
Abstract
Glioma is the most common primary tumor of the central nervous system (CNS). Glioblastoma (GBM) is incurable with current treatment strategies. Additionally, the treatment of recurrent GBM (rGBM) is often referred to as terminal treatment, necessitating hospice-level care and management. The presence of the blood-brain barrier (BBB) gives GBM a more challenging or “cold” tumor microenvironment (TME) than that of other cancers and gloma stem cells (GSCs) play an important role in the TME remodeling, occurrence, development and recurrence of giloma. In this review, our primary focus will be on discussing the following topics: niche-associated GSCs and macrophages, new theories regarding GSC and TME involving pyroptosis and ferroptosis in GBM, metabolic adaptations of GSCs, the influence of the cold environment in GBM on immunotherapy, potential strategies to transform the cold GBM TME into a hot one, and the advancement of GBM immunotherapy and GBM models.
Introduction
All tumors lacking IDH mutations with concomitant gain of chromosome 7 and loss of chromosome 10, EGFR amplification, or TERT promoter mutations are referred to as glioblastomas.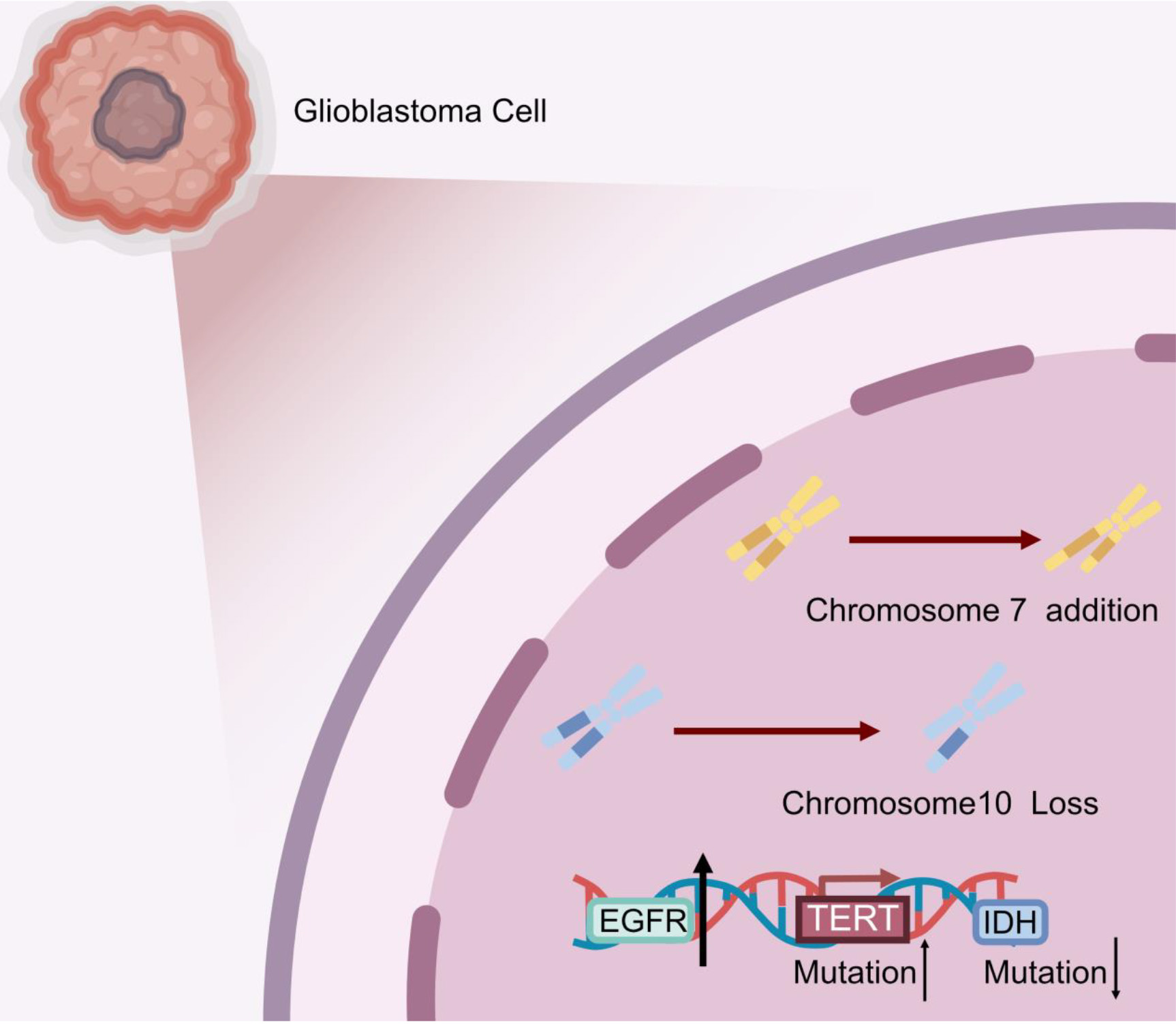
Glioblastoma multiforme (GBM) is the most common intracranial malignant tumor, and its prognosis has not made significant progress, despite the advances in treatments. In the 2021 edition of the WHO classification, gliomas lacking IDH mutations that have concomitant
TME and glioma stem cells
A tumor is a complex system comprising both tumor cells and various non-tumor cells, and the TME is a direct representation of this intricate system. The TME consists of cancer cells surrounded by diverse non-malignant cell types, such as cancer-associated fibroblasts, endothelial cells, pericytes, and other cell types that can differ based on the tissue, like adipocytes and neurons. Throughout various stages of tumor development, including initiation, progression, invasion, intravasation, metastatic dissemination, and outgrowth, the TME and its cells play a crucial role. Immune tolerance in the tumor microenvironment leads to immune escape from therapy, which is mainly due to the ability of tumor stem cells to remodel the tumor’s immune microenvironment [16]. Interaction of CSCs with their niche is critical for tumor immunosuppression and tumor recurrence. Moreover, it was demonstrated that a high-stemness signature related to a poor immunogenic response across 21 solid malignancies. Most notably, CSCs are able to recruit tumor-associated immune cells such as monocytes and macrophages, and these immune cells can play a role in promoting tumor progression due to the remodeling of the tumor microenvironment [17]. As a result, conducting systematic research on cancer stem cells and other related cells within the TME will be a vital approach in identifying new targets for treating malignant tumors [18].
