
Abstract
The outbreak of the newly emerging severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) all over the world has caused global public health emergencies, international concern and economic crises. The systemic SARS-CoV-2 disease (COVID-19) can lead to death through causing unrestrained cytokines-storm and subsequent pulmonary shutdown among the elderly and patients with pre-existing comorbidities. Additionally, in comparison with poor nations without primary health care services, in developed countries with advanced healthcare system we can witness higher number of infections per one million people. In this review, we summarize the latest studies on genes associated with SARS-CoV-2 pathogenesis and propose possible mechanisms of the virus replication cycle and its triggered signaling pathways to encourage researchers to investigate genetic and immune profiles of the disease and try strategies for its treatment. Our review shows that immune response in people with different genetic background might vary as African and then Asian populations have lowest number of affected cases compared with European and American nations. Considering SARS-CoV-2 pathogenesis, we put forward some potentially important genetic gateways to COVID-19 infection including genes involved in the entry and replication of SARS-CoV-2 and the regulation of host immune response which might represent explanation for its spread, severity, and morality. Finally, we suggest that genetic alterations within these gateways could be critical factors in influencing geographical discrepancies of the virus, so it is essential to fully study them and design appropriated and reliable therapeutic agents against COVID-19.
Introduction
Coronavirus disease 2019 (COVID-19) is the most recent pandemic threat to global public health, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1, 2]. Incubation period of the disease is 2–14 days and it associates with symptoms such as respiratory distress, dyspnea, dry cough, conjunctivitis, fever, diarrhea, acute kidney failure, and loss of taste and smell. Bats (Rhinolophus) are known as primary host and Malayan pangolin has been considered as possible intermediate host [3].
The virus belongs to the Coronaviridae family that are relatively large in size (50–200 nm in diameter) [4]. These viruses have a membrane which covers a long single-stranded positive-sense ribonucleic acid (RNA) genome (
Coronavirus classification/taxonomy
Coronaviruses are classified under the family Coronaviridae, that is the largest of three RNA virus families within the order Nidovirales [3]. The family is divided into two subfamilies, Orthocoronavirinae and subfamily Torovirinae [6]. The Torovirinae subfamily contains viruses that cause mainly enteric infections in cattle, horses, pigs, and cats, but they are not yet known to cause human infection. According to genotypic and serological characterization, members of the subfamily Orthocoronavirinae are subdivided into four genera including Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus [3]. Alpha and betacoronaviruses are harbored in mammals, particularly bat, causing gastroenteritis in animals and respiratory illnesses in humans. Gammacoronaviruses mostly circulate in avian species and a few mammals, whereas Deltacoronavirus infect birds and mammalian specious [3, 7]. Among all coronaviruses, seven of them cause human diseases. Four of these viruses cause common cold symptoms especially mild respiratory infection in immunocompromised individuals; these coronaviruses are HCoV 229E, HCoV OC43, HCoV NL63, and HCoV HKU1 [8]. The remaining three strains which cause severe respiratory distress/pneumonia and even death are SARS-CoV-1, Middle Eastern respiratory syndrome (MERS)-CoV, and SARS-CoV-2 [8].
