
Abstract
BACKGROUND:
Penile cancer (PeCa) is a rare disease, but its incidence has increased worldwide, mostly in HPV
OBJECTIVE:
To predict the main signaling pathways involved in penile tumorigenesis and its potential drug targets.
METHODS:
Genome-wide copy number profiling was performed in 28 PeCa. Integration analysis of CNAs and miRNAs and mRNA targets was performed by DIANA-TarBase v.8. The potential impact of the miRNAs/target genes on biological pathways was assessed by DIANA-miRPath v.3.0. For each miRNA, KEGG pathways were generated based on the tarbase and microT-CDS algorithms. Pharmaco-miR was used to identify associations between miRNAs and their target genes to predict druggable targets.
RESULTS:
269 miRNAs and 2,395 genes were mapped in cytobands with CNAs. The comparison of the miRNAs mapped at these cytobands and the miRNAs that were predicted to regulate the genes also mapped in these regions, resulted in a set of common 35 miRNAs and 292 genes. Enrichment pathway revealed their involvement in five top signaling pathways. EGFR and COX2 were identified as potential druggable targets.
CONCLUSION:
Our data indicate the potential use of EGFR and COX2 inhibitors as a target treatment for PeCa patients.
Introduction
Penile cancer (PeCa) is a rare malignancy; however, it is a public health concern in developing countries. In Brazil, it represents 2% of all male cancers, occurring more frequently in the North and Northeast regions. Maranhão state (Northeast region), where the patients of this study were obtained, presents with the highest PeCa rates in Latin America and worldwide [1]. The patients in this region are usually diagnosed with advanced tumour stages, and lymph node involvement, which invariably confers poor prognosis. Human papillomavirus (HPV) has been linked to nearly 50% of PeCa cases worldwide [2, 3]. In our region however, our previous study, revealed a higher frequency of HPV-related PeCa, in nearly 97% of the cases [4].
The available surgical treatment for advanced PeCa involves partial or total amputations of the penis and lymphadenectomy, which are associated with post-surgical complications and high morbidities [5, 6]. The combined use of chemotherapy was observed to marginally increase the survival rate of the patients [7, 8, 9, 10], with however the inherent toxicity of the chemo-agents. In addition, the rarity of this tumour makes it difficult to conduct large-scale clinical trials, so there are still no effective treatment strategies for advanced PeCa patients. Therefore, effective and less toxic treatments, such as the ones based on targeted therapy, are critically needed to treat PeCa.
MicroRNAs (miRNAs), non-coding RNA molecules, have emerged as one of the promising molecular markers for therapeutic use in several types of cancers [11, 12, 13]. These molecules are preferentially mapped in genomic regions with copy number gains and/or losses, as well as in regions of fragile sites [14, 15]. Further studies have confirmed that miRNAs play important roles in oncogenesis by regulating the expression of genes located within the regions affected by CNA, and/or miRNAs expression changes can be driven by CNAs within their own genomic regions [16, 17, 18].
A large number of integration strategies for distinct “omics” platforms have been described and have significantly contributed to the characterization of tumour molecular landscapes, such as the ones performed by the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA). Most of these strategies are based on computational pipelines, where the data is gathered from independent and public avaliable resources. In PeCa there is relatively limited information of the somatic alterations that occur at the distinct molecular levels and consequently of the integrated molecular signatures.
Therefore, and considering the prevalence of PeCa in developing countries, as well as the increasing number of cases worldwide, our main objective was to determine the predictive impact of copy number alterations in the miRNA/mRNA interactions, and to identify the potentially affected functional pathways in penile tumorigenesis. In addition, we aimed to determine whether the observed alterations and/or the signaling pathways involved, could be potentially targeted by the current clinically approved drugs.
Materials and methods
Experimental subjects
Patient accrual, social and clinical-histopathological variables
Tumour tissues of 28 PeCa patients, collected at the Hospital Aldenora Bello (São Luís, Maranhão, Brazil) were included in this study. The fresh tumour tissue samples were obtained at the diagnosis and before any cancer treatment. The clinical and histopathological variables were obtained from patients’ medical records. All patients signed consent form, which was approved by the Ethics Committee on Human Beings at the Federal University of Maranhão and by the National Research Ethics Commission (CONEP), Brazil (CAAE protocol number: 46371515.5.0000.5087).