In glioma, the TME includes not only tumor cells but also immune cells, endothelial cells, glial cells, and neuronal cells. GSCs can remodel the immune-tolerant microenvironment of gliomas regardless of tissue cell type, and immune-inflammatory cells in the tumor microenvironment are even capable of undergoing malignant transformation through the remodeling of glioma stem cells, which leads to changes in immune tolerance and heterogeneity of tumors by a mechanism that may be related to cell fusion [19]. Furthermore GSCs promote tumor angiogenesis and remodel the microenvironment of GBM by secreting histamine [20]. GBM has the ability to recruit normal cells from its surroundings to support its growth, maintenance, and invasion into the brain. Studies have demonstrated that the microenvironment in GBM varies depending on factors such as the isocitrate dehydrogenase status (mutated/wild type), the presence or absence of codeletion, and the expression of specific alterations like H3K27 and/or other gene mutations [21]. Recent investigations using Single-cell RNA sequencing (scRNA-seq) in high- and low-grade gliomas have revealed that intratumoral heterogeneity and dynamic plasticity across different cellular states are characteristic features of malignant brain tumors. As the tumor grade increases, there is an observed increase in the proliferation of malignant cells, larger populations of undifferentiated glioma cells, and a shift towards a higher expression of macrophage programs in the tumor microenvironment, compared to microglia expression programs [22].
Schematic diagram of GSCs and immune-related mechanisms: A. Tumor entities, including the hypoxic niche and cell necrosis niches caused by tumor cell pyroptosis and ferroptosis and the macrophage niche, involved in adaptive immunity in the tumor microenvironment. B. The jagged and vague tumor periphery mediates tumor cell invasion and dissemination and marginal ecological niches are colonized here. C. Inflammatory necrotic cells located in the tumor necrosis zone caused by pyroptosis and ferroptosis. D. Hippocampus-subependymal neural stem cell niche: Maintenance and expansion of hippocampal- and subventricular-derived neural stem cells follow both symmetric and asymmetric disaggregation patterns to maintain homeostasis of glial-associated downstream cells in normal brain tissue, which in the case of GBM are largely replaced by the associated tumor stem cell niche. At this point, tumor cells may reverse-differentiate into GSCs.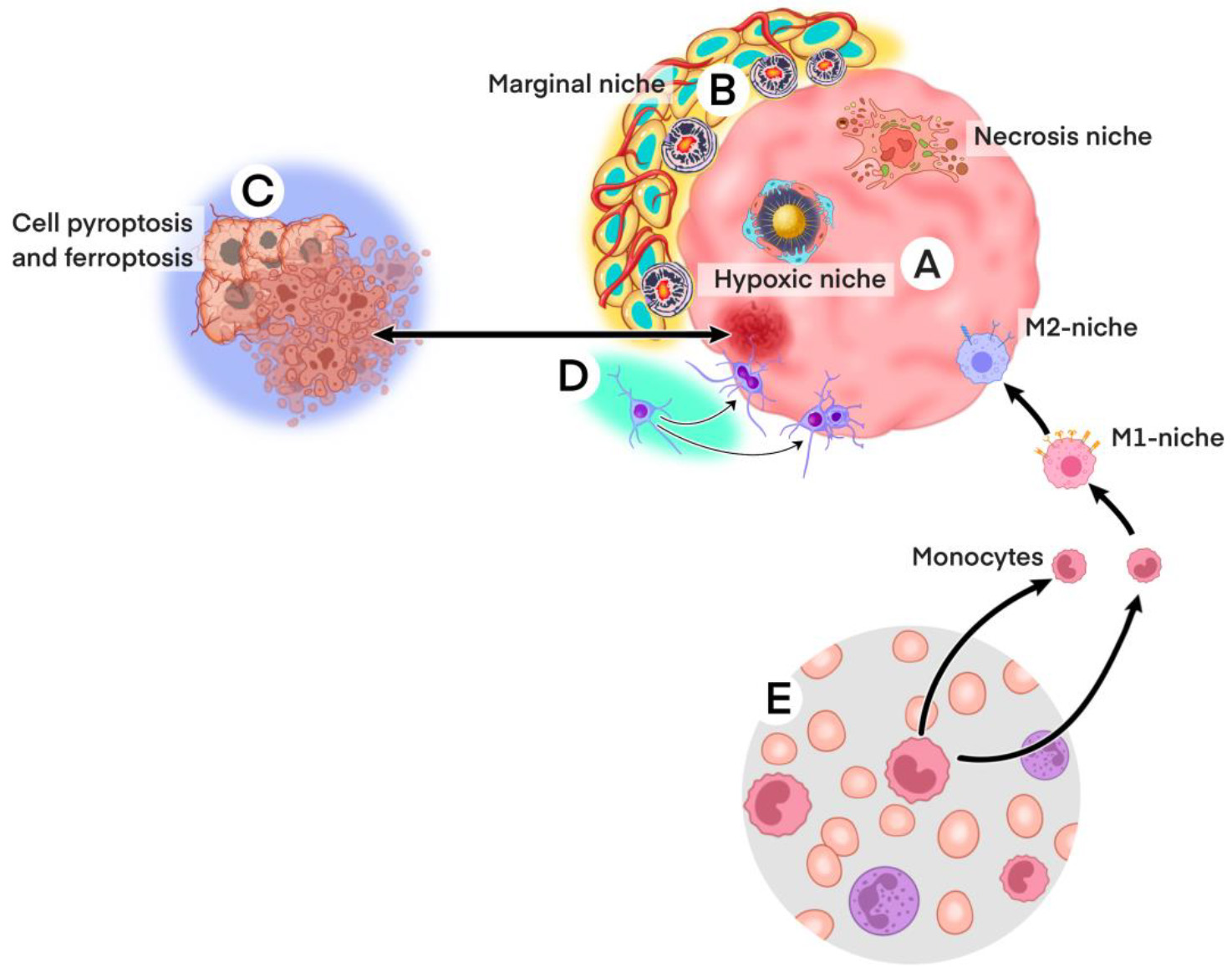
Human GSCs in adult and child were first reported in 2003 by Singh SK [23], and in 2006 by Quanbin Zhang, respectively [24], and their mysteries have not yet been fully unveiled. The existence of GSCs can be a subject of debate, and the answer to whether they exist or not depends on various factors and perspectives. The stem cell marker CD133 expressing cells which are identified as GSCs in experiments tend to express the progenitor marker Nestin simultaneously [24], thus they are actually progenitor cells that have initiated the differentiation process. Real GSCs are treatment-resistant, quiescent and pluripotent and reside in a niche determined by the adaptive GBM immune microenvironment (Fig. 2A and 1B). The mystery lies in the fact that if the same cells are traced by only CD133 single positive fluorescent staining but not by CD133 and Nestin double staining, they may be GSPCs, rather than GSCs [2, 25]. As of today, there are still cells that are discreetly referred to as GSC-like cells, rather than being explicitly labeled as GSCs. This distinction reflects ongoing debates and complexities in the field of glioma research [26]. In fact, as early as 2011, GSCs were defined as those cells capable of driving tumor formation and spreading by differentially labeling human GBM cell components in a xenograft model and following tumor development using a living microscope [27]. GSCs have also been reported as capable of differentiation into offspring cells which may reverse-differentiate into stem cells [24] (Fig. 2D). This is not consistent with the view of Singh SK [28], who cloned GSCs from pediatric GBM and stated that GSCs originated from resident neural stem cells (NSCs) of the host hippocampus or under ependyma and differentiate irreversibly [23]. Subsequent research appeared to provide evidence supporting the concept of reverse-differentiation in GSCs [24]. This suggests that GSCs may possess the ability to revert back to a less differentiated state, adding further complexity to our understanding of these cells and their role in glioma. Furthermore, new CD133+ cells were detected in the in vitro cell cultures of rat glioma C6 after all CD133+ had been removed and defined most C6 cells as GSCs [29]. The potential for C6 cells to reverse differentiate into GSCs now seems a more realistic possibility. Under the conditions at the time, this reverse differentiation observation was not comprehensive enough, and the potential stem cell microenvironment, especially the Niche, was proposed later and is still a hot topic today.