Structure of SARS-CoV-2
The RNA genome of the virus is around 30 kb in size, capped at the 5’ end, 3’-polyadenylated and contains two flanking untranslated regions (UTRs). It has multiple open reading frames (ORFs) which encode several genes. The order of the genes in the genome is a noncoding 5’-UTR, replicase genes (two overlapping ORFs (ORF1a and 1b)), structural proteins (spike (S), envelope (E), membrane (M), and nucleocapsid (N)) and accessory proteins-noncoding 3’-UTR [8, 9]. The ORF1a and 1b which are considered as the largest ORF, encode for 15 nonstructural proteins (nsp) (nsps 1 to 10 and nsps 12 to 16) [9]. These ORFs are translated into two polypeptides, pp1a and pp1ab that are identical at the N terminal while one of them has a C-terminus extension because of a frame-shifting event. Two cysteine proteases including nsp3 and nsp5 also known as a papain-like protease (PL2pro) and 3C-like protease (3CLpro), respectively, are produced as well. The nsp3 is responsible for cleaving between nsp1
The 3’-end of the virus genome encodes four structural proteins that are critical for binding to host cell receptor, assembly of the virion, morphogenesis, and release of mature virus particles from the host cell. The SARS-CoV-2 surface (S) protein is a transmembrane heavily glycosylated protein that has three major parts; S1 region contains a NH2-terminal domain (NTD) and a C-terminal domain (CTD) also known as the receptor-binding domain (RBD), S2 subunit or stalk fusion domain which is a transmembrane domain (TM), and a short cytoplasmic tail region (CP). The ectodomain of S protein forms homotrimeric spikes on the viral membrane surface and mediates viral entry into host cells. This protein is considered as a multifunctional molecular machine that has important roles in the early steps of viral infection through interaction with host susceptibility factors such as receptors and proteases [12, 13, 14, 15]. The transmembrane M glycoprotein is the most abundant protein in the viral particle. On the contrary, while the E protein is found in small quantities in the viral membrane, it is abundantly expressed inside the host cell, localized mainly in endoplasmic reticulum (ER) and Golgi complex of the infected cells [16, 17]. The difference in the number of these proteins suggests that M protein along with E protein is critical for arranging the assembly of the virus and forming its envelope [18]. In addition to other functions, the E protein as the smallest of all structural proteins is involved in release of viral particles from host cells [19, 20]. During viral assembly, the N protein is responsible for packaging of the viral genome into a helical ribonucleocapsid (RNP) through binding to the viral RNA [21, 22]. Furthermore, SARS-CoV-2 also has multiple subgenomic RNAs, including 3a, 3b, 6, 7a, 7b, 8b, 9b, and orf14 that are distributed along the structural genes and encode eight accessory proteins [9, 23].
SARS-CoV-2 entry
According to molecular modeling studies, RBD domain of S protein has a strong binding affinity to human angiotensin-converting enzyme 2 (ACE2) [24, 25]. ACE2 is generally localized in the surface of cells of the lung, kidney, heart, and small intestine, among others. Up to the present time, ACE2 has been suggested as the most possible receptor candidate for the virus. However, there is a need for further investigation of the receptor usage, ligand activation, viral pathogenesis and also the tropism of the virus. In a model study, it has been shown that CTD residues A475, N487, E484, and Y453 interact with S19, Q24, K31, and H34 of the
After binding of ACE2 to RBD, the trimeric S protein is cleaved by host cell proteases that make the S2 subunit of the virus to undergo some conformational changes, which subsequently leads to the fusion of the viral envelope with target cell membrane [28, 29, 30]. The S2 subunit of the virus has two heptad repeat regions 1 and 2 (HR1 and HR2) which are essential for the virus membrane fusion with the host cell. In different coronaviruses, the cleavage of S protein occurs at between the S1 and S2 domains (S1/S2 site) and also within the S2 domain proximal to the fusion peptide (S2 site) [31, 32]. It seems that both sites are needed to be cleaved for virus entry. Based on amino acid sequence in different viruses, different proteases are needed to cleave the S1/S2 site. The S1/S2 site of SARS-CoV-2 (AYT
Viral replication
After endosomal pH-dependent fusion of viral envelope with host cell membrane, the positive-strand genomic RNA is released into the cytoplasm and the translation of the replicase gene is started [29, 36, 37]. ORF1a cap-dependent translation generates replicase pp1a polyprotein. Meanwhile, the presence of a slippery sequence (5’-UUU AAAC-3’) and an RNA pseudoknot towards the end of ORF1a makes ribosomal frameshifting possible which leads to the production of a long pp1ab polyprotein. Both translated polyproteins undergo autoproteolytic cleavages which produce nsps, 1 to 11 and 1 to 16 for pp1a and pp1ab, respectively. At the next step, the replicase-transcriptase complex (RTC) mediates the production of full length negative-sense RNA copies of the viral genome which are used as templates for the production of full length positive-sense RNA genomes. Subsequently, multiple copies of subgenomic RNA (sgRNA) species are produced by a mechanism known as discontinuous transcription. sgRNAs encode for different viral proteins and only their nearest ORF to 5’ end is translated. After genome replication and sgRNAs translation, the structural proteins are entered into endoplasmic reticulum (ER) and migrate inside the secretory pathway within the ER-Golgi intermediate compartment (ERGIC). Concurrently, nucleocapsids assembled in the cytoplasm bud into ERGIC membranes that encompasses structural proteins. Through this process the life cycle of SARS-CoV-2 is completed and mature virions are released out of the cell via exocytosis [38, 39, 40].
