
Abstract
Sickle cell disease (SCD) is an autosomal recessive disorder. Although the molecular mechanisms at the origin of SCD have been well characterized, its clinical expression is highly variable. SCD is characterized by blood rheological abnormalities, increased inflammation and oxidative stress, and vascular dysfunction. Individuals with only one copy of the mutated β-globin gene have sickle cell trait (SCT) and are usually asymptomatic. The first part of this review focuses on the biological responses of SCT carriers during exercise and on the effects of combined SCT and diabetes on vascular function, several biomarkers and clinical complications. The second part of the review focuses on SCD and shows that the magnitude of red blood cell (RBC) rheological alterations is highly variable from one patient to another, and this variability reflects the clinical and hematological variability: patients with the less deformable RBCs have high hemolytic rate and severe anemia, and are prone to develop leg ulcers, priapism, cerebral vasculopathy, glomerulopathy or pulmonary hypertension. In contrast, SCD patients characterized by the presence of more deformable RBCs (but still rigid) are less anemic and may exhibit increased blood viscosity, which increases the risk for vaso-occlusive events. Several genetic and cellular factors may modulate RBC deformability in SCD: co-existence of α-thalassemia, fetal hemoglobin level, oxidative stress, the presence of residual mitochondria into mature RBCs, the activity of various non-selective cationic ion channels, etc. The last part of this review presents the effects of hydroxyurea and exercise training on RBC rheology and other biomarkers in SCD.
Keywords
Introduction
I feel deeply honored to receive the Fåhraeus Award 2023 from the European Society of Clinical Hemorheology and Microcirculation (ESCHM). I have started working in the field of blood rheology and microcirculation during my MSc in 2000 at the University of Montpellier and have been fascinated by red blood cells and blood. I am very grateful to my former supervisors, Jean-Frederic Brun, Corinne Caillaud and Jacques Mercier (University of Montpellier), and to my former mentors, Herbert Meiselman, Oguz Baskurt, Max Hardeman and Friedrich Jung, from who I learned about rheology, physiology and science in general. My first research works in this field focused on the impact of exercise on blood viscosity and its determinants in endurance athletes [1–3]. After my PhD, in 2003, I moved for one year in Lyon, where I started working on sickle cell trait (SCT) and the effects of exercise on blood rheology and other biomarkers. In 2004, I moved to Guadeloupe for 10 years and started working on sickle cell disease (SCD) using physiological, biophysical, cellular and molecular approaches: a disease I am still working on since I moved back to Lyon in 2014. I feel very lucky to have met exceptional people, most of whom have become friends, and with whom I continue to collaborate still today.
Sickle cell disease and sickle cell trait: Background
Sickle cell disease (SCD) is an autosomal recessive disorder and is the most frequent genetic disease in the world [4]. It is estimated that over 300,000 children are born each year with a severe inherited hemoglobinopathy, over 80% of these in low-or middle-income countries, and approximately 220,000 newborns are affected by SCD [4]. SCD is characterized by a single nucleotide mutation (adenine ->thymine) in exon I of the β-globin gene that leads to the presence of sickle hemoglobin (HbS) resulting from the substitution of valine for glutamic acid at the sixth position of the β-globin chain. The hydrophobic residue of valine associates with other hydrophobic residues causing HbS molecules to aggregate, forming fibrous precipitates when hemoglobin is deoxygenated. This phenomenon is called “HbS polymerization” and is responsible for the characteristic shape change termed “sickling” of red blood cells (RBCs). Sickle RBCs are poorly deformable and very fragile, leading to frequent vaso-occlusive episodes, chronic hemolytic anemia and other acute/chronic complications in affected patients.
People who inherit one sickle cell gene and one normal gene have sickle cell trait (SCT). While individuals with two βS-globin alleles (i.e., SCD) may develop severe clinical complications, those with SCT are partially protected from malaria [5, 6]. The places where malaria is most common are also the places that have the highest percentage of people with SCT. During infection, individuals with SCT have 50 to 90% fewer parasites in their blood than people with normal hemoglobin and also get rid of the parasites faster, which make SCT carriers to be more resistant to malaria than peoples with normal hemoglobin [7, 8].
Sickle cell trait
SCT and clinical risks
SCT prevalence can reach 8-10% for Afro-Americans and 10% in the Caribbean Islands. RBCs from SCT carriers are characterized by the presence of both HbS (less than 50%) and normal hemoglobin (HbA) but the excess of HbA slows HbS polymerization under deoxygenation, compared to a situation where no HbA is present into RBCs [9]. Indeed, SCT is usually considered as an asymptomatic and benign condition, compared with SCD, and reportedly does not affect growth and mental development [10–12]. However, it has been reported that SCT carriers are at higher risk than the general population for developing hyposthenuria, hematuria and renal medullary carcinoma [13, 14]. In addition, associations between SCT and albuminuria and chronic kidney disease (CKD) have been fairly well-established in retrospective analyses. Several large population-based studies, totaling over 25,000 African-Americans participants, have demonstrated a 1.5-2.0-fold increased risk of CKD among SCT carriers compared to non-carriers [15, 16]. The hypoxic, acidotic and dehydrated environment of the renal medulla is through to promote RBC sickling, even in SCT carriers, that may cause vaso-occlusion and irreversible damages into the kidney. Other research groups also observed that SCT would increase the risk for venous thromboembolism (and more particularly pulmonary embolism) of 1.5–2.0-fold among African Americans, compared to non SCT carriers [17, 18]. Indeed, although SCT is considered to be a benign condition, it seems that carriers are at risk for developing some clinical complications [19].
