
Abstract
BACKGROUND:
Uveal melanoma (UM) is a rare but deadly cancer. The main cause of death from UM is liver metastasis. Though the metastasis mechanism remains unclear, it is closely related to the immune microenvironment and gene expression.
OBJECTIVE:
This study aimed to identify the prognostic genes in primary and metastatic UM and their relationship with the immune microenvironment.
METHODS:
Primary and metastatic UM data from the GEO database included GSE22138 and GSE44295 datasets. Kaplan-Meier analysis, Cox regression models, and ROC analysis were applied to screen genes in GSE22138. TIMER2.0 was employed to analyze the immune microenvironment from gene expression. Prognostic immune gene correlation was tested by Spearman. The results were validated in the independent dataset of cohort GSE44295.
RESULTS:
Metastasis and primary differential gene analysis showed 107 significantly different genes associated with prognosis, and 11 of them were immune-related. ROC analysis demonstrated that our signature was predictive for UM prognosis (AUC
CONCLUSIONS:
The present study constructed an 11-gene signature and established a model for risk stratification and prediction of overall survival in metastatic UM. Since FABP5 and SHC4 are related to neutrophil infiltration in metastatic UM, FABP5 and neutrophil regulation might be crucial in metastatic UM.
Introduction
Uveal melanoma (UM) is the most common primary intraocular malignant tumor in adults and is characterized by insidious onset, rapid progression, high metastasis, and poor prognosis [1]. It represents approximately 85% of all ocular melanomas. Unfortunately, there remains a lack of effective indicators that can predict patients at high risk for metastasis and determine their prognosis, and the patient’s prognosis is generally poor once the primary tumor metastasizes [2, 3]. Currently, there is no effective treatment option available for metastatic UM patients, and approximately 50% of these patients will develop metastatic disease with only 6–12 months of survival from the diagnosis of metastasis [4].
Increasing evidence has demonstrated that immune cell infiltration (ICI) is crucial for predicting patient outcomes and therapeutic efficacy. Over the past several decades, targeted therapies and immunotherapy, especially immune checkpoint blockade (ICB), have become promising methods for the treatment of multiple types of cancer [5, 6]. Despite the remarkable progress of ICB therapy in metastatic melanoma and other tumors, UM remains immunotherapy-resistant [7]. Control rates for immune therapies in UM patients vary from 64% for combined immunotherapy (cytotoxic T lymphocyte antigen-4 [CTLA-4] plus anti-programmed cell death-1 [PD-1]) to 43% for tumor-infiltrating lymphocyte therapy. Moreover, one major limitation of immunotherapy is that a clinical response is only observed in 0–5% of patients [8]. Thus, it is crucial to identify novel therapeutic markers for immunosuppression in UM.
The tumor microenvironment (TME) is the internal environment which includes tumor cells, fibroblasts, immune cells, inflammatory cells, glial cells, and other nearby cells, as well as the surrounding areas [9]. The characteristics of hypoxia, low pH, and high pressure enables the tumor microenvironment to contain many growth factors, chemokines, and various proteolytic enzymes that can produce immuno-inflammatory reactions, which is very conducive to tumor proliferation, invasion, adhesion, and angiogenesis [10]. Tumors and the microenvironment are closely related, interdependent, mutually promoting and can jointly regulate the immune escape, growth, and metastasis of tumors. Previous research has found that the level of immune cell infiltration in the tumor microenvironment affects the prognosis of tumor treatment [11]. The activity of immune cells and stromal cells has been shown to predict the overall survival rate of patients with cancer [12]. However, as it is a tumor with low T cell immune infiltration, the current mainstream treatment using immune checkpoint inhibitors is not suitable for UM [3]. The development of a more prognosis-related immune microenvironment and corresponding gene signatures will be of great significance to the application of more therapeutic methods to improve the prognosis of UM patients.
Intrinsic tumor factors, such as genetic or epigenetic alterations, can affect the TME of UM. In previous studies, UM classification based on gene expression was found to be related to different immune components [13]. Therefore, identification of suitable prognostic markers of UM for early detection is a key challenge in the battle against the disease. It is also of great significance to accurately assess tumor prognosis, prolong patient survival time, and improve the quality of life. Thus, exploring biomarkers related to the immune cell infiltration of UM tumors as novel prognostic markers is necessary to provide critical guidance for immunotherapeutic selection. Consequently, there is an urgent need to increase our understanding of the relationship between the molecular and immune microenvironment of UM, which will aid in the development of novel prognostic biomarkers as well as individualized treatment regimens. Therefore, the aim of this study was to explore the prognostic-related differential expression of immune genes and their correlation and potential relationship with the immune microenvironment. Public datasets of UM samples were analyzed to study the differences in prognostic genes and immune microenvironments between primary and metastatic samples. We hypothesized that prognostic immune genes might lead to altered immune cell infiltration in the TME of UM, thereby affecting prognosis and metastatic potential.