Studies conducted on Drosophila have contributed to the introduction of the concept of the niche [30], and in many instances, niches have been observed to be located in close proximity to the endothelium of blood vessels [31]. The understanding of its function has improved with the deeper research. Our research of GSCs transdifferentiating into vascular endothelial cells [25, 32] was published in 2011, ahead of similar reports by Wang R [33] and Ricci-Vitiani L [34], and exciting commentary by Victoria L Bautch [35]. Nowadays, it is understood that this transdifferentiation process may occur within the hypoxic periarterial niche of GSCs [36]. The GSC niche may also be subdivided into perivascular, peri-hypoxic, immune extracellular matrix and GBM peri-invasive sectors [37, 38, 39, 40, 41], the functions of which remain obscure except as an adaptive GBM immune microenvironment. The niche regulates angiogenesis and protects the GSC from radiotherapy and chemotherapy, driving recurrent GBM (rGBM) [42, 43]. Macrophage niches are similar to the adaptive immune microenvironment of GBM.
Macrophage niche and tumor-associated macrophages
Researchers believe that the macrophage niche (mNiche) can be characterized by four fundamental functions: (1) providing a physical foundation or scaffold for the macrophage; (2) supplying nutritional factors to support the macrophage’s self-maintenance ability; (3) imparting the tissue-specific identity to the resident macrophage within the niche; and (4) the macrophages, in turn, should provide benefits to their niche. The mNiche plays an important role in tumor progression. mNiche is found throughout all mammalian organs. In addition to their role as immunesentinels, macrophages perform day-to-day functions essential to tissue homeostasis. mNiche maintains tissue homeostasis of macrophage, controls the macrophage population size and imprints their tissue-specific identity [41]. The mNiche has attracted attention for its potential therapeutic value. Previously, competition between macrophage precursors was proposed for development into resident macrophages in a limited number of niches [44]. Tight regulation ensures that monocytes differentiate into multiple heterogeneous macrophages only when niche space is available.
Nevertheless, the study of mNiche in tumors is still in its early stages, but significant progress has been made in understanding tumor-associated macrophages (TAMs). TAMs are the most abundant immune cells present in tumor tissues and are typically classified into two distinct subtypes: M1 macrophages and M2 macrophages [45].
M1 macrophages are known for their anti-tumor functions, whereas M2 macrophages have the opposite effect, promoting tumor development, metastasis, and inhibiting the anti-tumor immune response mediated by T cells. Additionally, M2 macrophages facilitate tumor angiogenesis and contribute to tumor progression. As a result, TAMs have become a promising target for tumor therapy [45].
In gliomas, similar to other solid tumors, the infiltration of TAMs is a notable characteristic. In GBM, TAMs are significantly elevated, as confirmed through bioinformatics studies. Higher levels of TAMs are associated with a decreased overall survival rate in glioma patients, suggesting that increased TAMs may be one of the mechanisms involved in immune escape in GBM. These findings indicate that TAMs-related signatures can serve as valuable prognostic biomarkers in GBM [46].
In addition to the presence of mNiche, the immune microenvironment of GBM is more complicated than in that of extracranial cancers such as the cold immune microenvironment.
The cold GBM immune microenvironment resists the immune response
Cold immune microenvironment of GBM
Cancers may be classified as “hot” when there is a large T cell and inflammatory response after immune checkpoint inhibitor treatment, “warm” or “cold” when there is little response to treatment [47]. For example, approximately 50% of melanoma patients respond to the combined blockade of the immune checkpoint PD-1 and CTLA-4, 75% of whom have a long-lasting response [48]. Thus, melanoma is a hot tumor type. Conversely, Glioblastoma is a cold tumor, mainly because of immune tolerance in the GBM microenvironment. Compared to other tumor types, glioblastomas have relatively few tumor-infiltrating lymphocytes (TILs), and those that are present have been shown to be highly expressive of exhaustion markers. The glioblastoma microenvironment is characterized by the presence of a large number of myeloid cells, such as microglia and macrophages, which have immunosuppressive activity. In addition, defects in antigen-presenting mechanisms can make the tumor cold in response to T-cell-dependent immunity. Finally, necrosis in glioblastoma plays an important role in weakening the anti-tumor immune response [47]. Only 10% of GBM patients have a short-lived response to immunotherapy [49, 50]. The concept of transforming a “Cold” tumor into a “Hot” one is a novel area of research in tumor immunotherapy (IO). However, the impact of intratumoral injection of tilsotolimod, an oligodeoxynucleotide Toll-like receptor 9 (TLR9) agonist, in patients with advanced melanoma has not been conclusively determined [51], suggesting that traditional research approaches still have limitations. Fortunately, quantitative systems pharmacology modeling in cancer immunotherapy holds great promise in addressing major challenges in the IO field [52].
Exploration for GBM cold environment
In the case of GBM, immunotherapy research has not stopped because of the cold immune microenvironment. Preclinical GBM models suggest Antigen-primed T cells could accumulate in brain tumors through healthy tissue tracking [53], and execute cytotoxic function with cellular precision [54], as well as adapt to a tumor’s evolving molecular profile via epitope spreading. Antitumor CD8 T cells can be controlled by PD-1/PD-L1 interactions [55]. PD-1 blockade augmented the anti-tumor CD8 T cell response, allowing the formation of memory T cells with the ability to prevent delayed tumor outgrowth [56]. In summary, data from preclinical models indicated the potential for GBM immunotherapy [56, 57, 58, 59, 60] but clinical trials have proved unsuccessful [61]. The phase III clinical trial of the anti-PD-1 monoclonal antibody, nivolumab, and the anti-growth factor VEGF-A monoclonal antibody, bevaci-zumab, for rGBM was terminated. However, Jackson, et al. considered that the cold nature of GBM may be converted into hot [62]. Recently, GBM cold tumors were divided into two subtypes with immune tolerance or immunodeficiency from data in the TCGA-GBM transcription database and the GEO dataset [63]. Tumor-associated macrophages were indicated as promising new therapeutic targets and GIPS as a biomarker for assessing the immune evasion mechanism, immunotherapy response and patient prognosis.