SCT and exercise
Large epidemiological studies also reported a higher risk of exercise-related idiopathic sudden death and exercise-induced rhabdomyolysis in SCT carriers than in the general population [20–23]. The link between SCT and these adverse events is not completely understood [24], and we decided to conduct several studies focusing on the effects of exercise on several biological parameters, which are known to be involved in the severe clinical complications observed in patients with SCD. The first study we conducted was in Cameroon and sportsmen with or without SCT performed a progressive and maximal exercise test of 12-15 min duration [25]. We found that SCT carriers had systematically greater RBC rigidity than those without SCT and we postulated that the slightly lower RBC deformability observed in SCT carriers could impact on microcirculation. We then confirmed these observations in several studies performed in Guadeloupe and using different exercise modalities [26–28]. We also investigated the effects of exercise on inflammation [27, 29–31] and coagulation parameters [32, 33], and observed slightly greater inflammation and coagulation activity in SCT carriers compared to subjects with normal hemoglobin. In 2009, we published a case study of a SCT carrier who developed a central retinal vein occlusion after having participated to a 138 km cycling race performed in tropical environment (temperature of 35°C and humidity of 65%) [34]. Several genetic factors known to increase the risk of thrombosis were screened but no other genetic mutation than SCT was found. We also measured blood viscosity and estimated RBC rigidity, and found that the values for these two parameters were very high compared to the values of the general population. In addition, the patient said that he felt very thirsty during and after the race. Indeed, we suspected that dehydration caused by the prolonged exercise conducted in extreme environmental conditions could have led to an excessive increase of blood viscosity, which would in turn trigger the occlusion of the central retinal vein. For these reasons, we conducted two studies in Senegal where we investigated the blood rheological responses of SCT carriers and non-carriers to a prolong exercise/physical effort performed in two conditions: a condition where subjects were allowed to drink water ad-libitum and a condition where subjects were deprived of water during exercise [35, 36]. The results showed that blood viscosity was higher in SCT carriers at rest, which confirmed previous studies [25, 37]. Exercise conducted in dehydrated condition caused a rise of blood viscosity in the two populations, but the magnitude of change was far greater in SCT carriers compared to the control group. Although hydration during exercise did not influence blood viscosity in the control group, it caused a large decrease of blood viscosity in SCT carriers and the values at the end of exercise were no longer different between the two groups [35]. These findings clearly suggest that hydration should be strongly encouraged in SCT carriers during exercise. A retrospective study of 47,944 black soldiers showed that with adoption of universal preventive measures, the risk of exercise-related sudden death attributed to SCT appears to be completely mitigated [38]. Figure 1 summarizes the results of several studies conducted by my research group and others.
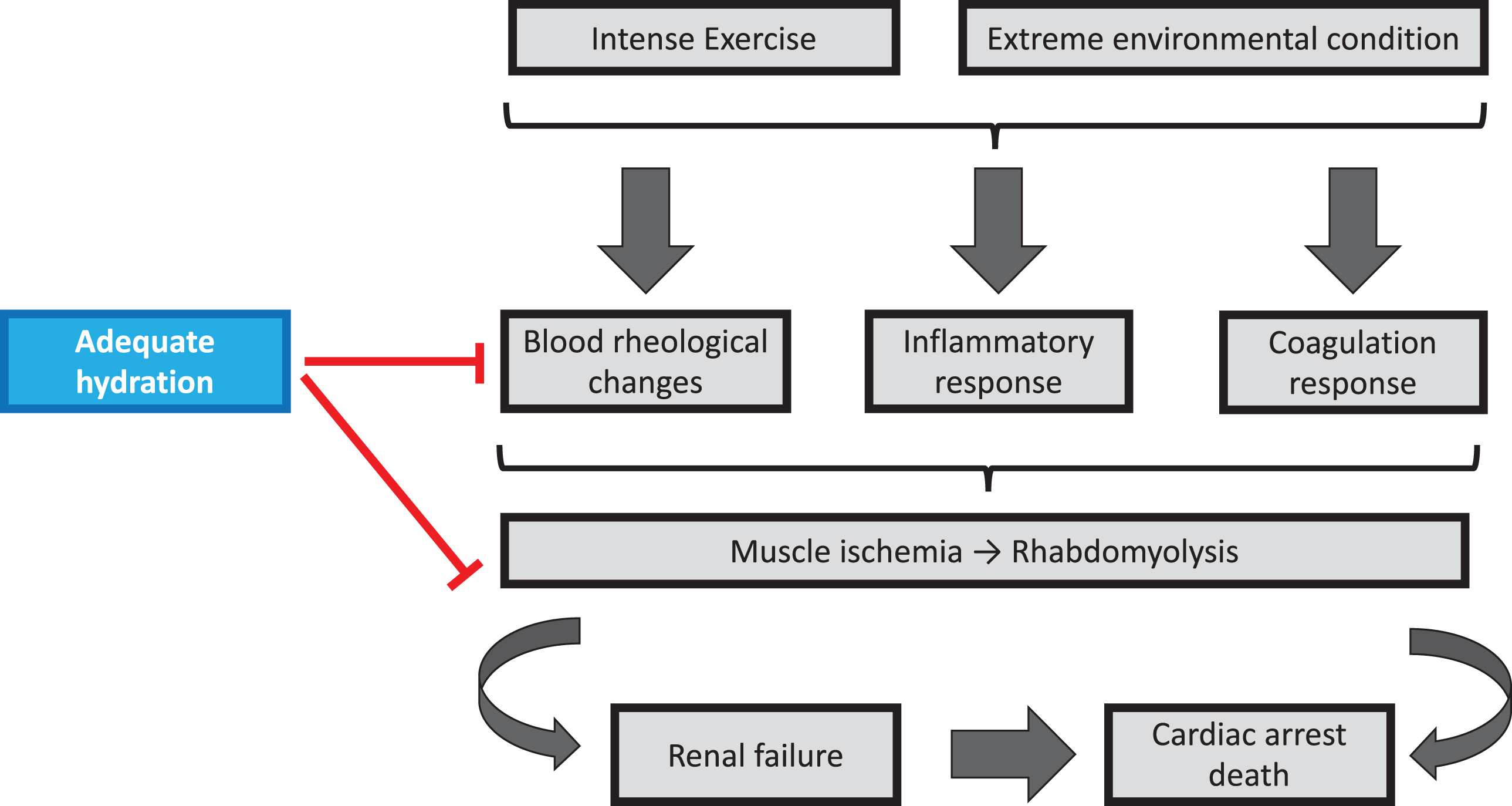
Effects of intense exercise, extreme environmental conditions and hydration on blood rheology, inflammation, coagulation and the risks of adverse events in SCT carriers.