The mean age at diagnosis of the patients was 64.6
The tumours were classified as Squamous Cell Carcinoma (SCC), presenting an average size of 3.6
Clinical-histopathological characterization of the PeCa patients evaluated in this study (
28)
Clinical-histopathological characterization of the PeCa patients evaluated in this study (
HPV genotyping was performed for all the patients by nested-PCR (according to BioGenetics Molecular Technologies (Uberlândia, Minas Gerais, Brazil) (patent number BR102017004615.0), and DNA sequencing, according to our previous protocol [4]. All the patients were HPV positive, of which 95.5% presented high-risk subtypes. HPV16 was the most common subtype, present in 63.6% of cases, followed by subtypes 35, 59, 74 (13.6%, each); 30, 44, 66 (9%, each); 06, 18, 53, 58 and 73 (4.5%, each). Multiple infections were detected in 50% of the cases (Table 1).
Copy number data of the PeCa tumours
All the patients were profiled for genome-wide copy number alterations (CNAs) by array-Comparative Genomic Hybridization (aCGH) analysis, as reported by Macedo et al. [4]. The aCGH analysis showed a total number of 2,314 CNAs, with a mean of 82.6
Integration analysis of CNA and miRNAs and mRNA targets
This analysis was performed by two different approaches: i. By the mapping of the genes and miRNAs at the main regions with CNA, called here CNA-genes and CNA-miRNAs, respectively, and ii. By the identification of common gene targets of the selected miRNAs above, that may be affected by both CNAs and miRNA expression alterations. The workflow of the study design and corresponding results are presented in the results section (Fig. 1).
Study design and corresponding results of the aCGH and miRNA data integration approaches, followed by the functional enrichment pathway and druggable targets predicted analysis. In one approach (269 CNA-miRNAs), 1,355 interactions were identified involving 65 CNA-miRNAs and 898 target genes (TarBase v.8.0). MiRPath v.3.0 was used for the enrichment of pathways considering two prediction algorithms, tarbase and microT-CDS. Five common pathways (14 CNA-miRNAs and 25 target genes) were identified comparing the two pathway methods. In addition, tarbase enrichment revealed an HPV-related carcinogenesis pathway (7 CNA-miRNAs and 13 target genes). In a second approach (2,395 CNA-genes), 1,358 interactions were identified involving 348 miRNAs and 292 CNA-genes. Comparing the interactions with the first approach, 115 common interactions were revealed, that is, between 35 CNA-miRNAs and 78 CNA-genes. Pathways enrichment was conducted with these interactions where it revealed 13 top pathways (10 CNA-miRNA and 13 CNA-genes). The Pharmarco-miR program was used to identify drugs targeting miRNA targets in both approaches.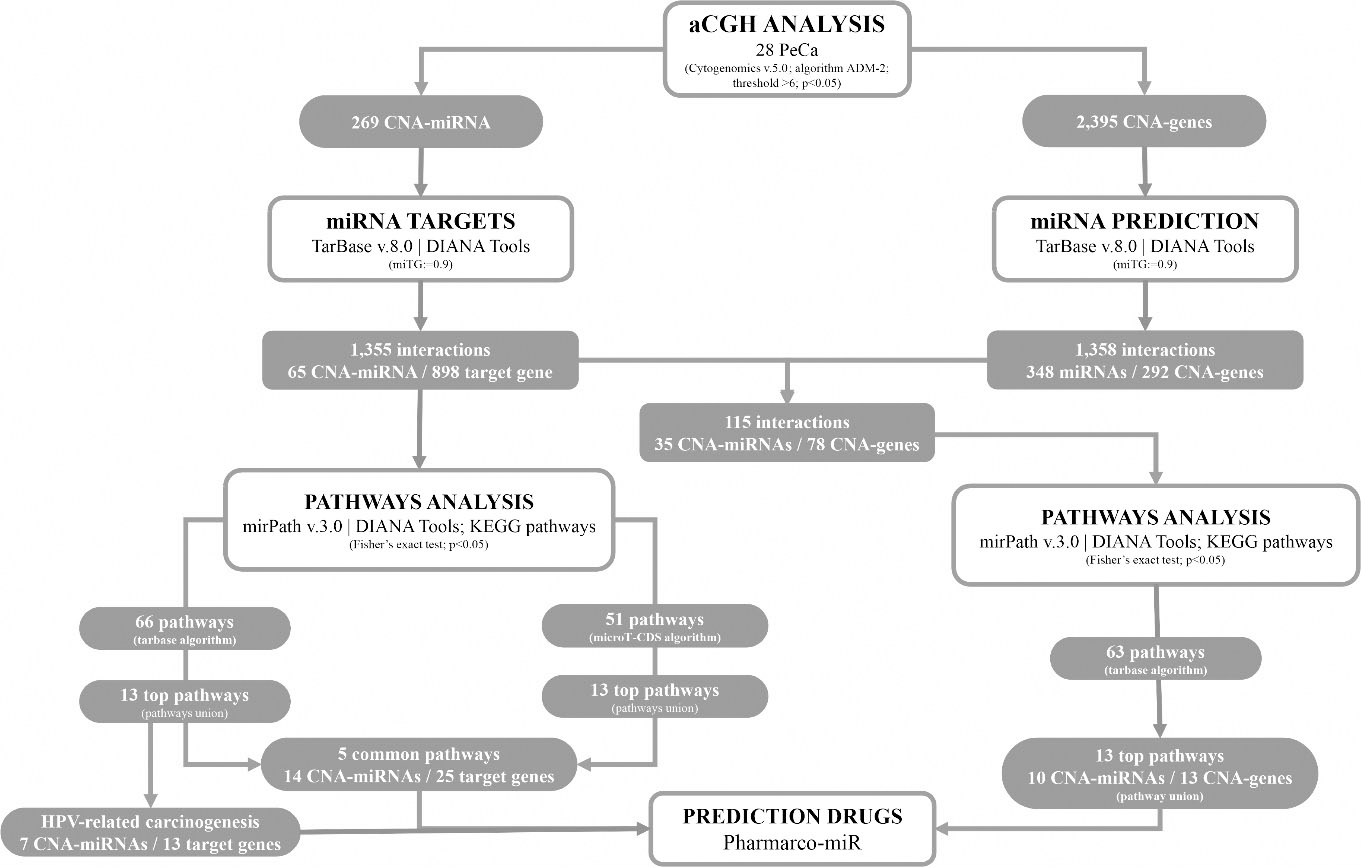