Can microglia/macrophages turn cold GBM hot?
Resident tissue macrophages (RTMs) proposed by Blériot C [64] appear to be much more reasonable than those of macrophages in the tumor tissue microenvironment simply divided into M1 and M2 proposed earlier [50, 65]. The heterogeneity of RTMs includes four characteristics: cell origin, local environment, inflammatory state and residence time in tissues that contributes to the resilient adaptation of macrophages to their dynamic environment [64]. Brain RTMs also present these characteristics, in addition to the blood-brain barrier [66, 67, 68] and the cerebral lymphatic system [69, 70, 71]. Microglia are a unique tissue-resident macrophage population that plays an important role in maintaining the tissue homeostasis of the CNS [72]. Its characteristics and functions are mediated by Sall1, SMAD2/3, IRF8, Nr4a1 (Nur77), Nr4a2 (Nurr1) and Nr4a3 (Nor1). Nr4a1 (Nur77) can downregulate the transcription of thyroxine-hydroxylase by recruiting the CoREST complex involving HDAC1 and HDAC2 enzymes in the TH promoter region [73, 74, 75, 76]. Mice lacking Nr4a1 had poor prognosis and had high concentrations of norepinephrine (NE), pro-inflammatory IL-6, and autoimmune effector T cells at the site of the affected tissue area in the CNS, which was also necessary for GBM to switch from cold to hot. Thus, we may deduce that if a similar experiment is performed in a GBM mouse model, transcriptomic sequencing of the tumor and myeloid precursor derived macrophages may enable identification turnoff factors responsible for turning cold GBM into a hot tumor. Appropriate sequencing targets would be those concerned with initiation of pyroptosis or ferroptosis, which can trigger an acute inflammatory response. Hence, there is a reason to be optimistic about the search for regulatory molecules that could potentially transform GBM from a cold tumor microenvironment to a hot one.
Pyroptosis and ferroptosis
Pyroptosis, PP
Thornberry NA [77] observed cysteine aspartase [caspase]-1-mediated programmed cell death, of a form morphologically distinct from apoptosis [AP], but of unknown mechanism in 1992. By 2015, PP effect is initially understood after gasdermin D (GSDMD) cleavage target of caspases-1 and -11 was discovered [78, 79]. PP was shown to be mediated by a pro-inflammatory caspase effect which caused cell death by cell membrane rupture and cell disintegration and was an anti-infective mode of inflammatory cell death against pathogens [63, 80, 81, 82, 83, 84, 85, 86, 87]. Chemical disruption of GSDMD was found to inhibit inflammatory cell death and activate IL-1 secretion by macrophages [88, 89]. More recently, methods to regulate its activity have recently been investigated. Succinate and disulfiram have been found to inactivate GSDMD to control PP and Ragulator-Rag complex has been found to be necessary for GSDMD pore formation and pyroptosis in macrophages [90, 91, 92]. Thus, mediation of PP centers around the inflammatory caspase substrate, GSDMD, which releases GSDMD-N and GSDMD-C domains on lysis, leading to PP by forming membrane pores. The extensive gasdermin family is composed of GSDMA, GSDMB, GSDMC, GSDMD, GSDME/DNFA5 and PVJK/GSDMF of which Gasdermin E shows promise as a potential target for disease therapy [93, 94].
Glioma pyroptosis (GPP)
Recent interest in GPP [95, 96, 97, 98, 99] has focused on TCGA and CCGA database bio-informatics-selection of genes and non-coding RNA (ncRNAs) associated with GPP and glioma prognosis [100, 101, 102]. Indeed, copy number variation and somatic mutation of 33 PP-related genes have been associated with GBM survival prognosis and a prognostic model constructed from 7 PP-related genes for validation in the CGGA cohort [95]. Moreover, CASP8, CASP4, CASP1, NLRP3, NLRP1 and NLRC4 have been identified as hub genes that divide gliomas into two subtypes with good and poor prognoses [96]. Fifteen scorch-death-related genes predicted overall glioma survival and nine pairs of target genes and drugs were identified. Genes encoding caspase 3 and IL-18 have been suggested as a potential prognostic biomarkers for overall survival of patients with diffuse gliomas [97]. Patients in the high-risk subgroup had shorter survival times than those in the low-risk subgroup. GSEA and ssGSEA showed the activation of immune-related pathways and the extensive infiltration of immune cells in high-risk subgroup. The prognostic value of PP-related gene expression in infiltrating immune cells has been indicated [98] in addition to glioma prognosis models of PP-related genes [99] and PP-related ncRNAs, including miRNA, lncRNA and circRNA, have also been implicated [100]. Most circRNAs are highly conserved and exon-derived with a few arising from intron cyclization. They may be classified as follows: exon circRNA (ecRNA), cyclic intron RNA (ciRNA), exon-intron circRNA (EIciRNA) and tRNA intron cyclic RNA (tri RNA) [103]. Expression of circRNA varies with developmental stage and is tissue-specific. Because circRNA is insensitive to nuclease and more stable than linear RNA, circRNA has obvious advantages in the development and application of new clinical diagnostic markers, such as the autophagy-associated circRNA, circCDYL [104] and other circRNAs have been linked to cancer cell ferroptosis [105], tumorigenesis [106], tumor metabolism [107] and drug resistance [108].
Ferroptosis and glioma immunity
Ferroptosis, similar to PP described above, is different from AP, but rather a recently highly concerned, new form of cell death that plays an important role in the occurrence and development of many diseases. The comprehensive introduction from the past, present and future of ferroptosis research written in 2020 lacked relevance to glioma [109] However, by 2021, Fe deficiency-related genes was proved to predict prognosis and immunotherapy in glioma., and the prognostic ferroptosis-related lncRNAs in glioma were associated with the immune landscape of glioma microenvironment and radiotherapy response [110, 111]. Furthermore, the characterization of a ferroptosis signature has been employed to assess the predictive prognosis and potential effectiveness of immunotherapy in glioblastoma [112], Additionally, a prognostic risk model has been developed using seven Fe deficiency-related genes for low-grade glioma (LGG), considering their implications for immunotherapy [113]. The utility of ferroptosis for GBM and LGG research is thus demonstrated.