SCT is highly frequent in Sub-Saharan African countries, with an estimated prevalence of 6-10% in Senegal for instance [4]. The prevalence of type 2 diabetes is increasing particularly rapidly in Africa, where urbanization, lifestyle changes, and an aging population contribute to an expected 162% increase in the disease by 2045 [39]. The high and increasing rates of type 2 diabetes in sub-Saharan Africa indicate that there is likely a large and growing population of individuals with both SCT and type 2 diabetes. In 2005, three published short letters questioned the fact that the coexistence of SCT and diabetes could have a major impact on health [40–42]. We decided to conduct a study in Senegal in 2014 to investigate the impact of SCT and diabetes on the vascular function, oxidative stress and blood rheology [43]. We demonstrated that individuals with combined SCT and diabetes, SCT only or diabetes only had impaired vascular function, as assessed by flow-mediated dilation technique and pulse wave velocity measurement, compared to a control group. However, the loss of vasodilatory capacity was the highest in subjects with combined SCT and diabetes. In addition, this same group was characterized by increased blood viscosity, oxidative stress, advanced glycation end products (AGE) level and inflammation compared to the three other groups. In a second study, we confirmed that individuals with combined SCT and diabetes had greater arterial stiffness and higher blood viscosity compared to healthy individuals, SCT carriers and patients with diabetes [44]. Human aortic endothelial cells were cultured with the plasma from each of the population and we observed that plasma from the combined SCT-diabetes group lead to a greater expression of E-selectin. The use of antioxidant and anti-AGE decreased the activation of endothelial cells. We also found that hypertension, retinopathy, and decreased renal function were more prevalent in individuals with combined SCT and type 2 diabetes than in healthy individuals, and subjects with type 2 diabetes or SCT only. These results suggest that SCT could exacerbate multiple factors that increase the risk of developing type 2 diabetes-related vascular complications, including arterial stiffness, blood hyperviscosity, AGE accumulation, and increased E-selectin expression. We then confirmed the negative impact of combined SCT and diabetes on blood viscosity and vascular function in an animal study where we fed Townes SCT mice with a high fat-high sucrose diet to make them diabetics [45]. These mice exhibited increased blood viscosity and lower pressure induced vasodilation, as assessed by laser doppler flowmetry, than mice with SCT or diabetes only, and control mice. From these studies, we concluded that physicians should pay greater attention to the double condition “SCT+diabetes” and that patients with combined SCT and diabetes should be monitored more frequently for retinopathy, nephropathy, and hypertension. Figure 2 summarizes the effects of diabetes only and of combined diabetes-SCT, on vascular function, several biological parameters and clinical complications.
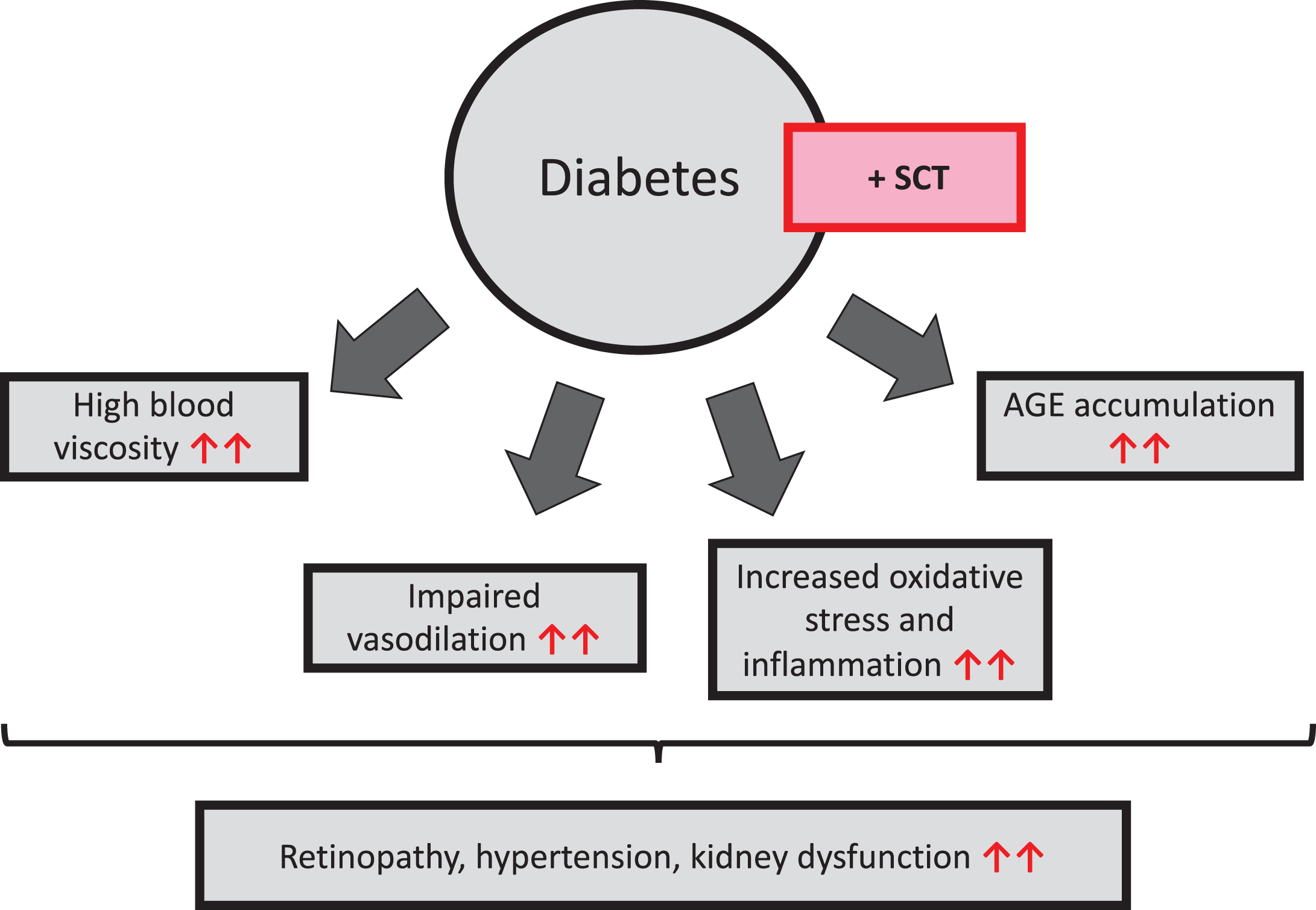
Effects of diabetes on blood viscosity, vascular function, oxidative stress, advanced glycation end products (AGE) and vascular complications. The presence of SCT in diabetic patients increases the frequency of vascular complications through its effects on several biological and vascular parameters (red arrows). SCT = sickle cell trait.