Schematic representation of the SARS-CoV-2 replication cycle, its driven signaling pathways and potential antiviral drugs. The life cycle of the virus in the target cells is shown with green arrows and signaling pathways associated with the host immune response are displayed with pink, blue, gray and brown arrows. As can be seen, the virus first enters host cells through binding to its known receptor angiotensin-converting enzyme 2 (ACE2) on the host cell membrane. Then the transmembrane serine protease 2 (TMPRSS2) processes viral spike protein (S) which leads JAK-2-mediated endocytosis and subsequently acidification of the endosome result in viral-host cell membrane fusion and release of viral single-stranded RNA (ssRNA) into the cytosol. Then host machinery translates the viral genome to produce the replicase and structural proteins. Initially open reading frame 1a (ORF1a) and ORF1b are translated by ribosomal frameshifting into polyproteins pp1a and pp1ab, respectively, that are cleaved by the proteases papain-like protease (PLpro) and 3C-like protease (3CLpro) into 15 mature nonstructural proteins (nsps). Structural proteins are expressed as subgenomic RNAs which are created by discontinous transcription. Both genomic and subgenomic RNAs are translated to yield only the protein encoded by the 5’-most ORF. After viral replication and ampli?cation by the RNA-dependent RNA polymerase (RdRp), the newly produced RNAs are encapsidated in the nucleocapsid (N) proteins in the cytoplasm and then transported to the ER-Golgi intermediate compartment (ERGIC) for further assembly. All structural proteins are inserted into the membrane of the rough ER (RER), from where they are shuttled to the ERGIC to interact with the viral RNA-N complex and assemble into viral particles. Finally, the budded vesicles containing the newly synthesized viral particle are transported to the cell surface for release (Green Arrows). Angiotensin II (Ang II) as a ligand of both AT1R and ACE2 is produced through ACE-mediated Ang I cleavage. By binding to AT1R pro-inflammatory response are activated and through attachment to ACE2, Ang II is converted to angiotensin 1-7 (Ang 1-7) that induce the Mas receptor associated anti-inflammatory response. Viral binding promotes shedding of proinflammatory cytokines and ACE2 receptors by some metalloproteases like ADAM17. The cleavage of ectodomains of these receptors makes them soluble which leads to loss of the protective function of surface ACE2 and may increase viral pathogenesis (Black Arrows). The virus activates nuclear factor 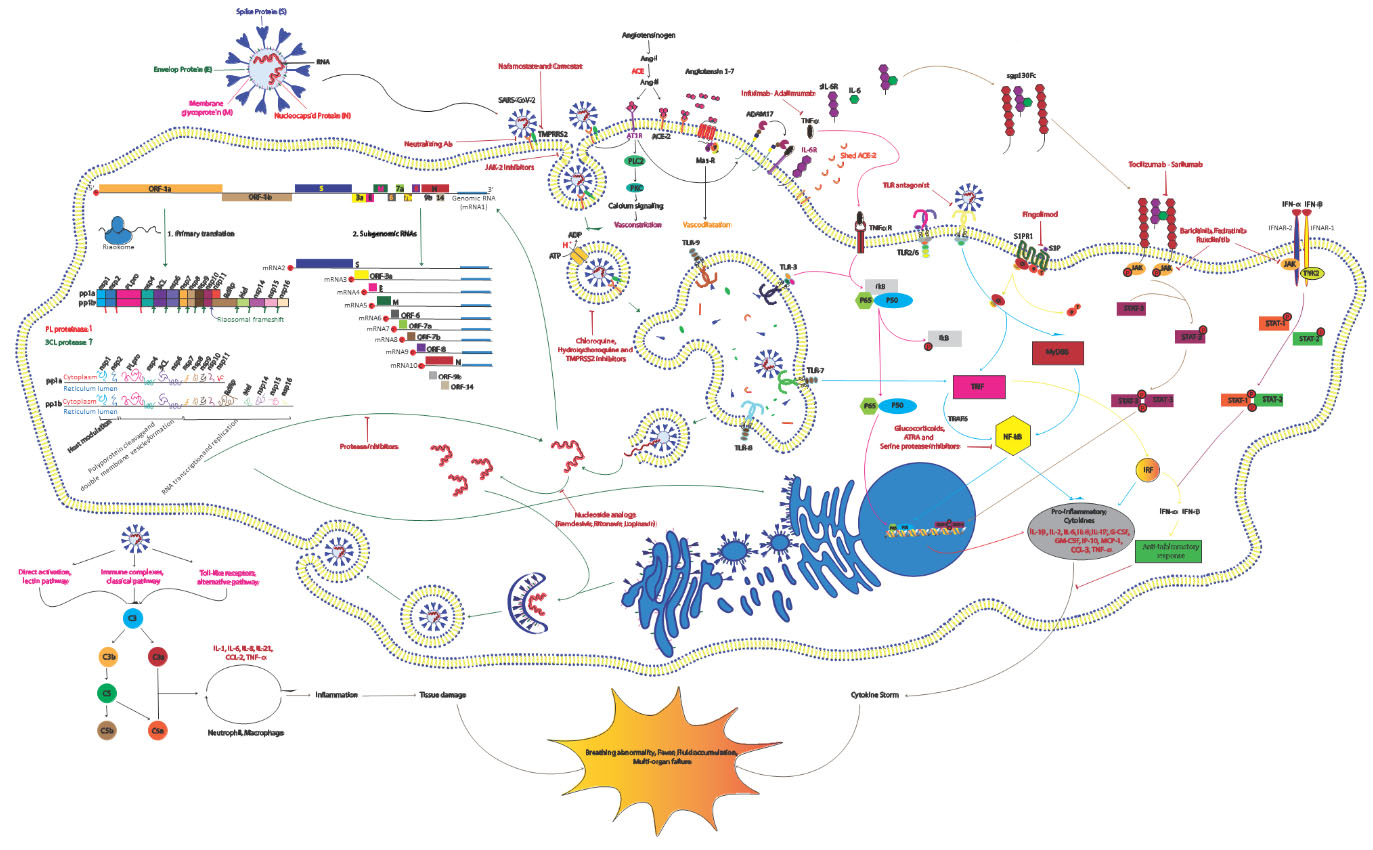
