotide binding, protein binding, ATP binding and hydrolase activity. The pathogen proteins of wHPI and cHPI networks were mainly located in the host cell cytoplasm, nucleus, and other intracellular parts. These ontology components were believed to be involved in both CVDs and other pathogenic infections.
However, certain biological processes, molecular functions and cellular components were found to be enriched exclusively for the proteins of cHPI network and were not enriched in the wHPI network. These ontology components characterize the nature of the proteins associated with CVDs and determine the particular processes and functions carried out by the proteins that comes in action to cause the CVD effects in the body rather than just usual pathogenic conditions. Such CVD specific enriched gene ontology components are shown in Fig. 4 (host proteins) and Fig. 5 (pathogen proteins). Taken together, the comparison of ontology analysis of the CVD associated host and pathogen proteins with the whole wHPI proteins indicates that during the CVD condition in the body, the pathogen proteins mainly perform single-organism metabolic processes, bind to nucleosides, and are present in extracellular vesicles and organelles. The host proteins are also involved in the single organism signaling, cell communication, perform cellular macromolecule metabolic processes, and are present in the organelles.

Gene ontology analysis of host proteins. a) The bar plot of enriched biological processes of host proteins; b) The bar plot enriched molecular functions of host proteins; c) The bar plot of enriched cellular components of host proteins. The blue bar represents the number of proteins; the green bar represents the reference p-value, and the red bar represents the p-value of the respective ontology term.

Gene ontology analysis of pathogen proteins. a) The bar plot of enriched biological processes of pathogen proteins; b) The bar plot enriched molecular functions of pathogen proteins; c) The bar plot of enriched cellular components of pathogen proteins. The blue bar represents the number of proteins; the green bar represents the reference p-value, and the red bar represents the p-value of the respective ontology term.
2) Pathway analysis
The aim of the pathway analysis was to identify the pathways that are exclusively involved and over-represented in CVD complications rather than the usual pathogenic infections. Hence, the pathway analysis of both whole cHPI associated host proteins as well as CVD associated host proteins was carried out to identify the pathways that are specific to CVDs during microbial infection. A total of 1787 and 1420 pathways were found to be enriched for the host proteins of wHPI and cHPI network, respectively with statistically significant p-value < 0.05. The commonly enriched pathways for the host proteins of both the networks were related to immune system, metabolism, signal transduction, post-translational protein modification and cytokine signaling. However, certain enriched pathways that were specific to the host proteins of cHPI network only were Signaling by NGF (R-HSA-166520), Epstein Barr virus infection (hsa05169), HTLV-1 infection (hsa05166), Fc epsilon receptor (FCERI) signaling (R-HSA-2454202), Herpes simplex infection (hsa05168), Viral carcinogenesis (hsa05203), Signaling by EGFR (R-HSA-177929), Cellular responses to stress (R-HSA-2262752), VEGFA-VEGFR2 pathway (R-HSA-4420097), ubiquitin-mediated proteolysis (hsa04120), and NGF signaling via TRKA from the plasma membrane (R-HSA-187037). These pathways are proposed to be involved in causing the CVD effects through microbial infections. The results indicate that the CVD complications can be specific to certain pathogens. However, other host specific pathways could also be identified and may be used by multiple pathogens.
The validation of the pathways enriched in host proteins from the cHPI network with the GEO datasets showed that similar pathways were enriched in common cardiovascular conditions induced by microorganisms. Interestingly, the maximum similarity of the pathways from the cHPI network was with that of the Human iPSC-cardiomyocytes infected with SARS-CoV-2. In this dataset of viral myocarditis, 84% of the significantly enriched pathways from our network were similar. Comparison with other conditions also showed overlap between pathways, i.e., 54% in endocarditis and 78% in pericarditis GEO datasets. In total, 232 such enriched pathways of the HP-PPIN were found to be present in all the three gene expression datasets of heart damage and were significantly enriched. The top 20 such enriched pathways are shown in Table 4 along with their respective p-values in each GEO dataset. A Venn diagram of the pathway overlap between the different datasets is shown in Fig. 6. This validates the findings with respect to enriched host response pathways identified from the HP-PPIN.