Materials and methods
Data collection and pre-processing
We searched the Gene Expression Omnibus (GEO) database (
The workflow of this study.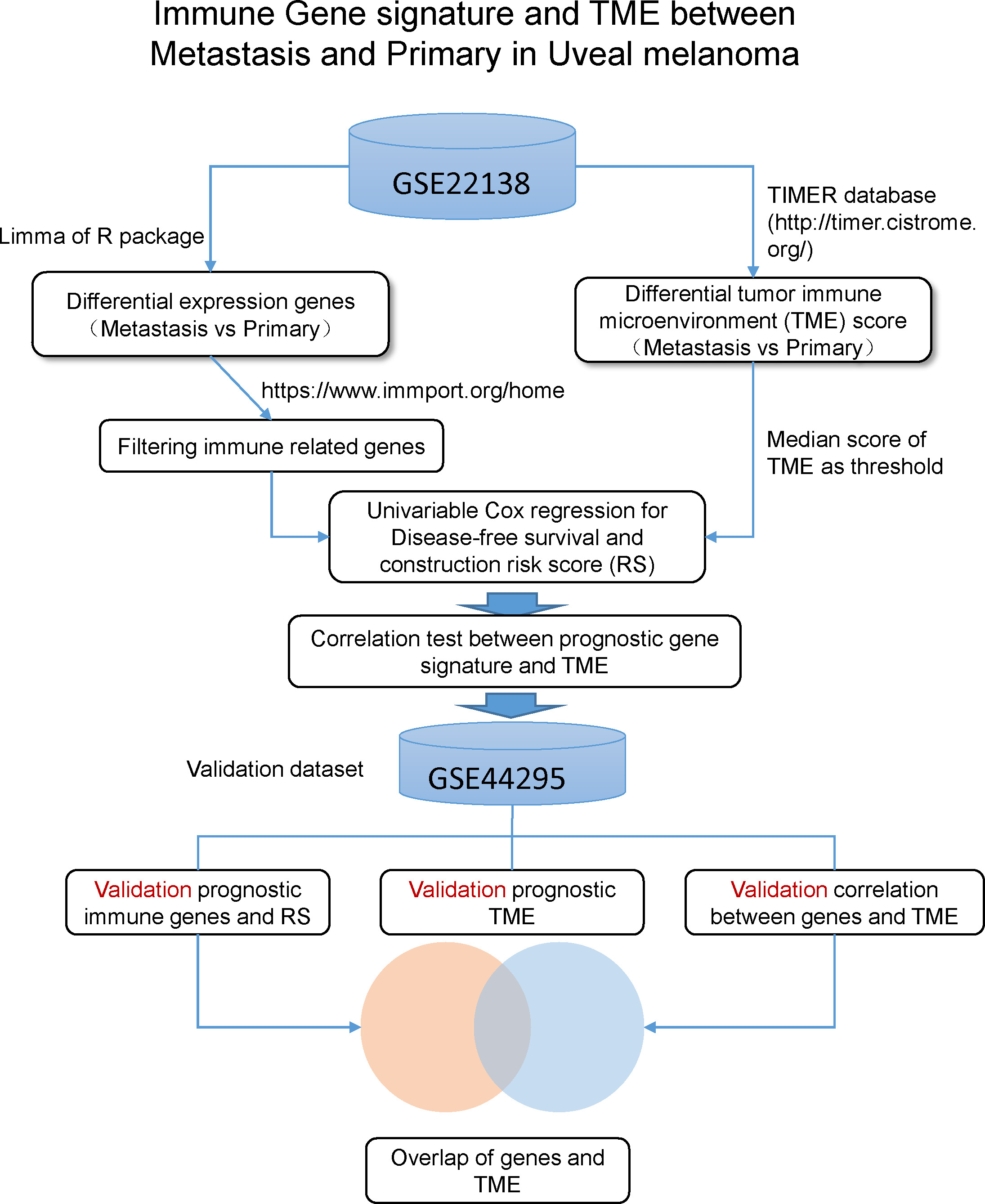
The GSE22138 dataset (platform GPL570, Affymetrix Human Genome U133 Plus 2.0 Array) containing 35 metastatic UM tissues and 28 primary UM samples was used for biomarker discovery for prognostic detection in metastatic tumors. The GSE44295 dataset (platform GPL6883 , Illumina HumanRef-8 Expression BeadChip, version 3.0) comprised 63 samples (24 primary samples without metastasis, 33 samples with metastasis, and 6 samples of unknown status) of classic UM. These two datasets included expression patterns between them. With log2 transform of GSE44295, the value range of GSE44295 data set is very close to that of the GSE22138 data set. Through the above conversions, the two groups of data became comparable. In this study, the data of 120 tissues were analyzed. The data analysis process is presented in Fig. 1.
R software (version 4.0.3) was used to extract gene expression data from the above datasets and received background adjustment and quantile normalization using affy [14] and simpleaffy [15] packages. Microarray information was transformed into gene expression information using the expression values of probes from the GEO dataset. Microarray annotation information was used to match the probes with the corresponding genes. Probes with more than one gene were eliminated, and the average value was calculated for genes corresponding to more than one probe. The samples with negative values were eliminated. The threshold value was determined by the number of genes with different expression thresholds.
Identification of differential expression genes (DEGs) was performed using the “Limma” R package [16]. Adjusted
Identification and validation of profiling differentially expressed IRGs (DEIRGs)
Immune-related gene (IRG) data were retrieved from the ImmPort database (
Kaplan-Meier survival analysis of DEIRGs
Univariate Cox regression analysis was performed to explore the impact of each DEIRG on replase-free survival (RFS). The DEIRGs of survival-related modules (
Kaplan-Meier curve analysis was conducted to evaluate the relationship between the risk score and RFS. The median value was used as the cut-off value. Factors with hazard ratio (HR)
Immune infiltration and correlation analysis
The Tumor Immune Estimation Resource 2.0 (TIMER 2.0;
Results
To explore the metastasis mechanism of UM, the primary and metastasis UM datasets, including GSE22138 and GSE44295, were retrieved from the GEO dataset. The clinical and pathological characteristics of each patient in the GSE22138 and GSE44295 datasets are summarized in supplementary Table S2. Specifically, the clinical characteristics of 120 patients, including age, sex, eye position, tumor location, retinal detachment, and survival time (days), were included in the datasets. As shown in Table S2, the median age was 62.12 in the training dataset. Of the 63 cases, 24 (38.10%) were female, and 39 (61.90%) were male. For 33 patients (52.38%), tumors were found in the left eye, and 30 (47.62%) occurred in the right eye. Patients with tumors on the equator (66.67%) and posterior to the equator (19.05%) made up the majority of the total, and the remaining patients had tumors that were all over the eye (1.59%), anterior to the equator (4.76%), and at unknown locations (7.94%). In the data, 36 (57.14%) patients had complicated retinal detachment, 22 (34.92%) had no retinal detachment, and the remaining five cases (7.94%) had an unknown retinal detachment status. For the survival time (days), the mean (SD) of survival days accounted for 1234 (30.57), while 1249.81 (696.51) were accounted for in the verification cohort.