Successful virus assembly is dependent on precise protein-protein interactions which are orchestrated by M and E proteins. N proteins seem essential for virus like particle (VLP) production, suggesting that nucleocapsid assembly and its fusion into ERGIC membranes promotes the viral particle formation. At this step, S proteins are incorporated into the particle and subsequent steps of the virions assembly is enhanced by M protein-nucleocapsid interaction [40, 41] (Fig. 1).
Disease severity among affected patients without certain underlying medical conditions proposes the involvement of genetic susceptibility factors which makes some people more prone to severe symptoms. Therefore, the role of both individual and population genetics can be pivotal in shaping the COVID-19 dynamics. As a result, host genes associated with the entry and replication of the virus as well as mounting host immune responses should be assessed for reaching genetic predisposition models to COVID-19 disease [42].
Population genomics role in COVID-19 pandemics
The highest number of infections almost one year after the pandemic in January 2021 have been reported in Andorra (107,656), followed by Gibraltar (86,882), Montenegro (82,203), Luxembourg (77,525) and other European countries. In this ranking system both number of affected people in a country and the number of cases per one million people is considered. So, the ranking system rules out population densities in some countries across continents. Although mentioned nations witnessed higher number of COVID-19 infection, most African and Asian countries even with wrecked health care system showed comparatively a smaller number of cases. With this regard, it seems that there is a geographical variation in infection to the virus, but the reasons behind it have been remained obscure. One of the proposing reasons for this pattern of infection can be differences in population genomics which provide susceptibility and protection against SARS-CoV-2 infection. Hence, understanding the host genetic background and differences between diverse populations are essential to investigate the risk and protective factors involved in developing the pandemic. Up to now, a new international COVID-19 host genetics initiative has been launched to give an opportunity to researchers all around the world for characterizing genetic determinants associated with COVID-19 susceptibility, severity, and outcomes.