From a simple molecular mechanism to clinical and blood rheological variability
As previously mentioned, SCD is genetic disease caused by a single mutation in the DNA sequence of the β-globin gene and the molecular/cellular/clinical consequences are well described: 1) polymerization of deoxygenated HbS, 2) RBC sickling at the origin of the increased RBC fragility and decreased RBC deformability, 3) chronic hemolytic anemia and occurrence of frequent vaso-occlusive like events. However, SCD is characterized by a very high intra- and inter-clinical variability [46–48]. SCD patients may develop different kind of complications, such as vaso-occlusive crises, acute chest syndrome, osteonecrosis, priapism, glomerulopathy, chronic kidney dysfunction, leg ulcers, stroke, pulmonary hypertension, and others, but it is difficult to predict which patient will develop one of these clinical complications or not. Moreover, the clinical course of a given patient may change over time: for instance, a patient can be exposed to frequent vaso-occlusive crises during childhood but not during adulthood; and this is unpredictable. Figure 3 shows the RBC deformability of 615 SCD patients of different ages and at clinical steady-state. The values are highly variable from one patient to another, with some patients having RBC deformability values close to the values of healthy individuals while others have a severe reduction of RBC deformability (less than 0.2). This RBC rhelogical variability led us to try answering two questions: 1) Is there a clinical relevance of this RBC rheological variability?; 2) What are the cellular and genetic modulators of this rheological variability?
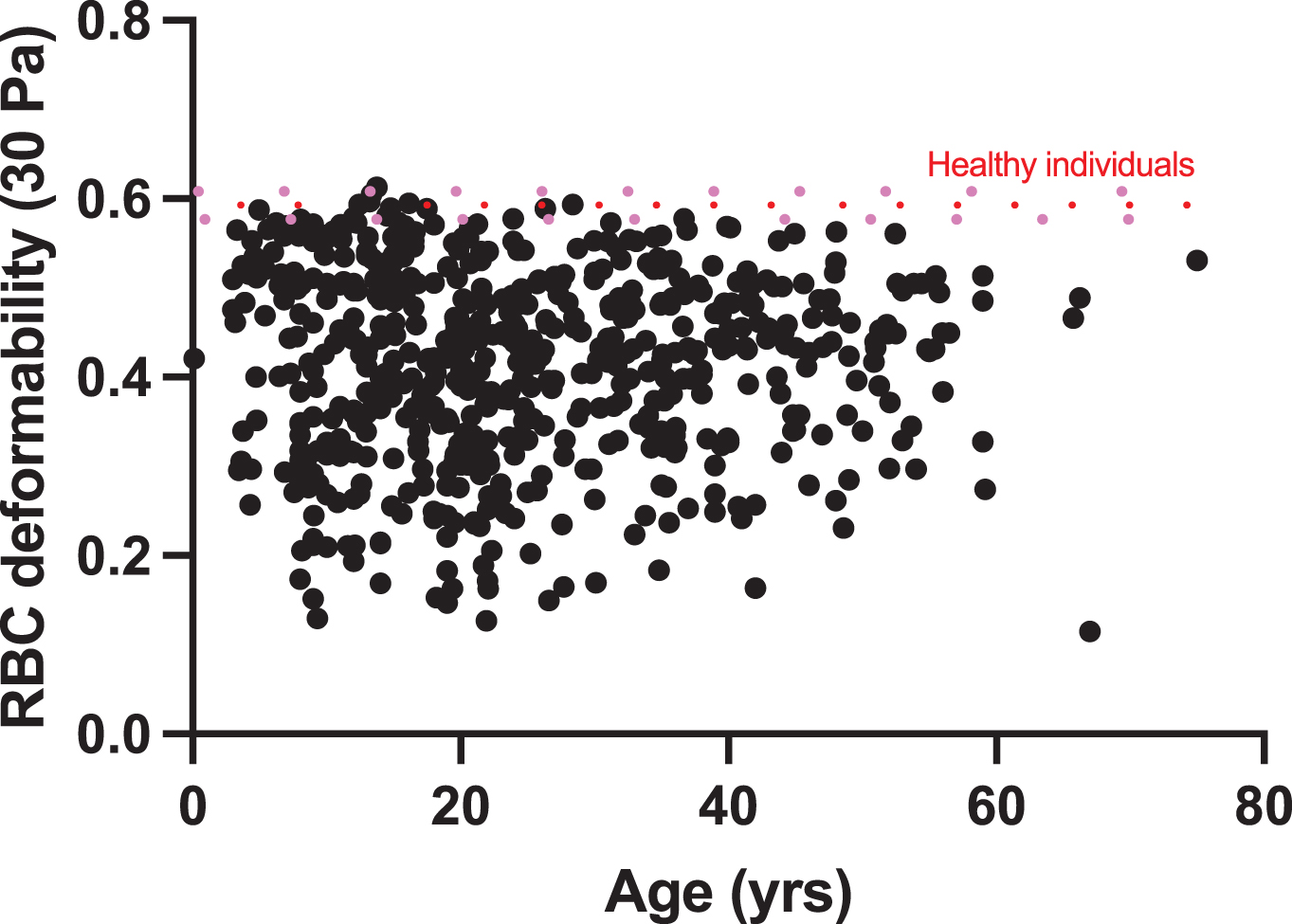
RBC deformability determined by ektacytometry at 30 Pa in 615 non-transfused patients with SCD (HbSS genotype) at clinical steady-state. Mean value (dashed red line)±95% IC (pink dashed lines) from 120 healthy controls.
We conducted several studies where we compared the blood rheological profile of SCD patients with one type of clinical complication to SCD patients without [49]. For priapism [50], leg ulcers [51], silent stroke [52], glomerulopathy [53] and pulmonary hypertension (unpublished observations), we found that SCD patients with one of these complications had systematically a lower RBC deformability than patients free of these complications. In addition, we reported a negative correlation between RBC deformability and the hemolytic rate (calculated from 4 biochemical/hematological parameters: plasma bilirubin, plasma aspartate aminotransferase, plasma lactate dehydrogenase and reticulocytes) in a large group of SCD patients, showing that the lower RBC deformability, the higher RBC fragility and hemolysis [54]. Indeed, it seems that there is a specific RBC rheological/biological phenotype in SCD patients prone to develop the complications cited above, which is characterized by the presence of very rigid and fragile RBCs that may disrupt easily into the blood circulation. The release of hemoglobin and heme into the plasma promotes inflammation and oxidative stress, and endothelial dysfunction [55–58].