Univariate and multivariate Cox regression (disease-free survival) analysis of clinical information of UVM
Univariate and multivariate Cox regression (disease-free survival) analysis of clinical information of UVM
Volcano plot and heatmaps of differential gene expression between metastatic and primary UM. (A) Volcano plot demonstrating 108 differentially expressed genes. (B) Heatmap demonstrating the 107 differentially expressed immune-related genes. (C) Heatmap demonstrating the 11 differentially expressed immune-related risk genes.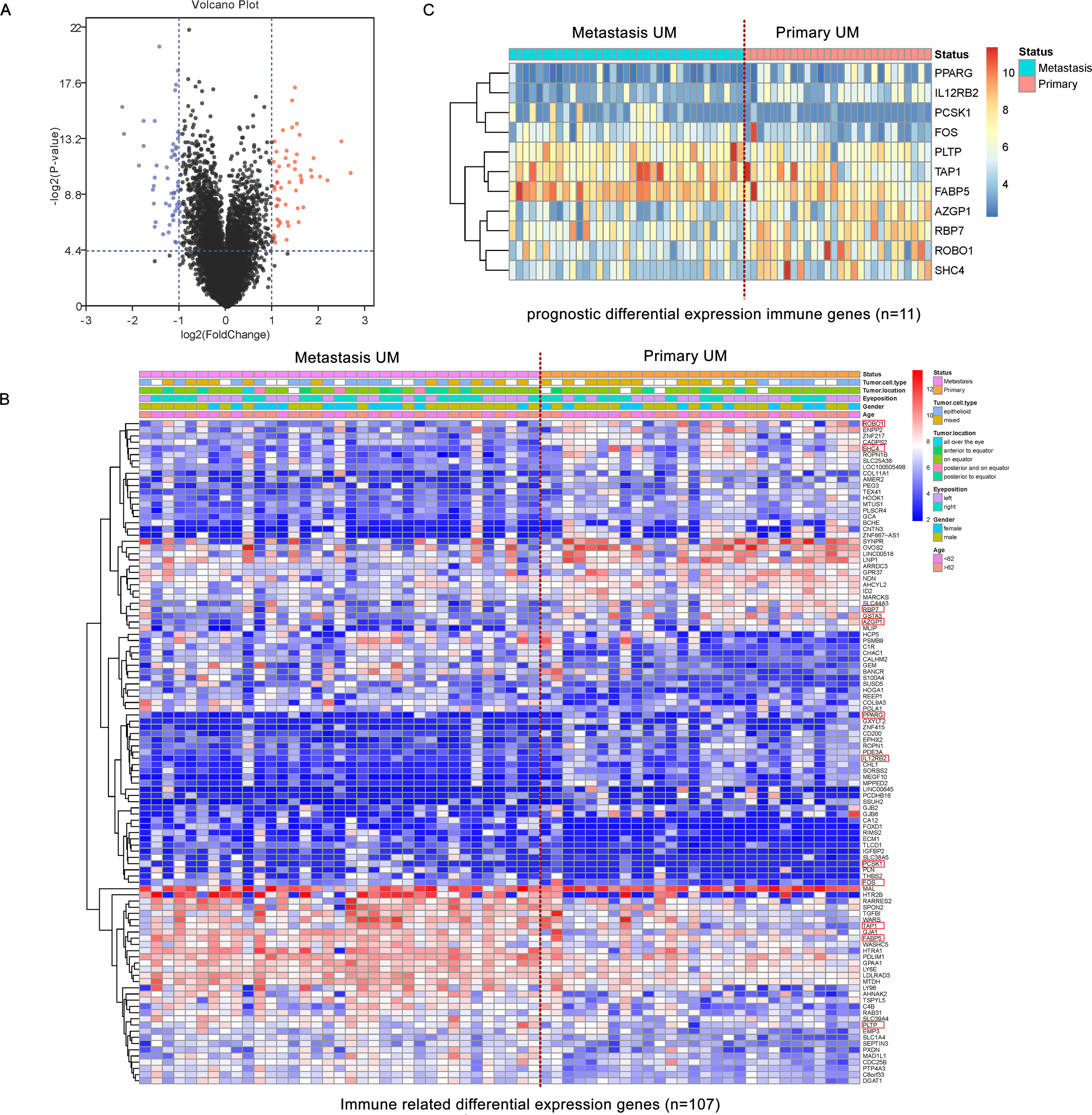
By using the GSE22138 dataset as a training cohort, 20530 mRNAs of 35 metastatic UM tumor samples and 28 primary tumor samples were included. According to the set thresholds (
Identification of prognostic immune-related DEGs
We performed univariate Cox regression analysis of the 107 DEIRGs based on the survival information (RFS) and screened out 11 RFS-related DEGs (
Forest plots of hazard ratios demonstrating the prognostic values of immune-related genes (IRGs). The dash line was used to mark the location of HR 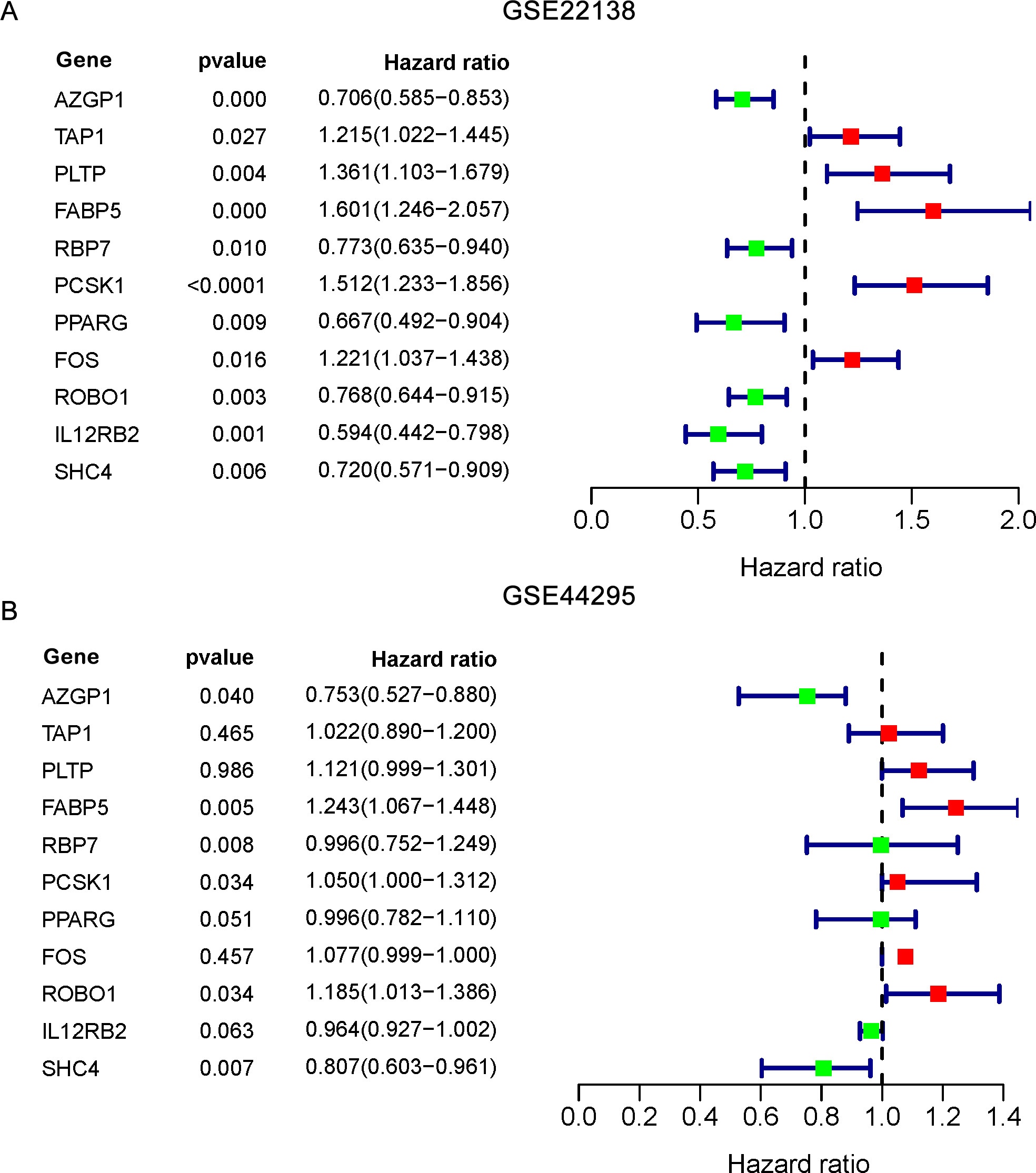
These RFS-related DEIRGs were used to further construct the prognostic signature by integration with common clinical factors, including age, sex, eye disease, tumor location, tumor diameter, tumor thickness, and retinal detachment. The factors were recorded in the GEO and analyzed by univariate and multivariate Cox regression analyses (Table 1).
The RFS risk score was calculated according to the median risk score for every UM metastasis patient [17]. Thus, if one’s risk score was higher than the median risk score, this patient would be assigned to the high-risk group. Kaplan-Meier curves (Fig. 4A) illustrate that the p-value of the log-rank test was
Tumor-infiltrating immune cells (TICs) analysis in UM