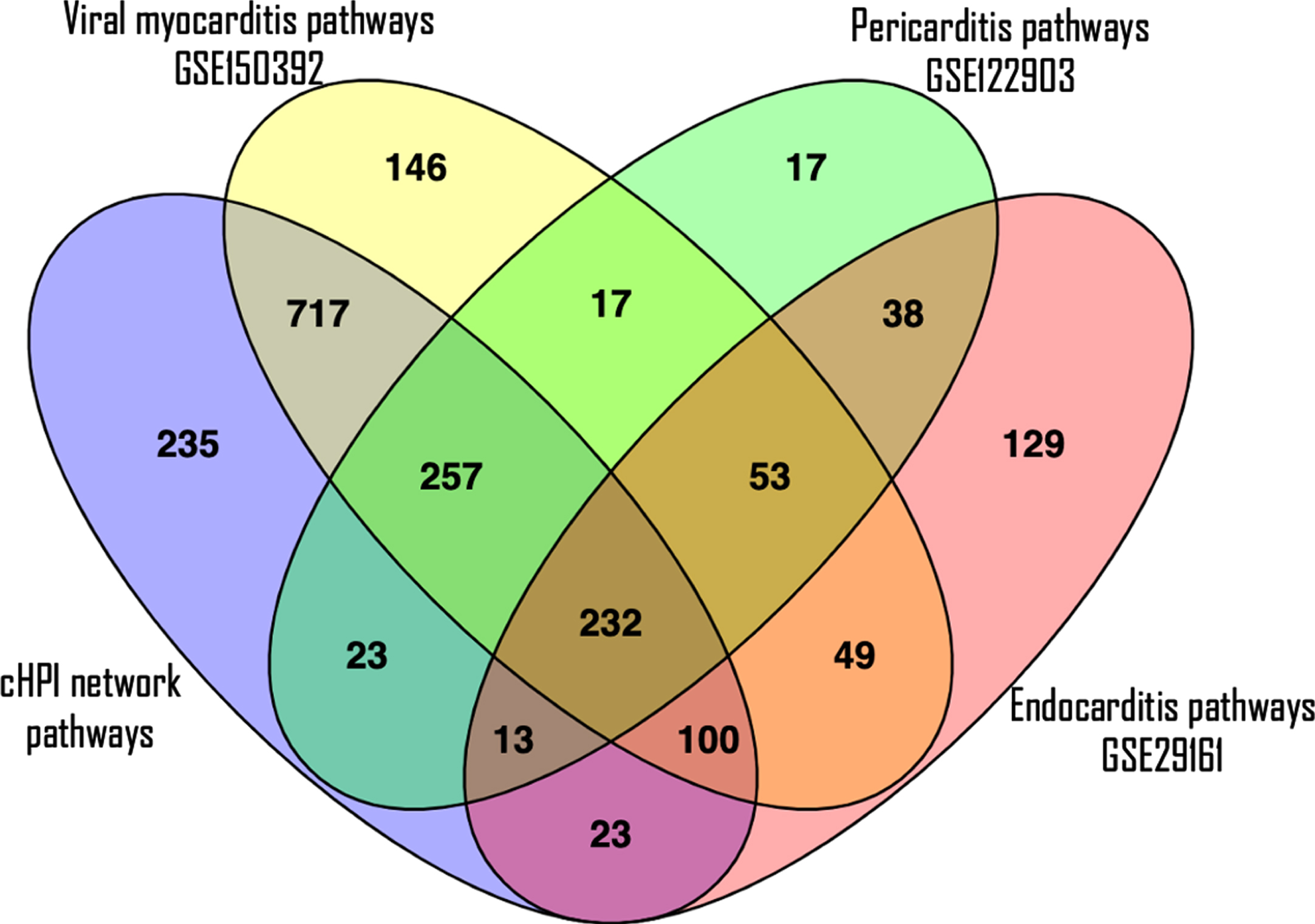
A Venn diagram to see overlapping of number of enriched pathways. The purple ellipse represents the total number of enriched pathways of cHPI network; the yellow ellipse represents the total number of enriched pathways involved in viral myocarditis related GEO dataset (GSE150392); the green ellipse represents the total number of enriched pathways present in pericarditis related GEO dataset (GSE122903) and the pink ellipse represents the total number of enriched pathways present in endocarditis related GEO dataset (GSE29161).