Initially, the complete aCGH data for all the 28 patients were analyzed to identify the most affected cytobands with CNAs. Only the CNAs present in at least 25% of the cases were considered. Next, the annotation of genes and miRNAs located in the cytobands with CNAs were obtained from the Agilent Cytogenomics v.7.0 interval base reports (Agilent Technologies, Inc.). The location of each miRNA was further confirmed using miRBase v.22 (
Association of genomic regions with CNAs and miRNAs with clinical and histopathological parameters from the patients
The association analysis of each clinical-histopathological variable and copy number alterations was performed using bivariate tests and
Identification of mRNA targets
The targets of the miRNAs mapped in the regions with CNA (CNA-miRNAs) were identified by the DIANA-TarBase v.8 (
Enrichment pathways analysis
To assess the potential impact of the miRNAs/target genes (that were mapped in the main cytobands selected above) on biological pathways associated with cancer, we used DIANA-miRPath v.3.0 tools (
Cytobands with CNAs in PeCa (
28) analyzed by aCGH in association with clinical-histopathological parameters
Cytobands with CNAs in PeCa (
Pharmaco-miR (
Results
The overall results of this study with the corresponding study design are presented in Fig. 1.
Copy number alterations and their association with clinical and histopathological parameters
Genome-wide copy number analysis in the 28 PeCa patients analyzed by aCGH revealed 38 altered cytobands with copy number alterations (CNAs), from which 89.5% were HPV integration sites in the host genome. The association analysis of the main affected cytobands with CNAs and the clinical-histopathological parameters from the patients revealed 15 cytobands (Table 2). All these altered cytobands were already described as HPV integration sites according to HPVbase. Gains on 14q12 presented association with HPV16 and multiple infections, while gains on 15q26.2-q26.3 were associated with age
Mapping of genes and miRNAs in genomic regions with CNA
According to the annotations of the CytoGenomics base interval report, 30 of the 38 affected cytobands harbored miRNAs (Fig. 1, Supplementary Table 1). Interestingly, 28 of these 30 cytobands (93.3%) are HPV integrated sites according to HPVbase. A total of 269 miRNAs were mapped in these cytobands (CNA-miRNAs), 211 in regions affected by copy number gains (78.4%), 27 by losses (10%), 13 by amplification (4.8%), and 18 by losses and deletions (6.7%).