In contrast, vaso-occlusive like complications seem to be characterized by a different RBC rheological/biological phenotype. Although an increase of RBC deformability is beneficial for blood flow and tissue perfusion in the healthy population, we and others reported that SCD patients with the highest RBC deformability have more frequently osteonecrosis [59] and acute painful vaso-occlusion [60, 61]. While RBC deformability decreases during vaso-occlusive events [62, 63], Ballas et al [62, 64] demonstrated that high level of RBC deformability during the recovery phase of a painful vaso-occlusive event was a predictor of a new painful crisis. This surprising finding may be explained by the fact that sickle RBCs with the highest deformability are also the most adherent RBCs to the vascular wall, thus decreasing the lumen of microvessels, slowing blood flow and initiating vascular occlusion [62, 64–66]. In addition, since RBC deformability impacts on RBC fragility, SCD patients with the greatest RBC deformability have the lowest hemolytic rate and less severe anemia [54], which may result in increased blood viscosity. Indeed, we found a positive association between blood viscosity and the frequency of vaso-occlusive crises both in adults [67] and children [60, 68] with SCD. In healthy individuals, any increase in blood viscosity does not systematically cause a rise in vascular resistance because the subsequent increase of wall shear stress stimulates nitric oxide (NO) production by endothelial cells, which promotes vasodilation [69–71]. However, in SCD patients, as we will discuss later, endothelial function is impaired and NO bioavailability is decreased [47, 48], which limit the possibility of the vessels to dilate and compensate for any increase in blood viscosity [68]. In summary, vaso-occlusive like complications seem to be characterized by a “increased RBC deformability-increased blood viscosity” phenotype. Of note, hydroxyurea treatment increases RBC deformability and decreases hemolytic rate, which result in increased hematocrit [72, 73]. However, the improvement of RBC deformability is so high that it compensates for the increase in hematocrit, which results in no change in blood viscosity [72, 73].
Genetic and cellular modulators of RBC rheology in SCD
Fetal hemoglobin (HbF)
There is a rapid switch from γ-globin to β-globin gene expression after birth, which leads to the replacement of fetal (α2γ2; HbF) by adult (α2β2; HbA) hemoglobin. This hemoglobin switch, however, is not total or irreversible, and all adults retain the ability to produce residual levels of HbF (less than 1% of total hemoglobin) [74, 75]. However, in SCD, there is a delayed switch from γ-globin to β-globin gene expression, and HbF levels remain above normal in most patients [76]. HbF prevents HbS polymerization because of its exclusion from the polymer [77]. Indeed, patients with the highest percentages of HbF are usually less severe than patients with low HbF level [76]. Hydroxyurea stimulates the production of HbF in SCD patients and decrease anemia and the clinical severity [78, 79]. Since HbF limits the polymerization of HbS, one could expect an impact of HbF on the rheological properties of RBCs from SCD patients. Figure 4 shows the positive association between HbF level and RBC deformability in 415 patients with SCD.
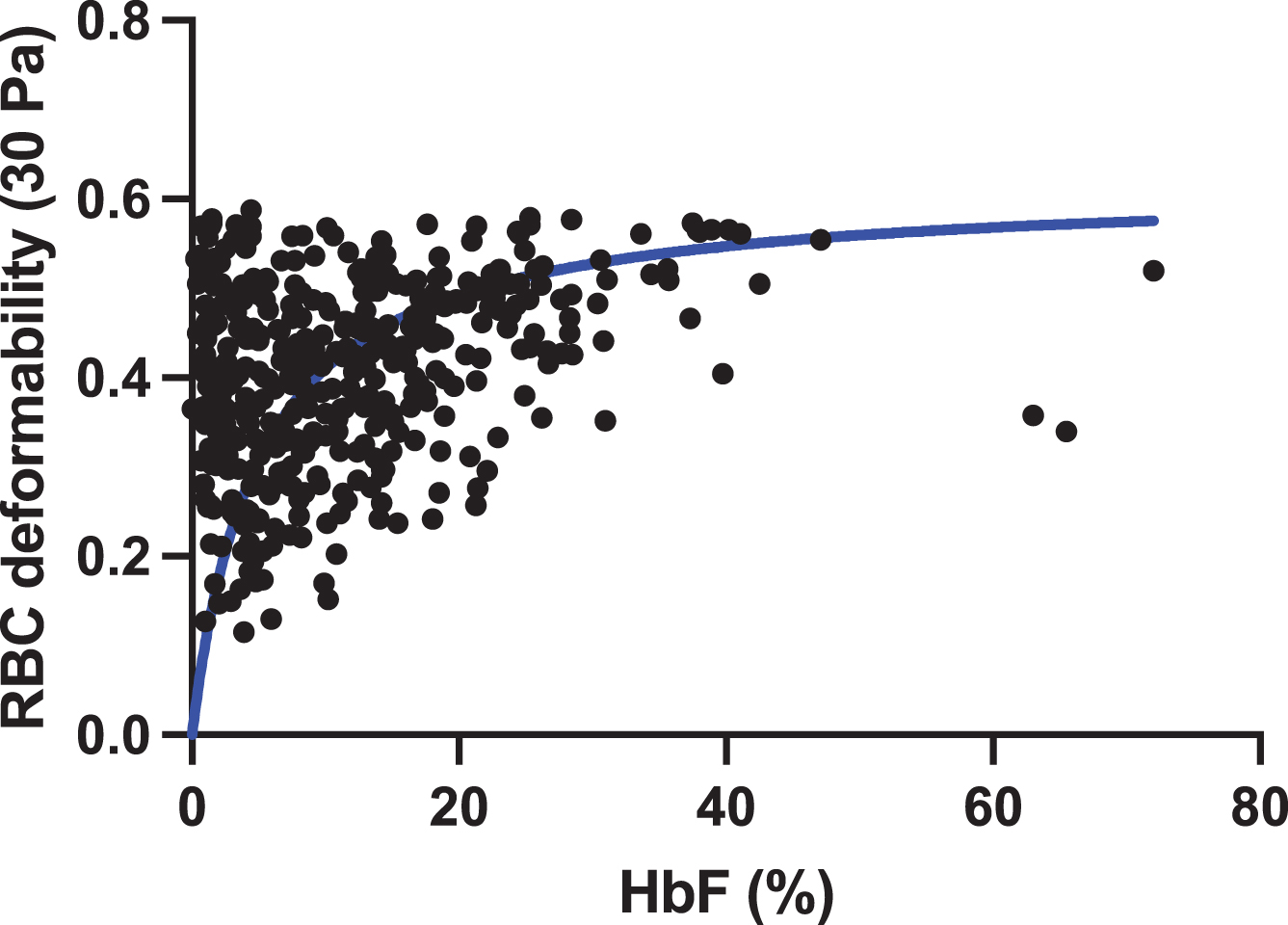
Association between HbF level and RBC deformability determined by ektacytometry at 30 Pa in 415 non-transfused patients with SCD (HbSS genotype) at clinical steady-state.