P-value comparison of top 20 enriched pathways of cHPI network with each GEO dataset
3) Enriched protein domains: Structural mimicry of host protein domains by pathogen
The pathogens utilize their domains homologous with the host protein domains for molecular mimicry to hijack the host machinery [34]. Hence, such homologous domains were identified for the pathogen proteins of cHPI network which they could use to imitate their counterpart, hijack the host cell and cause CVD effects. In case of viral proteins, 36 enriched homologous domains were identified that may be utilized to mimic the interactions with host proteins. In bacteria, 44 such enriched homologous domains were identified. The F-actin binding domain (FABD) and Interferon-regulatory factor 3(IRF3) were the top enriched viral domains. Alkaline phosphatase homologues (alkPPc) and Alpha-2-macroglobulin family (A2M) were the top enriched domains in bacteria. The list of all the enriched homologous domains of virus and bacteria along with fold enrichment and p-value are given in Supplementary Tables 3 and 4, respectively. Highly enriched domains that were shared with host in both virus and bacteria were DEAD-like helicases superfamily (DEXDc), Helicase superfamily c-terminal domain (HELICc), Protein tyrosine phosphatase, catalytic domain motif (PTPc_motif), Serine/Threonine protein kinases, catalytic domain (S_TKc) and ATPases Associated with a variety of cellular Activities (AAA). The domains identified to be utilized by both viral and bacterial proteins represent often used mechanisms to mimic the host protein interactions for rewiring the host cell machinery to cause CVDs.
A systems level understanding of interactions between pathogen and host proteins is a crucial step to establish a relationship between pathogen and host [24]. A collective study of interactions by multiple pathogens provides an insight about the mutual effect and strategy of pathogens to create the disease condition. Hence, a tripartite cHPI network was constructed in this work to analyze the network patterns and biological characteristics of all the HPIs leading to CVD. It was further compared with the wHPI network to distinguish the CVD specific proteins and pathways. A high number of HP-PPIs were observed from Papilloma virus, Herpes virus and Influenza A virus in both the networks. Interestingly, Saccharomyces cerevisiae, an opportunistic pathogen causing several disease like fungemia, endocarditis, pneumonia, peritonitis, urinary tract infections, skin infections, and esophagitis[35] was found to have a large number of HP-PPIs.
The top 10 pathogens of both wHPI and cHPI network were found almost similar expect 5 pathogens (shown in Table 1) that were amongst top pathogens in the cHPI network but not in the wHPI network. However, the large number of HP-PPIs reported for a particular pathogen does not indicate its central role in the disease network. For instance, Adenovirus and Hepatitis A/B virus have many HP-PPIs reported, as observed in the whole wHPIs but their proteins are not reflected amongst the central pathogen proteins of the cHPI network. Conversely, proteins from the Human Respiratory Syncytial Virus are not abundant in the whole wHPIs but they were central in the cHPI network. Both the network contained a large number of HP-PPIs and the computation of interactions between host proteins created a connected network. Given the important role of central hub proteins in the spreading phenomenon [36, 37], the hub proteins of these well-connected intra-species interactome were considered to be involved in the spread of the infection.
The most central in the intra-species cHPI network were Ubiquitin-60 S and 40 S ribosomal proteins and Polyubiquitin C&B proteins while the most central hub protein of the inter-species cHPI network were Alpha-synuclein, Breast cancer type 1 susceptibility protein, and TATA-box-binding protein. The ubiquitin-mediated proteolysis pathway was enriched in cHPI network as well as in the three GEO datasets examined. The proteins of ubiquitin system play a key role in fine-tuning the innate immune response of the host and can also be usurped by the pathogen to evade the innate immunity [38]. The viruses have been found to connect with the ubiquitin pathway at many levels to enhance viral replication [39]. Modulation of the host ubiquitin system by bacterial effector proteins inhibits innate immune responses and hijacks central signaling pathways [40]. Early inhibition of the ubiquitin-proteasome system in pathological hypertrophy restricted disease progression while enhancement of proteasome activities improved the outcome conditions like myocardial infarction caused by oxidative damage [41]. The highly central position of ubiquitin protein in pathogen interaction network as well as the host intra species network combined with pathway and domain enrichment analysis indicates the significant role of this protein in microbe induced heart disease.