To explore whether the 11 DEIRGs are involved in the process of immune infiltration in UM, we first evaluated the abundance of six tumor-infiltrating immune subgroups (Table S4), in which more than half of the cells had an expression abundance value equal to 0. Three cell types (CD8
After the univariate Cox proportional hazard regression analysis, neutrophil and myeloid dendritic cells were found to be related to prognosis (Table S5). The correlation between gene signatures and immune cells was analyzed by Spearman analysis.
As illustrated in Fig. 5D and Table S6, S7 and S8, the gene signatures AZGP1, FABP5, RBP7, and IL12RB2 were related to the infiltration of neutrophils; FABP5, ROBO1, IL12RB2, and SHC4 were related to the infiltration of myeloid dendritic cells; and FABP5 and IL12RB2 were related to the infiltration of both cell types. This indicates that the correlation between neutrophils and myeloid dendritic cells and these genes may be one of the factors contributing to their prognostic ability. In the validation dataset, the correlation analysis of immune cells and genetic markers showed that AZGP1, RBP7, and IL12RB2 were related to the infiltration of neutrophils; FABP5, ROBO1, IL12RB2, and SHC4 were related to the infiltration of myeloid dendritic cells; and FABP5 and IL12RB2 were related to the infiltration of both cell types.
Validation of the prognostic immune microenvironment
To test the prognostic ability of neutrophils and myeloid dendritic cells, the K-M curve was applied to analyze the GSE22138 and GSE44295 datasets. The median value of the two TME scores was used as a threshold to divide the data into high-risk and low-risk groups. The results showed that the neutrophil score and myeloid dendritic cell score were significantly classified into two groups in GSE22138 (
Validation of the correlation between immune gene signature and TME
Multivariable Cox regression for prognostic factors
Multivariable Cox regression for prognostic factors