Ferroptosis has also been shown to be responsible for glioma-associated immunogenic cell death [114, 115, 116]. The immunogenicity of ferroptosis in vitro and in vivo was first demonstrated by the induction of ferroptosis by RAS-selective lethal compound 3 (RSL3) in mouse fibrosarcoma MCA205 or glioma GL261 cells. Ironophils promoted bone marrow-derived dendritic cell (BMDC) phenotype maturation and elicited a vaccination-like effect in immunocompetent mice suggesting that the mechanism of immunogenicity is very tightly regulated by the adaptive immune system and is time dependent [117]. RNA-sequencing was used to construct a prognostic risk score model (FRGPRS) related to GBM overall survival from Fe deficiency related genes. Further comparison of genomic and clinical features, immune infiltration, enrichment pathways, pan-cancer, drug resistance and immune checkpoint inhibitor therapy in different FRGPRS subgroups showed that five Hub genes in the FRGPRS could be used to predict overall and progression-free survival of GBM patients. High FRGPRS was associated with strong immunity, higher tumor tissue ratio, good cytotoxic immunity and chemotherapy response in GBM patients [118]. The utility of ferroptosis for GBM treatment was also reported, and combination of Onofen and cold atmospheric plasmas could trigger AP, ferroptosis and immunogenic responses in GBM [119, 120]. Temozolomide was found to precipitate ferroptosis through dmt1-dependent pathways [121] and the ferroptosis inducer, disulfiram, could trigger lysosomal membrane permeability by upregulating ROS and enhanced the radiosensitivity of GBM cells [122]. Recently, scholars rediscovered from transcriptomic data that CYBB and SOD2 genes were significantly up-regulated in the mesenchymal subtype of GBM. In GBM cells that are resistant to the chemotherapy drug TMZ, they exhibit mesenchymal and stemness characteristics while also displaying resistance to ferroptosis, a type of cell death caused by iron-dependent oxidative stress. This resistance to ferroptosis is achieved through the activation of the CYBB/Nrf2/SOD2 axis. As a result, CYBB plays a crucial role in conferring ferroptosis resilience in mesenchymal GBM. The downstream compensatory activity of CYBB, achieved through the Nrf2/SOD2 axis, presents an opportunity for exploiting a potential strategy to overcome TMZ resistance by modulating ferroptosis. This finding holds promise for the development of new approaches to tackle drug resistance in mesenchymal GBM [123].
In summary, PP and ferroptosis in GBM are confined to the cell necrosis region, followed by immune adaptation (Fig. 2C). However, the immune cells come from the CNS lymphatic system (Fig. 2E), and the brain has traditionally been regarded as immune-exempt and lacking a lymphatic system, a view that may require updating.
Metabolic adaptations of GBM
The metabolic abnormalities in glioma involve disruptions in sugar, protein, and fat metabolism. Recently, more attention has been directed towards studying the glycosylation of post-translational modifications of proteins. The differential expression of glycosyltransferase genes determines the type of glycosylation and epigenetically regulates the progression of glioma. Hypoxia, a well-known factor in gliomas, has been found to induce GLT8D1, which enhances stem cell maintenance in glioma by inhibiting CD133 degradation through N-linked glycosylation [124]. As a result of these findings, various changes in the biology, biomarkers, and targeted therapies for glioma have emerged [125]. Comprehensive analyses have identified glycosyltransferase signatures and prognostic long non-coding RNAs (lncRNAs) related to glycosylation from databases such as TCGA and CGGA [126]. These analyses can be used to evaluate the prognosis of glioma patients and construct prognostic models for overall survival [127].
GSC-specific histamine secretion has been found to drive proangiogenic tumor microenvironment remodeling. Histamine, a metabolite secreted by GSCs, is produced due to MYC-mediated transcriptional upregulation of histidine decarboxylase (HDC) through GSC-specific H3K4me3 modification. GSC-secreted histamine promotes angiogenesis and GBM progression by activating endothelial cells through the histamine H1 receptor (H1R)-Ca2+-NFkB axis [128]. Interestingly, the role of histamine in the GBM microenvironment is opposite to that in the peripheral blood, where histamine triggers a positive immune response. The blood-brain barrier limits the entry of peripheral blood histamine into the GBM microenvironment, making the role of histamine-driven pro-angiogenic tumor microenvironment remodeling particularly noteworthy. Another important factor of concern is the MYC oncogene, which is often referred to as a “Superoncogene” due to its powerful role in regulating GBM metabolism [129]. The understanding of MYC has evolved over the years, and it is now known to control gene expression at multiple levels, including directly binding to chromatin and recruiting transcriptional coregulators, regulating RNA polymerase activity, and more. GBM is characterized by Myc deregulation and undergoes significant metabolic changes to meet the increased energy demand. Conversely, cancer metabolism disorders also impact MYC expression and function, making MYC a crucial link between metabolic pathway activation and gene expression. Ongoing and future studies will focus on controlling the Myc oncogene and exploring new treatments for GBM by targeting metabolic pathways to deprive tumor cells of nutrients through inhibiting MYC expression [129]. In summary, metabolic adaptations in GBM play a vital role in its malignant progression.
The immune system in the normal brain and the lymphatic system in GBM
Lymphatic vessels do not exist in human brain in medical cognition for a long time. However, as early as 2015, discharge of cerebral interstitial fluid and macromolecules by the dural lymphatic system and structure and function of CNS lymphatic vessels were described [130, 131]. Meningeal lymphatic vessels at the skull base were proved to involve in the clearance of cerebrospinal fluid (CSF) and neuroinflammation-induced lymphangiogenesis near the cribriform plate was showed to contribute to drainage of CNS-derived antigens and immune cells in 2019 [132, 133]. Furthermore, untill 2021, meningeal lymphatic vessels were found to regulate lymphatic drainage and immunity in brain tumors [134] and VEGF-c-dependent lymphatic drainage to participate in immune surveillance [135]. Finally, a complete CNS lymphatic system, encompassing arachnoid villi, periarangial pathways and dural lymphatic vessels and communicating with the cerebrospinal fluid has been proposed [136]. The view of immune exemption for the CNS has thus been considerably revised.
The situation is more complex in GBM and lymphatic outflow of cerebrospinal fluid in glioma is decreased [137]. Indeed, GBM cells inoculation proximal to the left ventricle (LV) in a mouse model disrupted the ependymal barrier and increased tumor-CSF interaction, negatively impacting immunotherapy. The author considered the occurrence of therapeutic targets in cerebrospinal fluid only if healthy ependymal membrane cells were present [138].