Genes involved in the entry and replication of SARS-CoV-2
ACE2 gene
The ACE2 is located on chromosome Xp22.2 and expressed in main target organs of the virus and also other organs with less involvement or even unknown role in the viral pathophysiology. Both lung alveolar and small intestinal epithelial cells highly express ACE2 and take part in COVID-19 transmission. According to immune-localization studies, the protein is present in smooth muscle cells, vascular endothelial cells, basal epidermal layer of the skin, as well as oral and nasal mucosa [43]. In the kidney, ACE2 is expressed weakly or moderately in podocytes and parietal epithelial cells whereas in the brush border of proximal tubular cells it exists in extremely high quantity [44]. The encoded enzyme mediates the conversion of angiotensin II to angiotensin [1, 2, 3, 4, 5, 6, 7] that is a vasodilator agent and has a pivotal function in the cardiovascular system. Based on comparative genetic analysis, the distribution pattern and influence of some single nucleotide polymorphisms (SNPs) in coding regions of the gene showed varying allele frequencies in various populations [45]. Distribution of pathogenic mutations in ACE2 differs among 9 populations, particularly around 40% and 55% of variants occur in African/African-American and Non-Finnish Europeans. Other populations including Latino/Admixed American, Finnish, East Asian, and South Asian carry 2–10% of such variants in ACE2 coding regions. On the contrary, prevalence of deleterious variants among Amish and Ashkenazi Jewish populations is zero [46]. These findings suggest that certain ACE2 alterations may provide potential resistance to COVID-19. Some variants which have the ability to inhibit ACE2-spike protein interaction may prevent virus entry into host cells. This is reported about alteration of residues p.Met383, p.Pro389 and p.Asp427 or in residues Arg708/710/716 that are located in the dimeric interface of the protein and are essential for TMPRSS2 cleavage [47, 48]. On the other hand, ACE2 pathogenic mutations can disrupt the renin-angiotensin balance and vascular functions which can subsequently cause hypertension and diabetes mellitus, the most common comorbid conditions of the virus [45]. Moreover, previous studies showed that ACE2 has pivotal role in inflammatory processes and its genetic deficiency results in overexpression of cytokines and induction of vascular inflammation in mouse model with knocked-out ApoE [49, 50]. Furthermore, the association of ACE2 expression with several immune signatures such as B, T and NK cells’ markers and even interferon response has been reported in different human tissues [51]. Consequently, the role of deleterious ACE2 variants in susceptibility to virus infection is largely conflicting with considering the receptor involvement in virus entry into host cell and also post-infection downstream processes like inflammatory responses.
One of the strongest risk factors for severe COVID-19 is age advancing [52]. Although higher complication and fatality rates among elderly can be justified by gradually decreasing innate and adaptive immune responses, expression of ACE2 in the lungs with age increasing can also provide good explanation to the higher disease severity and mortality among older patients with COVID-19 [53, 54]. Current research showed that men are more prone to COVID-19 infection than women [55]. One of the possible mechanisms for gender related susceptibility to COVID-19 is ACE2 localization on chromosome X, in which monoallelic versus biallelic presence of the gene put men at higher risk of infection and severity [46, 56, 57].
TMPRSS2 gene
The TMPRSS2 gene, also known as epitheliasin is located on chromosome 21q22.3 and encodes androgen-responsive serine protease which primes spike protein of SARS-CoV-2 and facilitates viral entry and activation [58, 59]. This protein is a 492 amino acid single-pass type II membrane protein and contains five domains including cytoplasmic domain, a transmembrane domain (aa 84–106), an LDL receptor class A (LDLRA, aa 113–148) domain (forms a binding site for calcium), a Scavenger receptor cysteine-rich domain (SRDR, aa 149–242) and a Serine protease domain (aa 255–492) [60]. Although the normal physiological function of the protein has been remained obscure, some studies have revealed that TMPRSS2 expression reduces the epithelial sodium channel and protein levels [60]. This enzyme is expressed in many tissues including lung, colon, kidney, cardiac endothelium, and small intestine; however, it is well known for its predominant expression in the prostate epithelium [61, 62]. These findings suggest that clinical manifestations of the virus such as acute myocardial injury, gastrointestinal complications and thrombosis as a result of endothelial dysfunction are associated with expression of TMPRSS2 in the mentioned tissues [63, 64]. Considering alteration frequencies of TMPRSS2 among different population, 35% and 59% of pathogenic variants are carried by African/African-American and Non-Finnish European populations, respectively. Meanwhile, only four deleterious variants are carried by each of the South Asian, East Asian and Finish populations [46]. A key residue for catalytic substrate binding of this enzyme is Asp435, which its substitution with Tyr has been seen with low frequency among Non-Finnish European population. These polymorphisms with diverse frequency provide potential explanation for differential genetic susceptibility to the virus.
Overexpression of this gene has been reported in association with some factors. Schuler et al. showed that in ciliated cells and type I alveolar epithelial cells (AT1) of human and mice there is an age-related expression pattern for TMPRSS2. They illustrated that expression level of the gene is increasing with age and suggested that developmental regulation of TMPRSS2 might be a good explanation for comparative protection of infants and children from the virus [65, 66]. Considering chromosomal location of the TMPRSS2 gene, it seems that Down syndrome patients might be at higher risk due to overexpression of the gene. Its oncogenic role in some cancers might explain poor outcomes in COVID-19 as well, particularly in individuals with prostate cancer [45, 49].