α-thalassaemia is inherited as an autosomal recessive disorder characterized by a microcytic hypochromic anemia, and a clinical phenotype varying from almost asymptomatic to a lethal hemolytic anemia. Having one or two α-globin gene deleted is pretty frequent in some places, such as in Jamaica [80], Carribean Islands [81], Southeast Asia [82] and West Africa [83]. Indeed, co-existence of SCD with α-thalassemia is not rare in countries where SCD is frequent [53, 81]. SCD patients with one or two α-genes deleted usually have higher RBC deformability than patients with no α-genes deleted resulting in lower hemolysis [48, 85]. We recently reported that the tendency of RBCs to sickle under deoxygenation is lower in SCD patients with α-thalassemia compared to those without [86], as a consequence of the reduction of the mean corpuscular hemoglobin concentration and less HbS polymerization. These RBC rheological impacts of α-thalassemia may explain why SCD patients with α-thalassemia have a lower risk of developing several complications where hemolysis plays a role, such as stroke/cerebral vasculopathy [87, 88] or glomerulopathy [53]. In contrast, patients without α-thalassmia are at greater risk of having frequent vaso-occlusive crises or osteonecrosis [59, 85] because the greatest RBC deformability results in less severe anemia (i.e., greatest hematocrit) that may increase blood viscosity.
Oxidative stress and NO
SCD is characterized by increased plasma oxidative stress and inflammation, and decreased NO bioavailability, mainly as a consequence of increased hemolysis [57]. The decreased NO bioavailability is mainly due to the NO consumption by the free plasma hemoglobin and the utilization of L-arginine (the precursor of NO) by the arginase released by RBCs [48, 89]. Asymmetric dimethylarginine (ADMA), an endogenous NO synthase inhibitor, is also abundant in RBCs and released during hemolysis [90]. Both ADMA and depletion of L-arginine can contribute to the uncoupling of endothelial NO Synthase, which in turn produces reactive oxygen species (ROS) instead of NO [91]. In addition, cell free hemoglobin, heme and iron react with hydrogen peroxide to form hydroxyl radical (Fenton reaction). It has also been demonstrated that the rate of auto-oxidation of HbS is increased, compared to HbA, which further increases the generation of ROS inside sickle RBCs [92]. Moreover, the repeated episodes of ischemia-reperfusion, such as those occurring during vaso-occlusive crises, are also a major source of oxidative stress [93]. In summary, there is an accumulation of ROS and a decrease of circulating NO into the plasma of SCD patients. We therefore decided to investigate the impact of oxidant/antioxidant molecules and NO on the RBCs. Nader et al [94] showed that sodium nitroprusside (a NO donor) increased slightly the deformability of RBCs from SCD patients. Incubation of RBCs from healthy and SCD individuals with t-butyl hydroperoxide (TBHP), a strong oxidant agent, has been shown to cause a reduction of RBC deformability and an increase of the strength of RBC aggregates but the magnitude of changes was higher with RBCs from SCD patients, as a consequence of the decreased antioxidant defense in the RBCs of SCD patients [95]. Incubation of RBCs from SCD patients with the antioxidant N-acetylcysteine increased RBC deformability [94]. Clinical trials performed in SCD patients showed that N-acetylcysteine was able to limit oxidative stress, hemolysis and RBC dehydration [96–98]. However, Sins et al failed to clearly detect a significant clinical impact of NAC therapy in SCD [99]. Further studies are needed to see how limiting oxidative stress and improving the rheological properties of RBCs could translate into clinical benefits in SCD patients.
Non-selective cationic ion channels
RBC deformability depends on several factors: membrane viscoelasticity (dependent on the cytoskeleton proteins and lipid bilayer properties), internal (cytosolic) viscosity (mainly determined by the mean cell hemoglobin concentration) and the surface-area-to-volume ratio, also called cell sphericity [100, 101]. Indeed, any change in one of these factors may impact on RBC deformability.
The volume of RBCs is tightly regulated by the activity of several ion channels located at the membrane level. The Gardos channel (KCNN4) is a Ca2 + sensitive, K+ selective channel present in several tissues including RBCs, where it is involved in cell volume regulation. Any increase in Ca2 + concentration may activate this channel, resulting in potassium, chloride and water loss, volume reduction and increased internal viscosity [102]. Mutations in the Gardos channel have been reported in patients with hereditary xerocytosis [103]: the Gardos channel hyperactivation is followed by RBC dehydration, a reduction in RBC deformability and hemolysis [104]. Several non-selective cationic ion channels may be involved in the entry of Ca2 + into RBCs [102]. Among them, Piezo1, a mechanosenstivive protein, is suspected to play a key role in the regulation of RBC volume, both in physiological and pathophysiological situations.