Prognostic effect and verification of the model. (A)–(C) Kaplan-Meier curves for risk score analysis in GSE22138 and GES44295 datasets. (B)–(D) K-M curves showing overall survival of high-risk and low-risk patients in GSE22138 and GSE44295 datasets.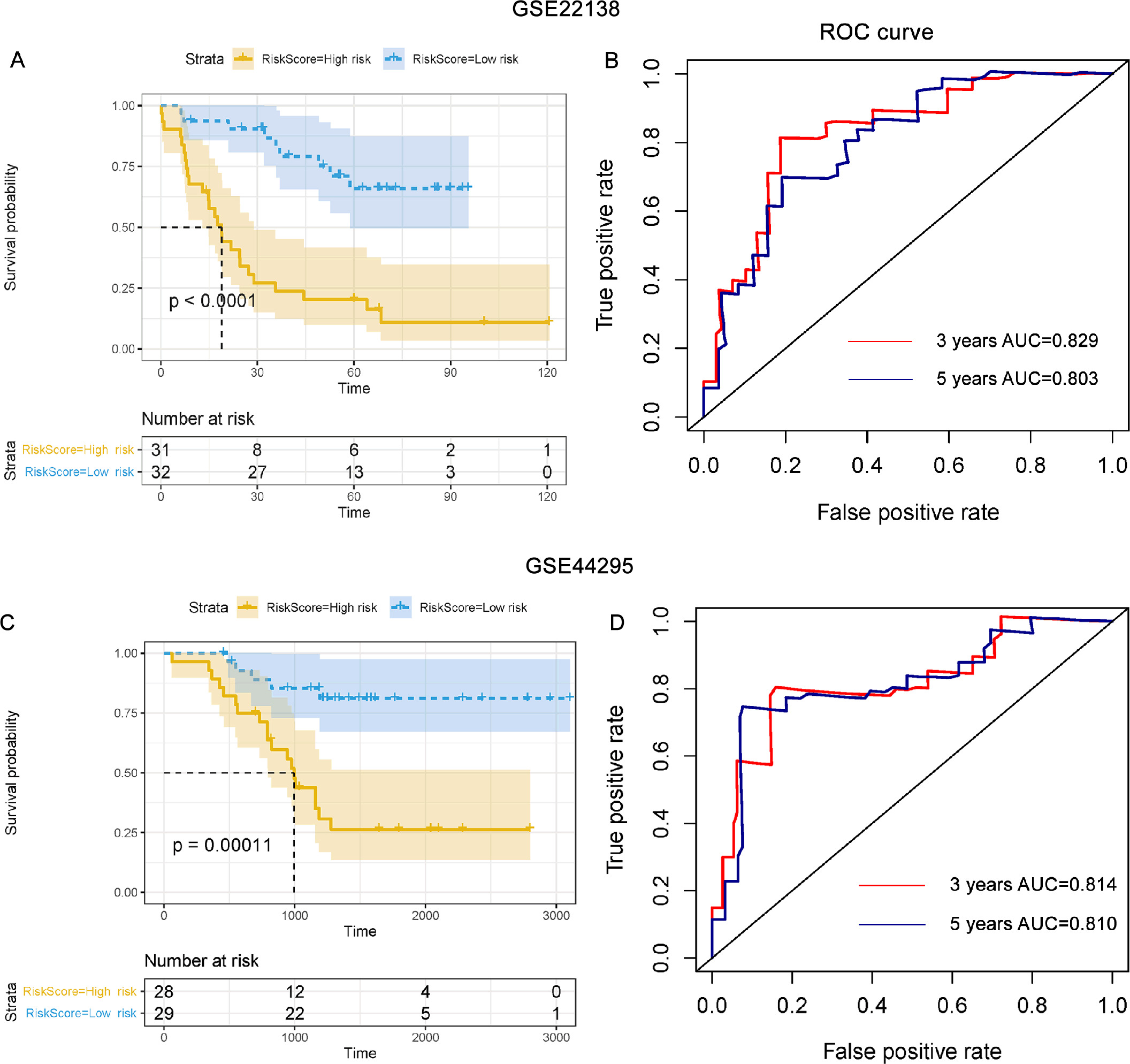
Correlation analysis between the 11-gene signature expression and the three types of infiltrating immune cells in UM. (A) The heatmap of the immune cells in UM samples. (B) The boxplot of the immune cells in primary and metastatic tumor samples. (C) The feature of clinical factors and CD8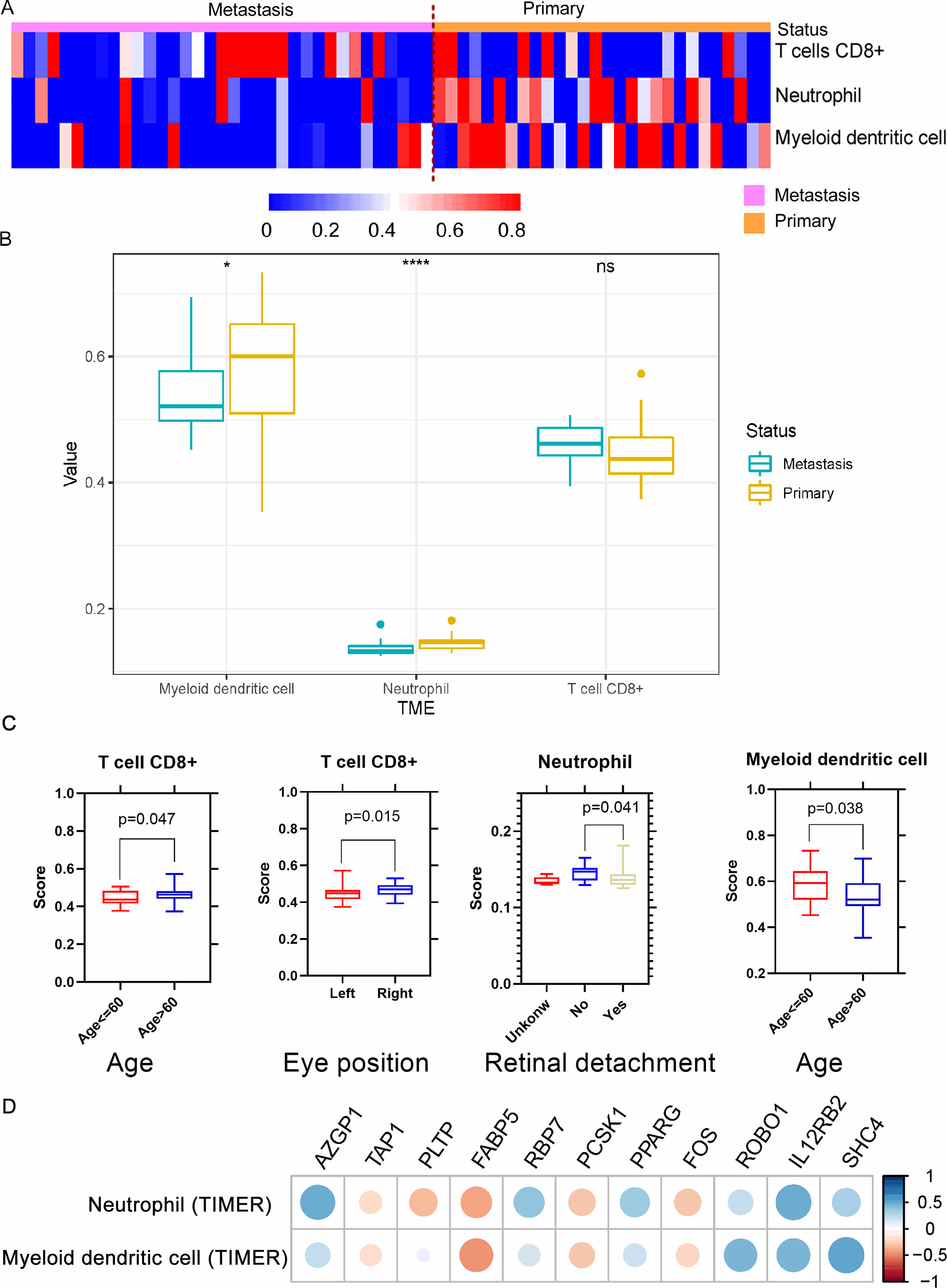
High abundance of neutrophils and myeloid dendritic cells are potential independent risk factors. (A) Kaplan-Meier curves for myeloid dendritic cell infiltration score analysis in the GSE22138 dataset. The survival rate of the high-risk group was significantly lower than that of the low-risk group (log-rank test 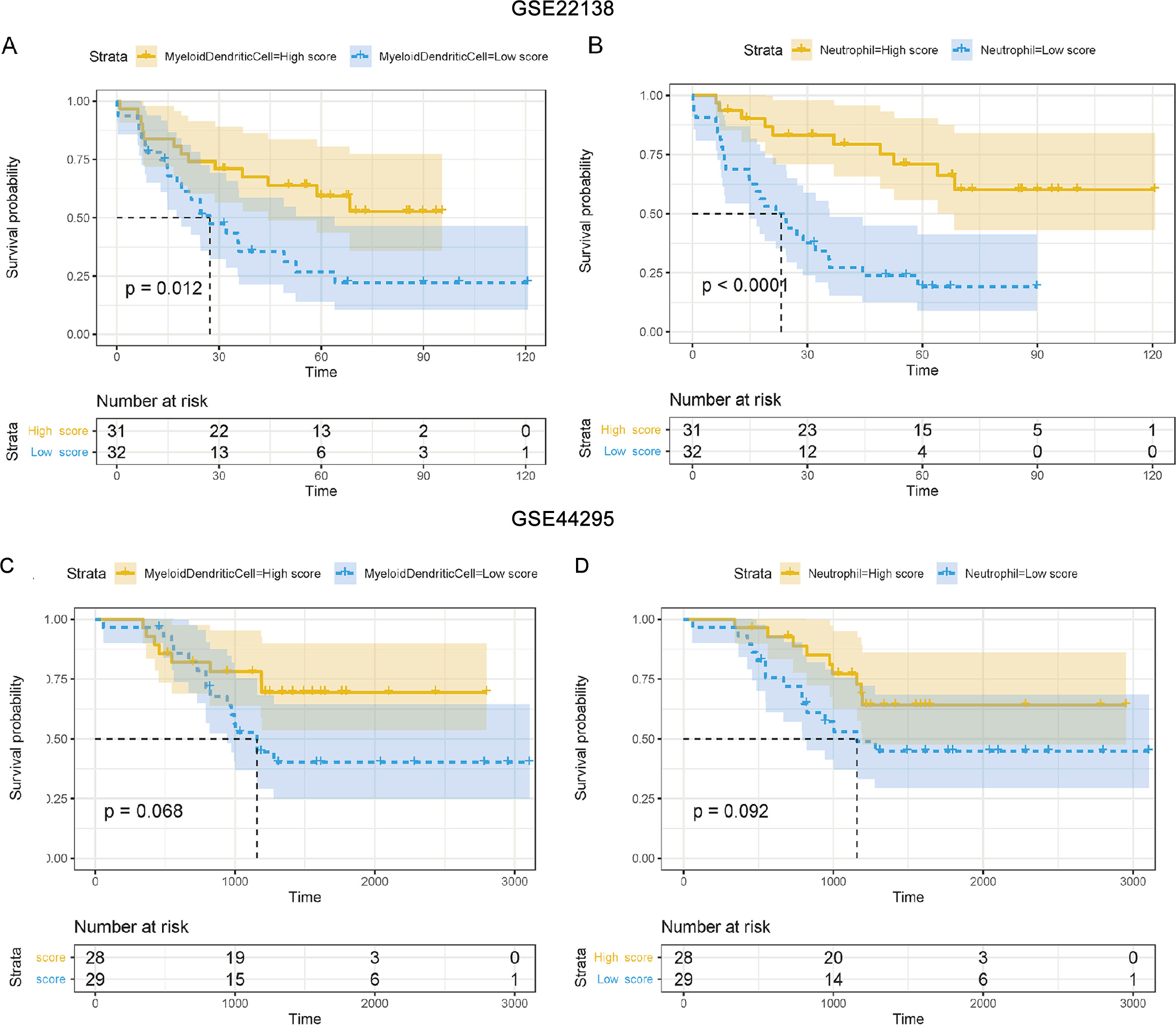
Correlation between gene signature and TME. (A)–(B) Correlation between gene expression and neutrophil infiltration in GSE22138. (C)–(D) Correlation between gene expression and neutrophil infiltration in GSE44295.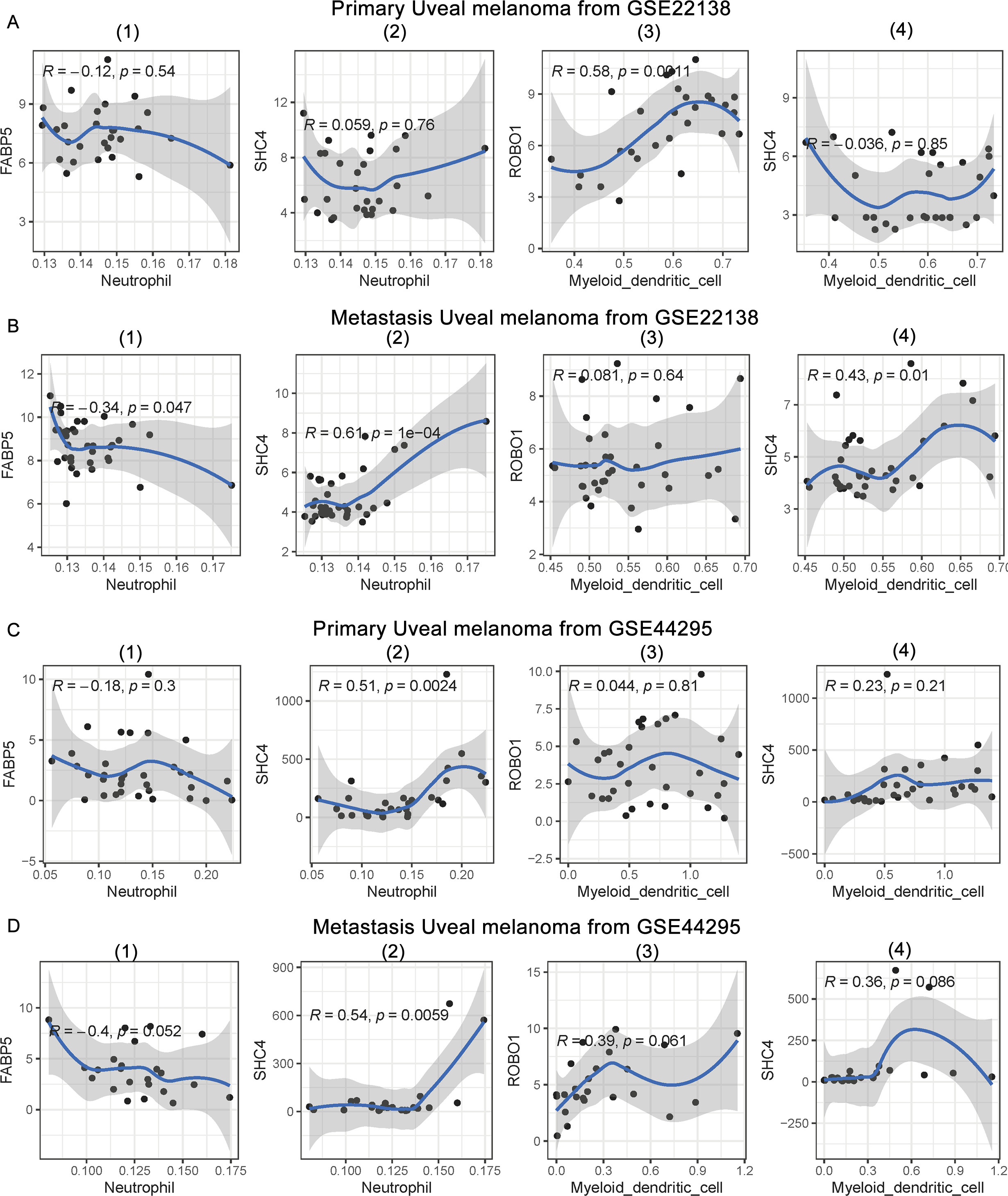
Although 11 genes were analyzed for correlation between the TMEs of the two data sets, the results could not directly reflect different correlations between metastasis and primary status. To further explore the mechanism of the immune microenvironment and metastasis, we calculated the correlation between mutually verifiable genes and the immune microenvironment in the two datasets. The results are shown in Fig. 7 and supplementary Table S9.
In the GSE22138 dataset, the results showed that FABP5 and SHC4 were not associated with neutrophils in primary UM tumors (Fig. 7A(1) and A(2)). However, FABP5 and SHC4 were significantly correlated with neutrophils in metastatic UM tumors (Fig. 7B(1) and B(2)). FABP5 was negatively correlated with neutrophils (