A second integration approach was based on the identification of miRNAs that potentially regulated the genes mapped at the main affected cytobands: 2,395 genes were identified to be mapped at the 38 cytobands with CNAs (CNA-genes). Based on the TarBase/Diana Tools, considering miTG
Prediction of mRNA targets in regions with CNA
Target prediction analysis (considering miTG
In addition, the identification of the genes mapped at the affected cytobands and that were predicted to be regulated by the 35 miRNAs mapped at the same cytobands, resulted in 78 mRNA targets (Fig. 1). From these miRNAs, miR-30b-3p, miR-30d-5p and miR-548d-3p were the ones to present the highest number of target genes, 19, 19 and 10, respectively.
Pathway enrichment analysis
The main biological functions related to the 65 miRNAs mapped at the 30 cytobands (CNA-miRNAs) were evaluated by KEGG pathway enrichment analysis using two distinct algorithms. The tarbase algorithm revealed 66 pathways, among the top 13: adherens junction (hsa04520), cell cycle (hsa04110), ECM-receptor interaction (hsa04512), fatty acid biosynthesis (hsa00061), fatty acid metabolism (hsa01212), glioma (hsa05214), hippo signaling pathway (hsa04390), lysine degradation (hsa00310), mucin type O-Glycan biosynthesis (hsa00512), prion diseases (hsa05020), proteoglycans in cancer (hsa05205), steroid biosynthesis (hsa00100), and viral carcinogenesis (hsa05203). The microT-CDS prediction algorithm revealed 51 main KEGG pathways, among the top 13: axon guidance (hsa04360), foxO signaling pathway (hsa04068), GABAergic synapse (hsa04727), hippo signaling pathway (hsa04390), long-term depression (hsa04730), lysine degradation (hsa00310), morphine addiction (hsa05032), mucin type O-Glycan biosynthesis (hsa00512), prion diseases (hsa05020), proteoglycans in cancer (hsa05205), signaling pathways regulating pluripotency of stem cells (hsa04550), steroid hormone biosynthesis (hsa00140), and TGF-beta signaling pathway (hsa04350). The significant pathways revealed by both of the miRNA prediction algorithms used are shown in Fig. 2 and Supplementary Table 3A and B.