GBM immunotherapy
Overview of immunotherapy modalities to glioblastoma
Overview of immunotherapy modalities to glioblastoma
Current immunotherapy modalities for the treatment of glioblastoma: 1. CAR T-cell therapy such as anti-IL-13R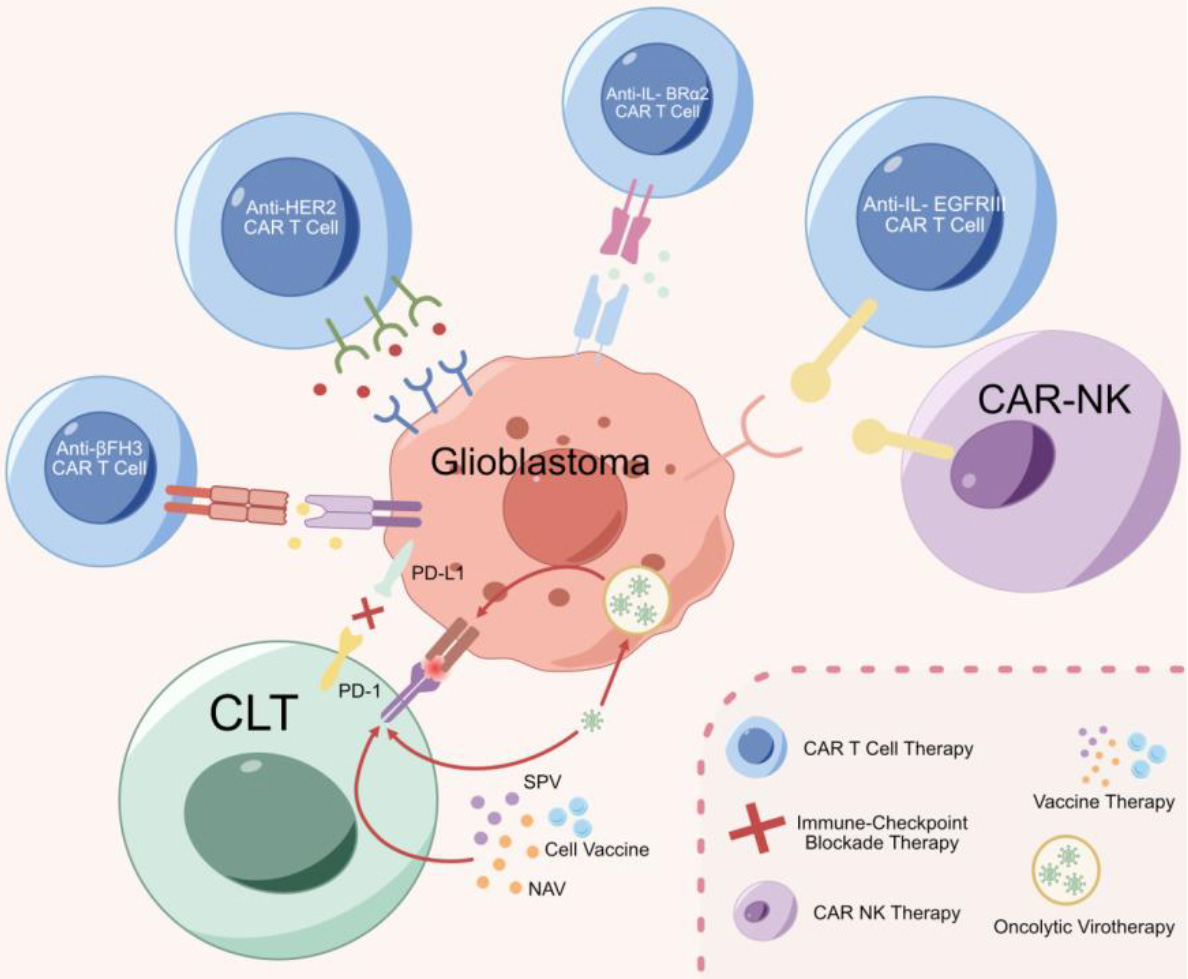
The failure of phase III GBM immunotherapy clinical trials has been attributed to the targeting of a single anti-tumor component, ignoring the acknowledged heterogeneity of the environment [139]. Further research progress has been widely concerned. Successful advances in immune checkpoint blockade therapy and targeting immunosuppressive proteins, such as programmed cell death protein-1(PD-1) and/or cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), have been reviewed [140], Initiating a paradigm shift in clinical and preclinical research and applied immunotherapy to solid tumors, which will be a potential breakthrough in the field of GBM drug treatment. However, resistance to GBM therapy has been ascribed to cancer stem cells (CSCs) and the inability of immunotherapy (IT) to completely eliminate CSCs results in failure to universally prolong patient survival [141]. A systematic IT approach to CSC elimination may provide a solution and progress has been made in CAR-T, immune checkpoint inhibitors, vaccination and oncolytic virus therapies for GBM (Fig. 3 and Table 1).
Chimeric antigen receptors (CAR) engineered T cell mediated adoptive immunotherapy (CAR-T) has made great progression in the treatment of hematological malignancies [142]. As far as GBM is concerned, as the peculiarities of the immune microenvironment described above, CAR-T has been of limited benefit for GBM, although preclinical models have furnished hope [143]. More research continues with the aim of improving CAR efficacy in GBM [144, 145]. The following three research approaches have been described.
IL13r
2 specific CAR-T
Interleukin 13 receptor subunit
EGFRvIII CAR-T and CAR-NK immunotherapy
The antitumor effects of EGFRvIII-specific CAR-T in in vitro and in vivo models of U87 cells were reported in 2013 [152]. It was later discovered that Infusion of CAR-modified T cell (CART)-EGFRvIII cells into ten recurrent GBM patients produced off-tumor toxicity or cytokine release syndrome and significant EGFRvIII -mediated CAR-T cells were found in peripheral blood [153]. Third generation EGFRvIII CAR-T (G3-EGFRvIII) increased IFN-
CAR-NK, a development based on CAR-T, is already a fourth-generation engineered cell, which has received as much attention as CAR-T, Fourth generation EGFRvIII specific CAR-NKs have been engineered [156]. Since EGFRvIII specific CAR-NK has been reported, a number of researchers [157, 158, 159, 160, 161] have demonstrated their results from different perspectives such as molecular mechanism and efficacy. Especially, MSCs can be home to GBM and not healthy brain cells, hence it serves as a tumour-specific drug-delivery system, including pro-apoptotic factors and tumor necrosis factor-related apoptosis-inducing ligands (TRAIL) [162]. Furthermore, the design of bi-functional MSCs expressing high levels of TRAIL and GD2 tCAR, which is associated with a robust anti-tumor activity against GD2-positive GBM cells, shows promise [163, 164].
HER2 or B7-H3 specific CAR-T therapy
HER2 is highly expressed on GBM ependymoma and medulloblastoma, but not in normal CNS tissues [165]. HER2-specific T cells, which target primary glioblastoma stem cells, have demonstrated promising preclinical effects in 10 GBM patients [166]. In clinical treatment of 17 HER2-positive, progressive GBM patients, there were no dose-limiting toxic effects, and CAR-T cells were detected in the peripheral blood for up to 12 months after infusion. However, despite these findings, there was no notable expansion of CAR-T cells or significant survival benefit observed in these patients [167].