Deoxygenation of sickle RBCs activates a cation permeability of unknown molecular identity (Psickle), leading to elevated intracellular Ca2 + and subsequent activation of the Gardos Channel. The subsequent RBC dehydration increases intracellular HbS concentration, facilitating its polymerization under deoxygenation and RBC sickling [105, 106]. We recently investigated the role of Piezo1 as a potential candidate for Psickle. We showed that Yoda1, a molecule used to activate Piezo1, caused greater RBC membrane hyperpolarization (due to Gardos channel activation) in deoxygenated condition than in oxygenated condition [107]. Activation of Piezo1 increased the tendency of RBCs to sickle under deoxygenation, decreased RBC deformability and increased RBC adhesion to laminin [107], while inhibition of Piezo1 by GSMTx4 caused the opposite. In addition, we found that SCD patients with a gain of function mutation in Piezo1 had increased tendency of RBCs to sickle under deoxygenation. These findings suggest that Piezo1 could be involved in Psickle but other channels might also play a role. TRPV2 channels share several features with Piezo1 such as mechanosensitivity as well as a strong permeability for Ca2 + [108], and may be activated by Δ9-tetrahydrocannabinol (Δ9THC) [109]. We recently demonstrated that in-vitro activation of TRPV2 channels by Δ9THC on the RBCs from SCD patients caused a greater membrane hyperpolarization during hypoxia compared to normoxia [110]. In addition, Δ9THC increased intracellular Ca2 + concentration and caused a decrease of RBC deformability [110]. Indeed, it seems that a group of ion channels, and not only one protein, could contribute to Psickle and modulate the rheological properties of RBCs in SCD.
RBC mitochondria retention
Recent studies reported the presence of mitochondria in mature RBCs in patients with SCD, which could be the result of a deficient mitophagy pathway throughout erythropoiesis [111, 112]. Moreover, SCD patients with a high percentage of mature RBCs containing mitochondria exhibited higher levels of reticulocytes and total bilirubin, suggesting increased hemolysis in these patients compared to those with a low percentage of mature RBCs retaining mitochondria [111]. Since hemolysis is related to RBC fragility, and RBC fragility is related to RBC deformability, we suspected that abnormal mitochondria retention and RBC deformability could also be related. Using flow cytometry, we quantified the percentages of mature RBC with residual mitochondria into a large group of SCD patients and found highly variable levels from one patient to another [113, 114]. We also investigated the functionality of these mitochondria as it was suggested that they could be not functional [111] and found that mature RBCs containing mitochondria from SCD patients were able to consume oxygen, and half of this oxygen consumption was devoted to ATP production [113]. Further, we observed that patients with the highest percentages of mature RBCs containing mitochondria exhibited greater hemolysis, lower RBC deformability and increased expression of RBC senescence markers. Figure 5 summarizes how the abnormal presence of still functional mitochondria could lead to oxygen consumption inside RBCs, leading to a reduction of the amount of available oxygen for HbS, resulting in HbS polymerization and RBC sickling [113].
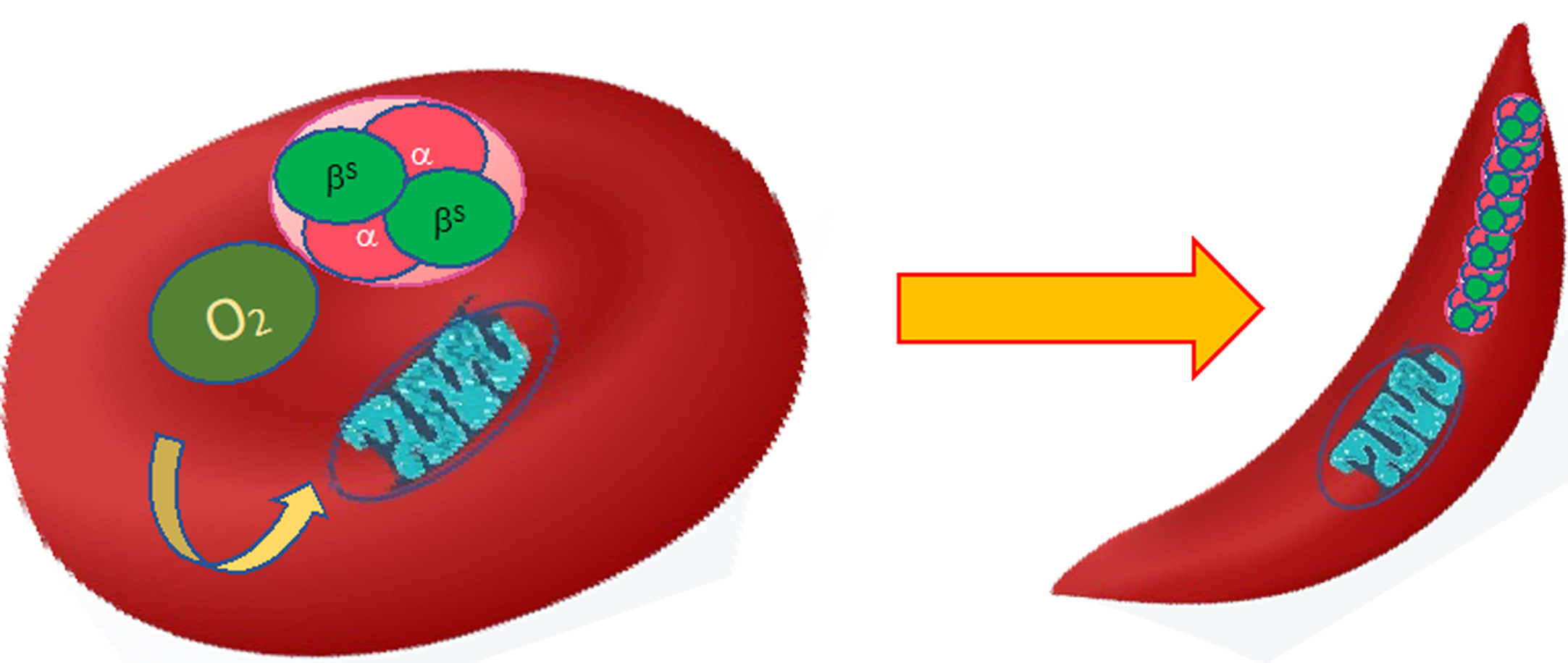
Impact of the presence of functional residual mitochondria into mature RBC from SCD patients on HbS polymerization and sickling. Mitochondria are thought to consume oxygen that may decrease the amount of oxygen available for HbS, precipitating its polymerization and RBC sickling.