The results showed that FABP5 was not associated with neutrophils in primary UM tumors (Fig. 7C(1)), but was almost significantly associated with metastatic UM tumors (
In the aforementioned results, we considered the relationship between clinical factors, gene expression, immune microenvironment, and metastasis. We then integrated multiple factors related to metastasis for further analysis. Finally, multivariable Cox regression was applied to analyze the gene signature, TME, and clinical factors. The results are presented in Table 2.
The results showed that retinal detachment, FABP5, and neutrophil scores were independent prognostic factors in the GSE22138 cohort. In the GSE44295 cohort, however, some clinical factors were absent from the raw data. Thus, we tested the immune gene signature and two TMEs using multivariable Cox regression analysis. The results showed that FABP5 and neutrophil scores were independent prognostic factors. External validation is crucial for validating the applicability of a gene signature. A total of 57 UM patients with independent effectiveness in the GSE44295 dataset were included for validation.
In this study, it was determined that neutrophils and myeloid dendritic cells were significantly associated with prognosis and metastasis. Of the 11 prognostic immune genes, three genes (SHC4, FABP5, and ROBO1) were significantly associated with the above two TMEs. SHC4 and FABP5 genes were significantly correlated with neutrophils in the metastatic tumor, but not in the primary tumor. ROBO1 was closely related to myeloid dendritic cells in primary tumors, but not in metastatic tumors. The results were validated in an independent dataset of UM.
UM is one of the most common intraocular malignant tumors occurring in adults, including local intraocular tumors and metastatic tumors. It is estimated that most patients develop metastases in the liver [2]. Once UM metastasis occurs, it is much more difficult to find a suitable treatment to cure the patient. At present, the mechanism underlying the occurrence and metastasis of UM remains unclear. Consequently, exploring the factors related to UM metastasis would provide an important theoretical basis for research on its diagnosis and treatment.