B7-H3 (also known as CD276) is a newly found molecule of B7 family. B7-H3 could promote the activation of T cells and the proliferation of IFN-
CAR-T research on both hematological and solid tumors has increased between 2009–2021 [173]. When it comes to GBM, including targets such as IL13Ra2, EGFRvIII, and HER2, there are challenges that need to be addressed. However, obstacles still exist, such as the high investment costs and a lack of cooperation among research units.
Immune checkpoint inhibitor therapy
Immunotherapy, involved in various immune checkpoint inhibitor molecules, has improved patients’ survival in different types of cancers. This is one of the most hopeful approaches for antitumor therapy. Glioma immune checkpoints including PD-1/PDL-1, Tim-3/Galectin-9, CTLA4, LAG3 and TIGIT/CD96, are targets for immune checkpoint inhibitor therapy [174]. The anti-PD-1 and anti-PD-L1 monoclonal antibodies approved by the US FDA- block distinct inhibitory signals that unleash T cells to aid tumor eradication. T cells, B cells, TAMs, myeloid stem cells (MDSCs) and natural killer cells (NK) all target the PD-1/PD-L1 pathway in GBM to trigger an anti-tumor immune response. Tumor that has been immunosuppressed is removed first and then immunotherapy is used to enhance the functions of the tumor infiltrating lymphocytes (TILs). Unfortunately, the administration of checkpoint inhibitor therapy has shown limited success in GBM clinical trials, primarily due to the challenges of successfully delivering the drugs across the BBB. Some progress has been made since PD-1/PD-L1 blocking therapy was predicted to be the future for cancer immunotherapy in 2019 [175]. PD-L1-mediated GBM immunosuppression has been reported to be related with infiltration and M2 polarization of TAM [176], suggesting targeting both TAMs and mNiche as a promising strategy [44]. Indeed, CD137 and PD-L1 targeted immunoviral therapy has been shown to induce a lasting anti-tumor immune response in a malignant glioma model [177]. Follicular helper T cells have been found to restore CD8 + -dependent anti-tumor immunity and anti- PD-L1/PD-1 activity [178]. For gliomas, the PD-1/PD-L1 axis and adenosine pathways have been found to be immunosuppressive [179] and TIGIT and PD-1 immune checkpoint pathways to be associated with prognosis and anti-tumor immunity [180]. Despite these promising results, we are still far from resolving the clinical challenges posed by the disease. Indeed, the prognostic value of bioinformatics in relation to immune checkpoint inhibition for GBM has been extensively studied [181, 182, 183]. Additionally, the inhibitory impact of engineered extracellular vesicle irradiation on GBM immune checkpoints has been reported [184], and all of these findings hold promise for potential clinical applications.
Vaccination: Cell, peptide and mRNA vaccines for glioma
Cell vaccines: In addition to CAR-T and CAR-NK regarded as T and NK cell vaccines [185], Dendritic cell (DC) fusion vaccine is the most important cell vaccine. Bone marrow-derived DC fusion vaccines have been given to tumor-bearing mice, alone or in combination with telimazolid, to prolong survival time [186, 187]. Glioma stem cell-targeted dendritic cells as a tumor vaccine against malignant glioma and DC glioma cell fusion as an antitumor vaccine in vitro culture have also been studied respectively [188, 189]. In a large phase III clinical trial of DC vaccine for GBM, 331 patients with GBM after standardized treatment were included, patients were randomized to receive temozolomide plus DC vaccine (
Synthetic peptide vaccine (SPV): TollR-3/poly-ICLC and TGF-
Nucleic acid vaccine (NAV): Both DNAV and mRNAV are safe and more easily manufactured than SPVs and aim to transmit genetic information encoding tumor antigens (Tas) to the host to generate an anti-cancer immune response [201, 202]. Although NAV is safe and easy to manufacture compared to SPVs, they have so far not been considered a viable alternative to SPVs. Judging from the situation that has been carried out, DNAV for cervical cancer, prostate cancer and breast cancer and mRNAV for melanoma, GBM and prostate cancer have been investigated. A DNA vaccine with a glioma antigen, SOX6 and a vaccine targeting IL13R
Oncolytic virus therapy
Oncolytic viruses (OVs) can replicate in cancer cells but not in normal cells, leading to death of the tumor cells. Oncolytic viruses therapy (OVT) uses intratumoral delivery of virus to TME for treatment, or causes direct cytotoxicity through viral infection and replication [212, 213]. The treatment induces immunogenic cell death (ICD) in infected tumor cells when destruction of tumor cells by OVT releases antigens into the TME, recruiting and activating local dendritic cells and specific T cells [213]. The research on oncolytic virus has never ceased. Earlier regimens involving the HSV1-tk gene with the antiviral drug acyclovir [212, 214] suffered from poor vector delivery and poor efficacy. However, HSV1G207, developed later, has been shown to be safe and effective in clinical trials. The advantage is that it allows conditional replication in tumor cells while preventing infection of normal cells [215], phase I clinical trials have been conducted, whether alone or in combination with radiotherapy GBM is effective and safe [216, 217, 218]. Furthermore, the new drug, HSV-rQnestin34.5v.2, is currently undergoing clinical trials, and it has demonstrated low toxicity to human beings [219, 220].
Summary and outlook
Plasticity of the GSC niche
The aforementioned GSCs Niche are almost ubiquitous in and around GBM entities, and their function has not been fully demonstrated. The perivascular niche (PVN) is considered to be a complex microenvironment containing endothelial cells plus astrocytes, pericytes, immune cells and other stromal cells that regulate GSC biology [221, 222, 223]. It is not clear how the various cellular components of PVN change GSC behavior, such as proliferation, quiescence, invasive dissemination, homing and chemoradiation resistance. Previous 2D and 3D in vitro cultures and tumor-bearing mouse models have inevitable limitations, and bionic models have received great attention and shown a bright future [224, 225, 226, 227, 228, 229, 230]. However, it seems that there are still many difficulties whether the wish of using bionic model to completely replace clinical cases can be achieved. Single-cell sequencing has been used to detect the interactions between GSCs and immune cells during tumorigenesis [13], analyze the inhibition of CD161 receptor by GBM infiltrating T cells [12], reveal functional heterogeneity of glioma-associated brain macrophages [11], and reveal the role of m6A-modified RNA in the glioblastoma microenvironment [231]. Single cell sequencing can detect the molecules of all single cell components from clinical specimens. In biomimetic models, the cells are often artificially introduced or stocked to mimic the natural environment, ranging from biomimicry to simulation, and even high simulation, eventually forming a realistic landscape resembling clinical GBM. However, such models come with potential risks that are difficult to achieve or replicate in reality.