Furin
Second gene that adds an additional level of complexity in SARS-CoV-2 entry is furin. This gene is located on chromosome 15q26.1 and encodes a type 1 membrane-bound protease which is expressed in many organs including brain, lung, liver, pancreas, gastrointestinal tract, and reproductive tissues [67]. Cleavage of a polybasic site at the S1-S2 boundary of spike protein by the furin protease is thought to facilitate the entrance of the virus into its target cells [14, 67]. Hoffmann et al. in a study on human lung cells described that the cellular protease furin is essential for S-protein-mediated cell-cell fusion and entry into host cells and they suggested that furin can be a potential target for therapeutic intervention [68]. Unlike TMPRSS2, furin is required for normal development, so its inhibition for prolonged time periods can have undesirable toxic effects [68, 69]. In contrast, in other studies several variants both in the furin cleavage site and furin gene which may affect COVID-19 entrance have been studied and their results showed that the furin cleavage site might not be required for the virus to enter human cells in vivo [67, 70]. With this regard, more studies are needed to clarify the exact role of furin in SARS-CoV-2 infection and also its polymorphisms effect on susceptibility to the virus among different populations.
Immunogenetics in COVID-19
HLA
The human leukocyte antigen (HLA) molecules are essential immune regulatory components with specific role in response to viral infections as receptors of viral peptides. These molecules are encoded by Major Histocompatibility Complex (MHC) genes and show extreme diversity and variations. Considering the fact that genetic differences at these genes result in various immune response against pathogens, HLA allele distribution among populations can provide a pattern for differential susceptibility to SARS-CoV-2 infection [71, 72].
A study on patients with severe respiratory distress, revealed that alongside with reduction of CD4, CD19 lymphocytes and NK cells, expression of HLA-DR was decreased to great extent, suggesting the involvement of HLA in COVID-19 progression [73]. A comprehensive in silico analysis for assessing binding affinity of all peptides of SARS-CoV-2 and MHC class I molecules across 145 HLA-A, -B, and -C genotypes revealed that individuals with HLA-B*46:01 alleles may not be vulnerable to the virus infection as this allele had the fewest affinity to the virus peptides. In contrast, HLA-B*15:03 had the greatest predicted binding peptides for SARS-CoV-2 suggesting probable role of the allele in increasing susceptibility to covid-19 [74]. Analysis of SARS-CoV and SARS-CoV-2 proteomes for prediction of the MHC class I epitope revealed that HLA-A*02:03 and A*31:01 are as effective antigen presenters for COVID-19 indicating that these alleles might provide protection for their carrier individuals, whereas HLA-A*03:02 seems to be a risk factor [75]. However, in other studies multiple HLA alleles such as HLA-A*02:01, HLA-A*02:02, HLA-B*15:03, HLA-B*40:01, HLA-C*12:03, HLA-DRA*01:01, HLA-DRB1*07:01, and HLA-DRB1*04:01 have been reported in association with some epitopes of SARS-CoV-2 [74, 76, 77]. Considering all this, the pivotal role of MHC in developing anti-viral epitope-based peptide vaccine is substantial.
Population-specific HLA alleles is another concept for describing susceptibility of different populations to COVID-19. For instance, protective HLA-A*02 alleles were shown to have high frequencies among Indian African populations [78, 79]. On the contrary, the susceptibility allele HLA-B*46:01 is highly distributed in South East Asia and rarely in Europe, while in Indian and African populations it is completely absent [80]. Another protective allele is HLA-B*15:03 which showed high frequency among African people, but it was absent in East Asian gene pool. Other allele that increases vulnerability to the virus is HLA-C*12:03. This HLA presumably is the most frequent allele among the European people [45]. These findings can be a good explanation for less risk and severity of the virus in Indian and African populations than European and other world populations and implicate that in people from different backgrounds, susceptibility according to HLA loci can vary significantly.