In this study, we used GEO data to identify and analyze 11 UM prognostic genes related to the tumor microenvironment. By screening the differentially expressed genes between the primary and metastatic samples using Kaplan-Meier and univariate Cox analysis, and intersecting with the immune gene list provided by and downloaded from the official website of ImmPort, we obtained 11 UM prognostic genes related to immunity. Specifically, AZGP1, RBP7, PPARG, ROBO1, IL12RB2, and SHC4 indicated good prognosis, while TAP1, PLTP, FABP5, PCSK1, and FOS were associated with poor outcomes. In addition to PLTP, PCSK1, and FOS, the remaining genes showed evidence of correlation with the progression of various types of cancer. For example, reduced AZGP1 expression was found to be an independent predictor of early prostate-specific antigen (PSA) recurrence [18]. RBP7 is associated with tumor invasion in colon cancer [19]. PPARG reverses breast cancer-associated skeletal muscle mitochondrial dysfunction and lipid accumulation [20]. ROBO1 inhibits the proliferation of esophageal cancer cells [21]. Reduced IL12RB2 expression is associated with the development of lung adenocarcinoma [22]. SHC4 regulation is related with prostate cancer progression [23]. Down-regulation of TAP1 in squamous cell carcinoma of the oral tongue is an indicator of improved survival [24]. FABP5 promotes proliferation, invasion, and metastasis of cervical cancer cells [25]. The mechanism by which PLTP, PCSK1, and FOS influence cancer progression can be further studied and discussed. In addition, we explored the relationship between seven common clinical factors, including age, sex, eye disease, tumor location, tumor diameter, tumor thickness, and retinal detachment, with 11 gene signatures.