For a long time, SCD was considered to be a disease of red blood cells only. However, accumulating evidence in the past decades showed that vascular function is also impaired. Belhassen et al [115] showed that flow-mediated dilation of the brachial artery of SCD patients is decreased compared to healthy individuals. Using cutaneous laser doppler flowmetry techniques, we showed decreased microcirculatory vasodilation in SCD patients compared to healthy subjects [68, 116]. More recently, using sublingual video microscopy, we reported higher percentages of small microvessels (less than 6 μm diameter) in SCD than in healthy individuals, which may be problematic as SCD RBCs are less deformable than RBCs from healthy controls [117].
Several factors may be at the origin of the chronic vasculopathy described in SCD. Autonomic nervous system dysfunction with marked sympatho-vagal imbalance probably plays a role in the loss of vasodilatory capacity in SCD [67, 118–122]. Hemolysis is also a key factor of vascular dysfunction as accumulation of free hemoglobin and heme into the plasma promotes inflammation and oxidative stress, and causes a reduction of NO bioavailability [48, 123–125]. RBC-derived microparticles are also though to play a role in the vascular dysfunction of SCD [126, 127]. A high percentage of RBCs are characterized by externalized phosphatidylserine (PS) in SCD, demonstrating advanced RBC senescence [58], which is accompanied by a release of PS positive microparticles into the plasma [128–130]. We demonstrated a positive association between the plasma concentration of RBC-derived microparticles and pulse wave velocity, a marker of arterial wall stiffness, in SCD [94]. We then isolated and purified RBC-derived microparticles from the plasma of SCD patients and added them to cultured human aortic endothelial cells, which resulted in increased ICAM-1 expression and production of pro-inflammatory cytokines [94]. In another study, we found that neutrophil adhesion to endothelial cells was increased when pre-incubating these endothelial cells with RBC-derived microparticles from SCD patients compared to those from healthy individuals, and the increased neutrophil adhesion was mainly dependent on VCAM-1 expression [131]. The heme content of these microparticles and the externalized PS at the surface of these microparticles are thought to be involved in the activation of endothelial cells and the development of vascular dysfunction [126, 131]. This pathway might provide new targets for the therapeutic preservation of vascular function in SCD [126, 131].
Sickle cell disease and treatments
The last part of my Fåhraeus lecture dealt with some treatments in SCD, and I choose to present few results on the effects of hydroxyurea and physical activity. For further information on other therapies, the reader may refer to other reviews [132–136].
Hydroxyurea
Hydroxyurea is mainly used to stimulate the production of HbF in SCD [79]. Charache et al [78] and others [79] demonstrated that hydroxyurea treatment reduces the frequency of vaso-occlusive crises and acute chest syndrome, and decreases the severity of anemia. Indeed, some physicians were afraid that hydroxyurea could, in some cases, increase hemoglobin to high concentration, that could increase blood viscosity and cause vaso-occlusive like complications. We therefore conducted a two-years follow-up study where we investigated the changes in blood rheology in SCD patients starting hydroxyurea [72, 73]. We demonstrated that hematocrit and hemoglobin concentration increased over the 2 yrs period but without any changes in blood viscosity. The lack of change in blood viscosity was attributed to the improvement of RBC deformability and the decrease of RBC aggregates strength, which compensated for the effects of the increase of hematocrit on blood viscosity. This study demonstrated that hydroxyurea therapy does not increase blood viscosity and suggest that it could also be used in patients with HbSC disease, who are characterized by blood hyperviscosity, even at steady-state [72, 73].
Physical activity
It was long thought that sickle cell patients could not participate in physical activity because the acute metabolic and physiological changes that may occur during exercise (acidosis, hyperthermia, hypoxemia, dehydration) may promote HbS polymerization and RBC sickling [137]. However, more than 10 years ago, we performed several studies in Guadeloupe and Ivory-Coast where we looked at the effects of a single short (less than 20 min duration) and moderate (below or at the first ventilatory threshold) cycling exercise on various biomarkers known to be involved in vaso-occlusive crises [138–140]. These studies demonstrated that this type of endurance exercise did not cause major changes in RBC deformability, blood viscosity, inflammation or oxidative stress, and of major importance, did not trigger vaso-occlusive crises or other clinical complications. We therefore decided to investigate the effects of chronic regular physical activity in SCD as it has been shown that exercise training may have cardiovascular, muscle, metabolic, respiratory, blood rheological and inflammatory beneficial effects in other chronic diseases [137]. We conducted studies in mice using two models of SCD (SAD and Townes mice) and demonstrated that 8 weeks of regular physical activity decreased blood viscosity and inflammation [141, 142]. Since acute short and moderate endurance exercise did not cause any clinical adverse events in SCD patients, and that training improved the biological phenotype of sickle cell mice, we decided to test the effects of a light training protocol composed of 15-30 min exercise sessions, twice a week, for 6 weeks in children with SCD [143]. Plasma NO increased, RBC aggregates strength decreased and ventilatory efficiency increased at the end of the 6 weeks-training. These findings reinforce the results of another study conducted in SCD adults and showing improved exercise tolerance and muscle benefits after 8 weeks of training at moderate intensity (3 sessions of 45 min per week) [144–146]. All these studies are now motivating the SCD medical and scientist community to evaluate the long-term effects of regular physical activity on the clinical severity of the patients.
Conclusion
SCD is the prototype of a blood rheological disease. However, the various experimental results accumulated in the 3 last decades show that SCD is highly complex with various cellular and molecular mechanisms being involved in the modulation of the clinical expression of the disease. The identification of new pathophysiological mechanisms sometimes leads to the development of new therapies to decrease the clinical severity and improve the quality of life of the patients.
Footnotes
Acknowledgments
I would like to thank all the great scientists I had/have the chance to collaborate with: many of them became very good friends. I am also grateful to my previous and current PhD students and post-doc who contribute actively to the field of hematology, red blood cell, hemorheology, vascular biology and sickle cell disease, and participate to the wonderful spirit of the “Vascular Biology and Red Blood Cell” team: Julien Tripette, Mona Hedreville, Xavier Waltz, Yann Lamarre, Keyne Charlot, Anaïs Mozar, Mor Diaw, Berenike Mockesch, Celine Renoux, Laurence Beral, Sarah Skinner, Elie Nader, Emeric Stauffer, Mélanie Robert, Sofia Esperti, Romain Carin, Marie Martin, Claire Bordat, Franciele Lima. I am very grateful to my wife, Kati, my kids, Chloé and David, and my parents, Marcelle and Christian, who always accepted my passion and off-hours work for hemorheology.