SARS-CoV-2-driven signaling pathways
Cytokine storm is an exacerbated systemic inflammatory response to COVID-19 which results in death due to multiple organ failure particularly lung, heart, kidney and liver [81, 82]. This response occurs mainly because immune effector cells release a huge amount of pro-inflammatory cytokines and chemokines such as IL1-
Previous investigations revealed that polymorphisms of TLR genes or components of TLR pathway affect SARS-CoV infection. In animal models with deficiency of TLR3, TLR3/TLR4 adaptor TRIF and Toll Like Receptor Adaptor Molecule 2 (Ticam2), susceptibility to SARS-CoV infection has increased [92, 93]. Variations in TLR genes result in different immunological redundancy of TLRs and distinct expression pattern in each population [94]. So, proposing TLR genes as a crucial determinant of differential vulnerability to COVID-19 could be logical. However, this concept should be evaluated empirically.
As components of innate immunity, complement hyper-activation has been reported in COVID-19 patients [95]. In null C3 mice with SARS-CoV infection, levels of cytokines and chemokines in both the sera and lungs were low and respiratory dysfunction was not severe [96]. These reports implicate pivotal role of complement components in the induction of the cytokine storm and susceptibility to SARS-CoV-2 infection and disease severity. Although there is no evidence regarding the impact of complement gene polymorphisms on the risk and severity of SARS-CoV-2 infection, association between variations of the genes and susceptibility to diseases has been documented about other infectious diseases [45]. Further investigations are required to shed light on the importance of gene polymorphisms in innate immunity role-players regarding increased or decreased susceptibility to COVID-19.
Other genes that are involved in the control of inflammatory responses are the components of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. The protein families of mammals involved in this pathway include JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2) that interact with the cytoplasmic domain of type I and II cytokine receptors and through phosphorylation of the receptors’ subunits promulgate cytokine signaling [97, 98]. Tyk2 is part of the type I and type III interferon signaling pathways and has critical role in transduction of IFN-
Host genetic signatures of COVID-19 guide personalized treatment
To date, no approved effective medication against SARS-CoV-2 infection has been presented, but some potentially repurposable agents such as hydroxychloroquine, melatonin, and chloroquine are investigated by several national and international research groups and some vaccines have been produced [102, 103]. However, none of the vaccines provide 100 percent immunity against the virus.
The primary mechanism of preventing COVID-19 infection is to inhibit virus entry through targeting the endosomal pathway. Two known drugs involved in this process are hydroxychloroquine and chloroquine known to increase the pH of endosomes and inhibit membrane fusion [103, 104]. Second way of inhibiting viral and host cellular membrane fusion is differential glycosylation of both ACE2 and the S protein. Additionally, several variants of ACE2 which inhibit ACE2 and the S protein interaction may affect the clinical efficiency of chloroquine or hydroxychloroquine, explaining the reason of diverse response of patients to mentioned medicines among different populations [104, 105]. Another target associated with viral-host cell membrane fusion is TMPRSS2, which is involved in S protein conformational change. Its blockage by inhibitors such as camostat mesylate in the face of cellular increased pH environment could be a substantial approach in virus replication inhibition [59, 106]. It has been proposed that inhibitors of endosomal acidification like CatB/L, E-64D and hydroxychloroquine/chloroquine may have more effect on TMPRSS2-absence patients and might not work or work less for the patients with normal TMPRSS2 enzyme [106]. Considering this, it seems that African and Non-Finnish European populations who carry deleterious variants on TMPRSS2 gene may be more sensitive to the endosomal acidification inhibitors.