Integrated ontology and pathway analysis showed the activation of immune system including innate immune system, complement system and cytokine signaling in response to the pathways of microbial infection. The immune response proteins were high in fraction in the central nodes of the intra-species network, indicating its highly significant role. The inferred significance of immune response due to the collective effect of multiple microbial infections in CVDs is in agreement with our ontology and pathway analysis results. Chronic inflammatory cardiomyopathy has been attributed to the triggering of heart autoimmunity by cytopathic effects of the microbes [42]. The role of immune mediated inflammatory response in cardiac damage has previously been reviewed [43]. The innate immune system interacts with metabolic disturbances in pathogenesis of CVDs [44]. A definitive link between CVD and abnormalities of immune activation has already been reported in HIV infected individuals [45, 46]. SARS-Cov-2 infection activates innate immune response in pluripotent stem cell derived cardiomyocytes [47]. Bacterial infection has been found to impair the endothelial function by circulating endotoxins, induce proliferation of smooth muscle cells and local inflammation, and activate the innate immune response [48].
The enrichment analysis for enriched protein domains shared between pathogen and host proteins showed protein helicase domains like DEXDc and HELICc. The direct interaction of the host and pathogen proteins containing DEXDc and HELICc domains with the same host protein was verified from the HP-PPIN. The helicase domains are a part of proteins like Retinoic acid inducible gene I (RIG-I) and Melanoma differentiation associated gene 5 (MDA5) that are key cytosolic PRRs for detecting nucleotide PAMPs of invading viruses [49]. The DEXDc domain senses the viral RNA and activates the caspase recruitment domain (CARD) via IRF-3, NF-κB, Type I Interferon and Interferon stimulated genes that directly inhibit viral replication. Several viral proteins are known to selectively abrogate the signaling by RIG-I and MDA5 to inhibit the innate immune response [50]. The RIG-1/MDA5 constitutes a surveillance system conserved across vertebrates [51]. Apart from enrichment of DEXDc and HELICc among host and pathogen domains, the constituent proteins of this innate immune response were also the top enriched ontology terms of pathogen proteins in the HP-PPIN, namely - host type I interferon mediated signaling pathway, host IRF-3 activity, and host MDA5 activity. Taken together, both findings highlight the mechanisms for the activation of the host innate immune response and devious methods of its inhibition by the pathogens.
Conclusion
Construction of cHPI network of pathogens proteins interacting with host proteins in microbial CVDs has allowed us to define the biological role of its constituent entities. The comparison of cHPI network with the wHPI network has overcome the bias due to availability of the experimental HPIs and aided in identifying the proteins and pathways that are central and specific to CVDs. This work identifies the main organisms, host proteins and pathways specifically involved in pathogenesis of microbial CVDs. Our analysis paves the way for future identification of novel therapeutics based on network topology and biological characteristics.
Materials and methods
Network construction
All the experimentally determined HP-PPIs related to the human host and all pathogens were gathered, housed in several HPI databases: Reactome [52], HMDAD [53], PHI-base [54], OrthoHPI [55], VirusMINT [56], MatrixDB [57], BioGrid [58], HPIDb [59], MINT [60], IMEx [61], IntAct [62], UniProt [63], MPIDB [64], VirHostNet [65], I2D [66], InnateDB [67], DIP [68] and PHISTO [69]. These databases are exclusively dedicated for HPIs, however, there are some other databases that serve as data repositories to search and collect protein/gene interaction data, provide the information of PPIs of a single host [70]. These include STRING, HPRD, GeneMANIA and PINA. The extracted raw data was pre-processed to ensure that there was no discrepancy in the data. The following pre-processing steps were carried out - Filtration and removal of the data pertaining to the interactions between pathogen proteins and hosts other than humans. Conversion of different protein IDs collected from different sources into UniProt accession numbers in order to maintain the uniformity in the data. Transformation of pathogen names into a single uniform format on the basis of the same UniProt Taxon identifier to remove the differences in syntax/nomenclature. Any kind of duplicate records were removed from the data to prevent redundancy.
For this HP-PPI data between host and whole pathogens, a tripartite wHPI network was constructed between pathogen with its proteins interacting with the host proteins. This network was constructed to compare the attributes of cHPI network with the wHPI network. To construct the cHPI network the information contained in the previously reported MorCVD [71] database was used. A tripartite network of pathogen and their proteins interacting with the host proteins involved in CVDs was created. Cytoscape 3.5.1 [72] was used for constructing the networks.