The dynamic nature of CSCs implies plasticity of GSCs [232], reinforcing the message of our recently published review “GSCs and Their Microenvironments: Docking and Transformation” [233]. In short, GSCs change according to the microenvironment and therapeutic signals.
A cure for GBM
Standard care for GBM only prolongs the patient’s very short lifespan and the prognosis is particularly severe for unresectable GBM [234, 235, 236, 237, 238]. Immunotherapy promises to be less than ideal [239, 240, 241]. Future treatment direction pays more attention to combination strategies. For example, the bispecific antibodies targeting two different antigens has proven to be a valuable approach, [242, 243] but the BBB excludes most macromolecular monoclonal antibodies [244, 245]. Fortunately, novel cyclic peptides that modulate BBB functions have been reported to enhance monoclonal antibody delivery to the brain [244] and focused ultrasound-mediated BBB disruption has been showed to improve the delivery of anti-CD47 monoclonal antibodies [246]. Alternatively, intratumoral administration is very valuable for improving drug distribution and sustained release. For example, PLGA nanoparticles which have been found to enhance the penetration of paclitaxel in brain tissue, including some other implants, can improve the therapeutic effect [247, 248, 249, 250, 251]. In addition, nanoformulation has been used to transform “cold” GBM tumors into “hot” and promote immune cell infiltration [252, 253]. Intranasal administration has also been proposed as a potential delivery method [254, 255]. However, most of the mentioned approaches are still in the preclinical stage, and more research is needed to explore their potential effectiveness and safety for further investigation.
Botanical medicines, such as leaf extract of Terminalia catappa L. inhibited tumor cell migration and invasion in a human GBM PDX [256, 257], artemisia annua had an in vitro anti-cancer effect and resveratrol inhibited the proliferation of dendritic cells induced by human GBM GSCs [258].
In short, there is hope to improve GBM, especially the survival prognosis of rGBM, which is currently in the stage of in vitro or in vivo experiments in animals, and there is still a painstaking research process on when incurable GBM can be turned into a treatable one.
A new model of GBM immunotherapy
GBM heterogeneity of cell composition, gene expression and phenotype means that some experimental models involved in the above preclinical studies are over-simplified, such as spheroids which represent a random aggregations of cells without a tissue-like structure, extracellular matrix or neighboring non-tumor cells. Heterogeneous tumor spheres that better meet the requirements of clinical research are being studied, including heterospheres from co-culture of cancer and stromal cells, producing spheroids containing NK cells [259] or grown in the presence of osteoclasts and probiotics, increased cytotoxicity to CSCs [260]. Moreover, an immunocompetent cancer stem cell model that recapitulates tumor heterogeneity, invasiveness, vascularity, and immunosuppressive microenvironment in syngeneic immunocompetent mice was developed and used for tested a genetically engineered oncolytic herpes simplex virus that is armed with interleukin 12 (G47-mIL12). The results showed G47
Organotype tissue sectioning models involve culture of surgically removed tumor tissue, maintaining inter- and intra-tumor heterogeneity and tumor structure [262, 263, 264]. This technique does not involve selective growth of tumor cells may be used for personalized treatments and to evaluate individual sensitivity to invasive and patient-specific effects of anti-invasive drugs [263]. An in vitro brain slice model for targeting of brain metastases of breast cancer has also been constructed [265]. Such a model is expected to contribute to immunotherapy studies of solid tumors, including GBM.
Currently, one of the most cutting-edge areas of research is focused on organoid models. Organoid models have the ability to replicate the structure and function of original organs, and in the long-term, they hold the potential to replace patient-based studies [266, 267]. They have potential for basic cancer research, drug screening and personalized susceptibility studies and may bridge the gap between in vitro and in vivo cancer models [266, 268]. The GBM organoid model, generated by traditional 3D culture, genetic engineering and co-culture, shows promise, preserving the phenotype and 3D TME of the original tumor [269, 270, 271, 272, 273, 274, 275, 276, 277, 278]. These methods can also be used to produce other organoid models of brain tumor such as medulloblastoma and brain metastasis. It has been widely used in basic research and clinical transformation research, especially in immunotherapy research, which has considerable potential. Combining innovative technologies, such as 3D bioprinting and 4D real-time imaging, are likely to produce realistic modeling of brain tumor organoids although structural and genetic fidelity aspects remain unclear [279].
In summary, the path towards transforming incurable GBM into a curable condition has come closer, but there is still a considerable distance to cover. Nevertheless, there is hope as a recent seminar, co-organized by the National Brain Tumor Society and the Parker Institute of Cancer Immunotherapy, has brought together experts who have highlighted potential future directions for GBM therapy [280, 281, 282].
Conclusions
Glioma microenvironment, which is remodeled by GSCs, is different from other cancers. In addition to the unique characteristics mentioned above, the heterogeneity of GSCs and TME is the key to be clarified in the future. For example, Driving factors of GSC plasticity and heterogeneity (such as reprogramming transcription factors and epigenetic modifications) has been proved to be related to the induction of immunosuppressive cell states, which may lead to therapeutic opportunities for GSC-intrinsic mechanisms [283]. Another example is the interaction between tumor-associated microglia/macrophages and GSCs in TME [284]. We have only verified that SU3 (GSCs) can trigger the malignant transformation of macrophages into cancer cells [285]. However, if we can elucidate the molecular mechanisms underlying this transformation, we may be able to manipulate the related molecules and revert the transformed macrophages back to the M1 state, which could potentially inhibit GSCs.
Data availability
No underlying data was collected or produced in this study.
Author contributions
Conception: HQ, WAM, ZWY, WY.
Interpretation or analysis of data: FXF, WJ, THY, YK, ZYD, JDY, CHC, CH, XXT.
Preparation of the manuscript: FXF, WJ, THY, HQ.
Revision for important intellectual content: HQ, WAM, ZWY, WY.
Supervision: HQ, WAM, ZWY, JDY, CHC.
All authors agree to be accountable for the content of the work.
Funding
This work was funded by grants from the Natural Science Foundation of China (No. 81101909; 81172400; 81272793; 30200335;30672164; 30872654; 81302196; 81302180; 82073873; 82072798), the Health Talent Training Project of Gusu (GSWS2020122), the Youth medical talent Foundation of Jiangsu (QNRC2016217), Suzhou Medical, Health Technology Innovation Project (SKY2021028) and Science and technology planning project of Suzhou (SS2019050) and Nanjing health science and technology development project (YKk18106).
Footnotes
Acknowledgments
The authors would like to express their gratitude to EditSprings [
Conflict of interest
The authors declare that they have no competing interests.