Although there are limitations in our understanding of SARS-CoV-2 associated immune signaling pathways, studying and investigating pathways that are involved in excessive inflammatory responses can give us clues on how to overcome the infectious outcomes driven by COVID-19. So, final approach against SARS-CoV-2 infection might be to target pathways triggered by the virus. Targeting intracellular molecules instead of viral proteins shows potential advantage as antiviral drugs could be negated by mutations in the virus genome while producing drugs targeting specific signal transducers which is similar in most virus infections can result in effective therapy particularly for the management of COVID-19 [107, 108]. Considering structural similarities of SARS-CoV-2 with pathogenic SARS-CoV, it seems that the infection mechanisms of this group of viruses might be identical and may induce the activation of shared intracellular pathways. It is well stablished that viral RNAs are detected by the pattern recognition receptors such as the family of TLRs (TLR3 and TLR7/8), the cytosolic RNA sensor, and retinoic acid-inducible gene (RIG-I)/MDA5 [109]. Virus recognition result in activation of downstream transduction pathways such as IFN regulatory factor-3 (IRF3), nuclear factor
NF-
The JAK/STAT signaling pathway has roles in transducing extracellular signals associated with cytokines, lymphokines and growth factors. Notably, the cytokine IL-6 promotes the downstream activation of JAK/STAT and in turn the activation of this pathway stimulate IL-6 expression [118, 119]. Abnormal expression of IL-6 promotes the secretion of other cytokines and causes inflammation in the vascular system that is observed in COVID-19 patients [120]. The inflamed vessels produce angiotensin II which induces the synthesis and secretion of IL-6. Activation of JAK/STAT pathway by Angiotensin II receptor type 1 (AT 1 receptor) has been revealed in previous researches and this pattern establishes a positive inflammatory feedback loop [121, 122]. The S protein of SARS-CoV downregulates ACE2 expression and causes over-production of angiotensin II. It is speculated that this might also be true in SARS-CoV-2 and its S protein may be involved in overproduction of IL-6 in AT 1/JAK/STAT-dependent manner and subsequently result in vascular inflammation and lung injury [123, 124]. Additionally, both NF-
The sphingosine-1-phosphate (S1P) 1 as second messenger during inflammation play critical role in many immune response processes such as cytokine and chemokine production, lymphocyte trafficking and vascular integrity by binding to five G-protein coupled receptors (S1PRs 1–5) [128]. The S1P 1 receptor which is activated by Ras/ERK, PI3K/Akt/eNOS, and PLC/Ca 2
Vitamin D and COVID-19
Vitamin D has been found to exert immunomodulatory effects and contribute in induction of anti-inflammatory responses, especially in viral infections [131, 132]. Moreover, vitamin D can induce expressions of receptors of innate and adaptive immune systems in the respiratory system [133, 134]. Low levels of this vitamin in patients with COVID-19 have been shown to be linked with the severe disorder and higher and mortality [135, 136]. The immunomodulatory and anti-inflammatory effects of optimal serum concentrations of 25-hydroxy vitamin D have been shown to be beneficial in COVID-19 patients [137]. However, a recent study has shown that administration of high-dose parenteral vitamin D3 did not affect in-hospital mortality rate of critically ill COVID-19 patients with vitamin D deficiency [138].
Role of non-coding RNAs in COVID-19
Non-coding RNAs, particularly those with regulatory roles have important roles in the pathoetiology of both immune-related and infectious disorders [85, 139, 140]. A number of long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), particularly those regulating host cell cycle, apoptotic pathways and immune functions have been found to be differentially expressed in COVID-19 patients versus healthy subjects [141]. Moreover, expressions of numerous lncRNAs in SARS-CoV-2-infected cells have been suggested to be controlled by IRF1, IRF4 STAT1 and STAT3. Notably, these lncRNAs may participate in antiviral responses [142].
Conclusion
COVID-19 pandemic has affected different populations with diverse pattern, as the number of total affected cases per one million people in some countries with developed health care system is more than other geographic locations with under developed health care systems. Moreover, susceptibility of people to the virus within populations and even among members of a family is different as some patients show mild symptoms of the diseases, while others experience moderate symptoms and some cases are critically ill. These evidences suggest that several factors such as multiple virus variants and host genetics might play critical role in making people more prone/resistant to the infection. In this study we tried to cast light on the SARS-CoV-2 replication cycle, its driven signaling pathways and their associated proteins with the aim of understanding mechanisms involved in the disease pathogenesis. We propose that targeting specific proteins associated with virus entrance and replication as well as host immune response can accelerate development of therapeutic agents and subsequently reduce mortality rate among different populations and ethnic groups. However, intense and profound studies are needed to investigate each angle of proposed approaches in managing the pandemic and confirm such a conclusion.
As an active area of investigation, variants in SARS-CoV-2 and its associated host genes should be classified in different categories such as asymptomatic, moderately symptomatic and severely symptomatic with urgent hospitalization. Providing such a classification and identifying genomics and genetic pathways associated with SARS-CoV-2 susceptibility may help clinicians in managing further waves of the pandemic. At the end, we encourage all scientists to assess gene expression and also variants among individuals with severe COVID-19 using novel massive parallel sequencing methods and other new approaches.
Footnotes
Future perspectives
Future high throughput sequencing strategies as well as genome-wide association studies in SARS-CoV-2-infected individuals and assessment of association between putative genetic variants/differentially expressed genes and disease course would facilitate identification of the role of genetic factors in this regard.