The interactions between the host proteins of wHPI and cHPI networks were also computed using STRING v11 [73] tool to examine the intra-species connections between host proteins. The confidence mode of this tool was used to compute the high confidence interactions with score≥0.7 and by enabling all prediction sources. To get a holistic set of HP-PPIs, these inter-species and intra-species sets of wHPI and cHPI networks were merged using Union operation of Set Theory in Cytoscape 3.5.1. Hence, the resulting networks were a tripartite graph which contained host protein and pathogens and their proteins with three types of edges: a) edges between pathogen proteins and host proteins, b) edges between pathogen and their proteins and c) edges between two host proteins.
Network randomization and validation
The biological validation of the networks was carried out by fitting the network to the power law using the Network Analyzer module of Cytoscape 3.5.1. The statistical validation was done by constructing randomized networks using the Erdos-Renyi [74] approach implemented using the “Igraph” package [75] in R statistical computing environment (https://www.r-project.org/). Briefly, the number of nodes and edges was preserved to construct 1000 random networks. The average clustering coefficient of the random networks was compared with that of the wHPI and cHPI networks.
Topological analysis of the networks
Several topological measures have been proposed to explore the specific features of complex networks [76]. Topological analysis of transcriptional regulatory and metabolic networks helps to identify essential nodes that possess vital functional activity in microorganisms [77, 78]. Initial studies suggested that highly connected nodes (having high degree or “hubs”) are essential [79, 80]. It was also shown from network analysis of a diverse set of 20 organisms that degree and betweenness centralities show significant correlation with lethality [81]. Degree and eigenvector centrality are also positively correlated. Additionally, hubs play important roles in structural and functional properties of a network [82]. In case of protein networks these nodes may tend to form protein complexes or module like structures having important functional roles [83]. It was found that 10–100 from the top high degree proteins have been listed as central in biological network studies [84, 85]. However, as we have used the top selected nodes for analysis of enrichment of biological properties, a more specific criterion was used. The unique property of the biological networks is that they follow the power law, which distinguishes them from the non-biological networks. Therefore, the significance of the hubs is indicated by the exponent of the power law with smaller exponent values signifying higher significance. Hubs possessing an exponent value < 2 usually have important roles in cellular systems and are considered as central hub nodes [86–88]. Therefore, in this study, the nodes that had degree exponent < 2 were denoted as central proteins in both the networks. A comparison of the mean degree of central and non-central nodes in the network has also been shown in Supplementary Table 1. The degree, eigenvector centrality and betweenness centrality of the central nodes was much higher than the average value of these parameters for the networks. These three topological parameters were calculated using Igraph package of R studio. The code for network validation and the topological analysis is available from the GitHub link: https://github.com/nirupmajadaun21/Network-biology.
Functional enrichment analysis
Functional enrichment was done to identify the enriched pathways and processes in the wHPI and cHPI network and those unique in the cHPI network. It was done using gene set enrichment analysis tool of KOBAS 3.0 [89], a web server for annotation and identification of enriched pathways, diseases and gene ontologies. The latest version of this server integrates the information of approximately 5000 species from databases including BioCyc, Gene Ontology, KEGG Disease, OMIM, NHGRI GWAS Catalog, PANTHER, Gene Ontology Slim, Reactome and KEGG pathways. This server uses machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. Therefore, the identification of statistically significant enriched pathways and gene ontologies was done using this tool and only those having p-value < 0.05 were considered. Recent studies have reported the use of molecular mimicry by pathogen proteins to hijack host cellular pathways [90]. Therefore, we looked for similar enriched domains between the host and pathogen proteins of cHPI network. These were computed using the UniProt database profile in FunRich tool [91].