Previous studies have suggested that the progression and metastasis of UM seem to depend on a variety of signals mediated by the inflammatory microenvironment [26]. In addition to neoplastic UM cells, there are many other factors, including lymphocytes, neutrophils, and macrophages, that work together. Hence, the development of tumors is not a single and isolated event. To judge the treatment regimens and predict the prognosis of the tumor, we should not only focus on the tumor cells, but also pay attention to the different infiltrating cells around the tumor.
We conducted an immune infiltration analysis of 11 genes and 22 main TICs to clarify the relationship between these prognostic genes and the immune microenvironment. The results showed that the correlation between neutrophils and myeloid dendritic cells and the 11 genes may be one of the factors affecting prognosis. Both neutrophils and myeloid dendritic cells had good predictive values for the prognosis of UM patients in the training cohort. The low score of the two TICs showed a better prognosis. The results indicated that neutrophil and myeloid dendritic cell infiltration decreased in metastatic UM.
Neutrophils are important immune cells that constitute the first line of defense against inflammation and infection [27]. Neutrophils were closely associated with tumor metastasis in a previous study [28, 29]. The immune system not only supports the human body to resist foreign invasions, but also helps to fight against the body’s own renegade cancer cells. It is generally believed that neutrophils have dual functions of promoting tumor progression and inhibiting tumor growth. Specifically, neutrophils can promote tumor cell inflammation, proliferation, invasion, and angiogenesis, while inhibiting T cell activity to promote tumor immune evasion. In contrast, neutrophils can promote the anti-tumor effect of T cells and produce corresponding cytokines to participate in tumor suppression [27, 30, 31, 32]. In this study, neutrophils were found to be a protective factor, and neutrophil infiltration was associated with a good prognosis, which is in line with those of previous studies and may be related to its anti-tumor mechanism. Moreover, the potential use of neutrophils for targeted immunotherapy in a variety of tumor models has been confirmed [33, 34]. However, the regulation of neutrophils remains unclear. In this study, FABP5 was found to be a negative factor for regulating neutrophils (Fig. 7A(1–2) and 7B(1–2)), which is a relationship that has rarely been reported. As far as we know, Gally et al. found that FABP5
Myeloid dendritic cells are considered as a critical factor in antitumor immunity [37]. Also known as classical dendritic cells, or DCs, they are a heterogeneous group of antigen-presenting cells (APCs) with specialized functions. These functions serve as a central link between initial immunity and secondary immunity and can activate the initial T cells and B cells to exert anti-viral and anti-bacterial immune effects [38]. Studies have found that DCs can recognize and infiltrate tumors, secrete soluble factors that regulate the tumor microenvironment, and present tumor-associated antigens to induce T cell responses that promote cancer immune function [39, 40]. DCs play a key role in the induction and maintenance of breast cancer anti-tumor responses, owing to their unique abilities [41]. This is in accordance with our observations, which showed that DC presence is a protective factor, and DC infiltration is associated with a good prognosis, which provides ideas for further research. Previous study indicated that the loss of ROBO1 expression predicts a high risk of recurrence, metastasis, and a poor outcome in prostate cancer [42]. The relationship between ROBO1 and myeloid dendritic cells has not yet been reported. SHC4 is positively associated with myeloid dendritic cells, indicating that low expression of SHC4 and a decrease in DC infiltration are closely related to tumor metastasis.
The current study found that FABP5 and SHC4 are associated with the infiltration of neutrophils in metastatic UM tumors. ROBO1 is related to myeloid dendritic cell infiltration in primary UM tumors, but not in metastatic UM tumors. In contrast, SHC4 is related to myeloid dendritic cell infiltration in metastatic UM tumors, but not in the primary UM tumor. One interesting finding is that FABP5 was found to be a high-risk factor. A possible explanation for this might be that there are other ways which are more dominant for FABP5 to regulate UM progression.
This study had several limitations. First, the sample distribution in the GEO database may differ from the actual clinical population. In addition, the relationship and mechanism of the action between the 11 genes and the prognosis and immune cell infiltration of UM require further elucidation. Although FABP5, SHC4, and ROBO1 were validated in an independent dataset, further insight is needed for the 11 DEIRGs obtained from the datasets in this study.
Conclusion
The gene signatures (AP1, PLTP, FABP5, PCSK1, FOS, AZGP1, RBP7, PPARG, ROBO1, IL12RB2, and SHC4) could accurately identify patient prognosis and interact closely with the tumor immune microenvironment. FABP5, SHC4, and ROBO1 showed different correlations in primary and metastatic UM tumors, which may help to better understand the mechanism of UM metastasis and provide metastatic UM patients with personalized prognosis prediction and treatment options.
Author contributions
Conception: K Shi, JT Tang.
Interpretation or analysis of data: LY Yuan, SW Zhou, JT Tang, W Ran, and ZM Wang.
Methodology: K Shi, JT Tang, LY Yuan, W Ran, and SW Zhou.
Preparation of the manuscript: K Shi, JT Tang, SW Zhou, and ZM Wang.
Revision for important intellectual content: K Shi, JT Tang, W Ran, and ZM Wang.
Supervision: K Shi.
All authors read and approved the final version of the manuscript.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210427.
sj-xlsx-1-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-2-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-2-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-3-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-3-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-4-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-4-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-5-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-5-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-6-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-6-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-7-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-7-cbm-10.3233_CBM-210427.xlsx
sj-xlsx-8-cbm-10.3233_CBM-210427.xlsx - Supplemental material
Supplemental material, sj-xlsx-8-cbm-10.3233_CBM-210427.xlsx
Footnotes
Acknowledgments
This work was supported by the Chongqing Natural Science Foundation (cstc2019jcyj-msxmX0423). This work was also supported by the Multidisciplinary Joint Foundation of Gansu Provincial People Hospital (20GSSY2-3). Immune genes were downloaded from
We thank the Advanced Energy Science and Technology Guangdong Laboratory supercomputing platform. We also thank the supercomputing administrators, Jizheng Duan and Qiong Yang. Moreover, we thank the Gene Expression Omnibus for making massive genomic data of UM patients available. The authors declare no conflict of interest.