Biological characterization of network proteins
1) Host proteins
Several biological characteristics were taken into consideration to correlate the important network topological parameters of the proteins with their biological significance. The host proteins were primarily characterized based on the following biological characteristics: Essentiality: Essential proteins are those that are indispensable for the survival of an organism, and therefore are considered a foundation of life [92]. The essential host proteins of the networks were identified using the updated DEG 10 [93]. DEG database includes essential genes identified by genome-wide essentiality screens determined under diverse conditions for survival, pathogenesis, and antibiotic resistance. Immune-relatedness: Immune-related proteins in the networks that regulate the innate and adaptive immune response along with cytokine signaling response were identified using the proteins extracted from the Reactome database [94] related to immune system pathways (Adaptive Immune System, Cytokine Signaling in Immune system and Innate Immune System). Host-factor role: Some proteins are utilized by the pathogens at multiple stages of their life cycle i.e. adhesion, invasion, replication, growth and multiplication[95]. Host factors were identified using the vhfRNAi database [96]. Extracellular location: The cellular location of a protein plays a major role in case of HPIs. Proteins exposed to the extracellular environment, both cell surface receptors and secreted proteins are required for initial invasion and serve as entry points. The entry points of the HPI play a key role in pathogen recognition and subsequent immune-regulatory processes [97]. Therefore, extracellular host proteins present in the networks were characterized with the help of gene ontology annotation done by KOBAS server.
2) Pathogen proteins
The pathogen proteins were characterized on the basis of virulence on the basis of sequence characteristics with the VirulentPred server [98] using bilayer cascade support vector machine based approach. Unlike viruses, bacteria usually do not insert their genome inside the host cell, rather express a wide range of secretory molecules that bind to host cell targets and facilitate a variety of host responses [99]. Therefore, secretory proteins of bacteria present in the networks were also characterized through GO annotation.
Validation of enriched host pathways with expression data from cardiac damage studies
The gene expression datasets for the most commonly known cardiovascular conditions namely myocarditis, endocarditis and pericarditis were collected from the GEO database [100] to validate the enriched pathways of cHPI network.
Three gene expression datasets with GEO accession ids were processed as follows: GSE150392 – RNA seq of Human iPSC-cardiomyocytes infected with SARS-CoV-2. The DEGs were extracted from the published supplementary dataset of the original study [101]. GSE122903 - RNA-Seq data for global analysis of circRNA-associated ceRNA network for investigating underlying pathogenesis of constrictive pericarditis. This raw dataset was processed in R Studio computing environment using DESeq2 [102], a Bioconductor package. GSE29161 – The whole genome microarray analysis of circulating gene expression profile to investigate the host response during Infective Endocarditis and identify potential biomarkers. The fold change from this whole genome microarray dataset were computed using GEO2 R tool of GEO.
The DEGs from all the datasets were extracted based on fold change values of at least±1.5 and corrected p-value < 0.05. Further, the enriched pathways were identified for the DEGs of each GEO dataset using KOBAS server. The number of common enriched pathways with corrected p-value < 0.05 was computed. It was ensured that the p-values of the top 20 enriched pathways of the cHPI network was compared across the three datasets examined.
Appendices
CVD: Cardiovascular diseases; HP-PPI: Host-pathogen protein-protein interactions; HPI: Host-pathogen interactions; HIV: Human Immuno-deficiency virus; SARS-CoV-2: Severe Acute Respiratory Syndrome-Corona Virus 2; ACE2: Angiotensin converting enzyme; NGF: Nuclear growth factor; EGFR: Epidermal growth factor receptor; HTLV1: Human T-cell leukemia virus type 1; DEXDc: DEAD-like helicases superfamily; HELICc: Helicase superfamily c-terminal domain; MAPK: Mitogen Activated Protein Kinase; GTPase: Nucleotide guanosine triphosphatase; ATP: Adenosine triphosphate; VEGFA-VEGFR: Vascular endothelial growth factor - vascular endothelial growth factor receptor; TRKA: Tropomyosin receptor kinase A; iPSC: Induced Pluripotent Stem Cells; cHPI: CVD associated host-pathogen interactions; wHPI: whole host-pathogen interactions; GEO: Gene Expression Omnibus; DEG10: Database of Essential Genes; DEGs: Differentially expressed genes; PRRs: Pattern recognition receptors; PAMPs: Pathogen associated molecular patterns; KEGG: Kyoto Encyclopedia of Genes and Genomes; OMIM: Online Mendelian Inheritance in Man; NHGRI: National Human Genome Research Institute Home; GWAS: Genome Wide Association Study; circRNA: circular RNA; ceRNA: Competing endogenous RNA.
Footnotes
Acknowledgments
NS would like to acknowledge Ms. Tanya Mehrotra for helping in the extraction of the data.
Funding
This research work was funded by Council for Scientific and Industrial Research (CSIR) by providing the research fellowship to NS (09/836(0021)/2016-EMR-I) during the course.