Targeted KEGG pathway clusters/heatmap regulated by the miRNAs mapped at the cytobands mostly affected by CNAs in the cases studied. A: Top pathways identified by the tarbase algorithm; B: Top pathways identified by the microT-CDS algorithm (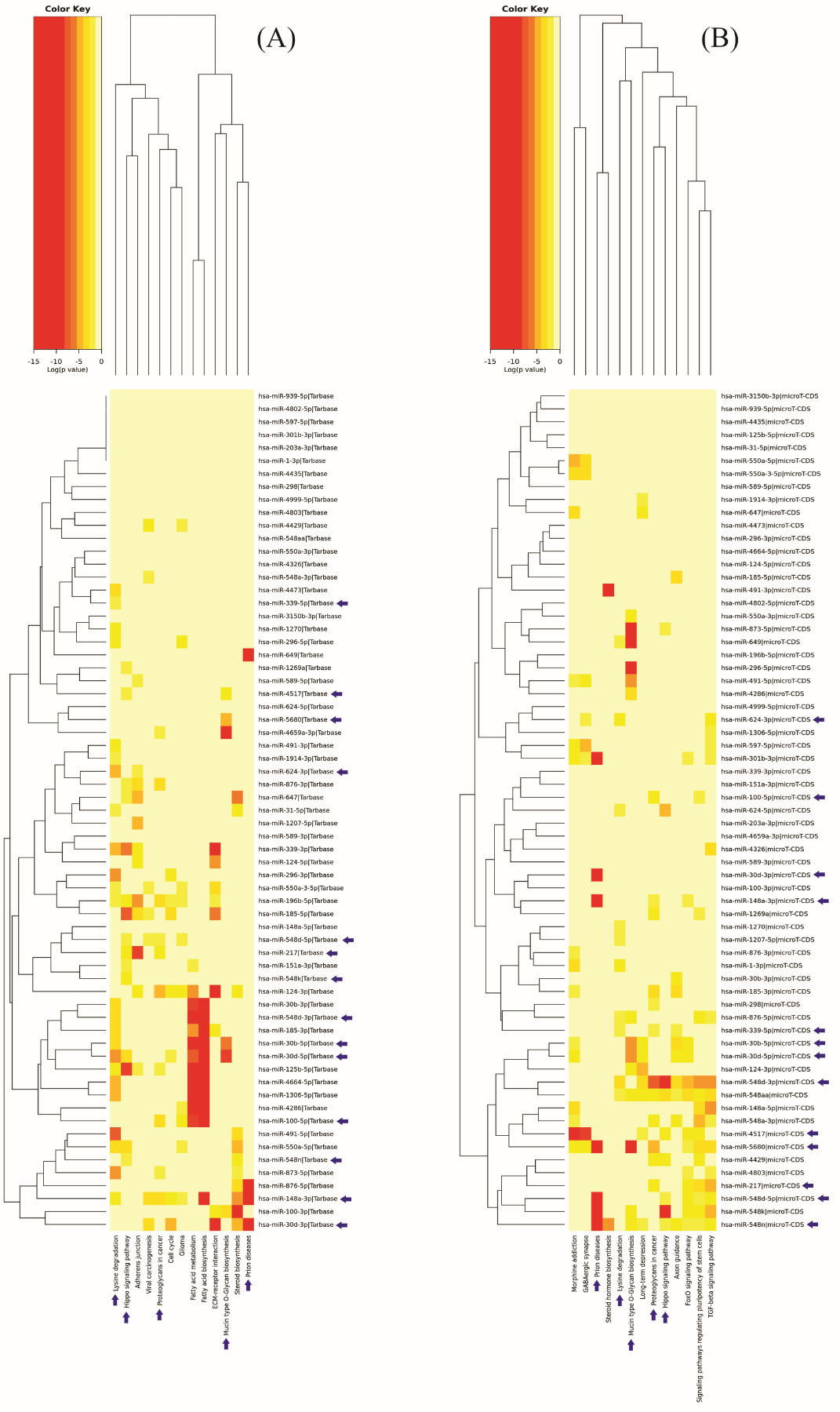
Target genes identified as potentially regulated by miRNAs mapped in regions with gains or amplifications and also described as negatively controlled by HPV oncoproteins (DIANA-miRPath v.3.0; KEGG: hsa05203)
DIANA-miRPath v.3.0; KEGG: HPV-mediated viral carcinogenesis pathway – hsa05203.
Notably, five common pathways were identified by both algorithms: hippo signaling pathway (miR-548d-5p, miR-548k, miR-548n and miR-4517), lysine degradation (miR-339-5p, miR-548d-3p and miR-624-3p), mucin type O-Glycan biosynthesis (miR-30b-5p, miR-30d-5p and miR-5680), prion diseases (miR-30d-3p and miR-148a-3p) and proteoglycans in cancer (miR-100-5p, miR-148a-3p and miR-217). Furthermore, considering that all the tumours were HPV positive, we also investigated the HPV-associated carcinogenesis pathway, in which 13 genes were identified as potentially regulated by miRNAs mapped in regions with gains or amplifications (tarbase algorithm). Interestingly, these genes have also been described as negatively controlled by HPV oncoproteins (KEGG- hsa05203) (Supplementary Fig. 1, Table 3).
Pathway enrichment analysis using the two algorithms (tarbase and microT-CDS) was also conducted considering the common genes (
Top 13 signaling pathways with the involvement of 13 genes observed with gains of copy number and also targeted by miRNAs mapped at the main affected cytobands (Tarbase algorithm)
Integrative network analysis showing the main pathways and the druggable targets predicted by Pharmaco-miR analysis. The network was generated by PathVisio v3.3 and Biorender softwares using the data generated through the Diana-Tools, KEGG and Pharmaco-miR.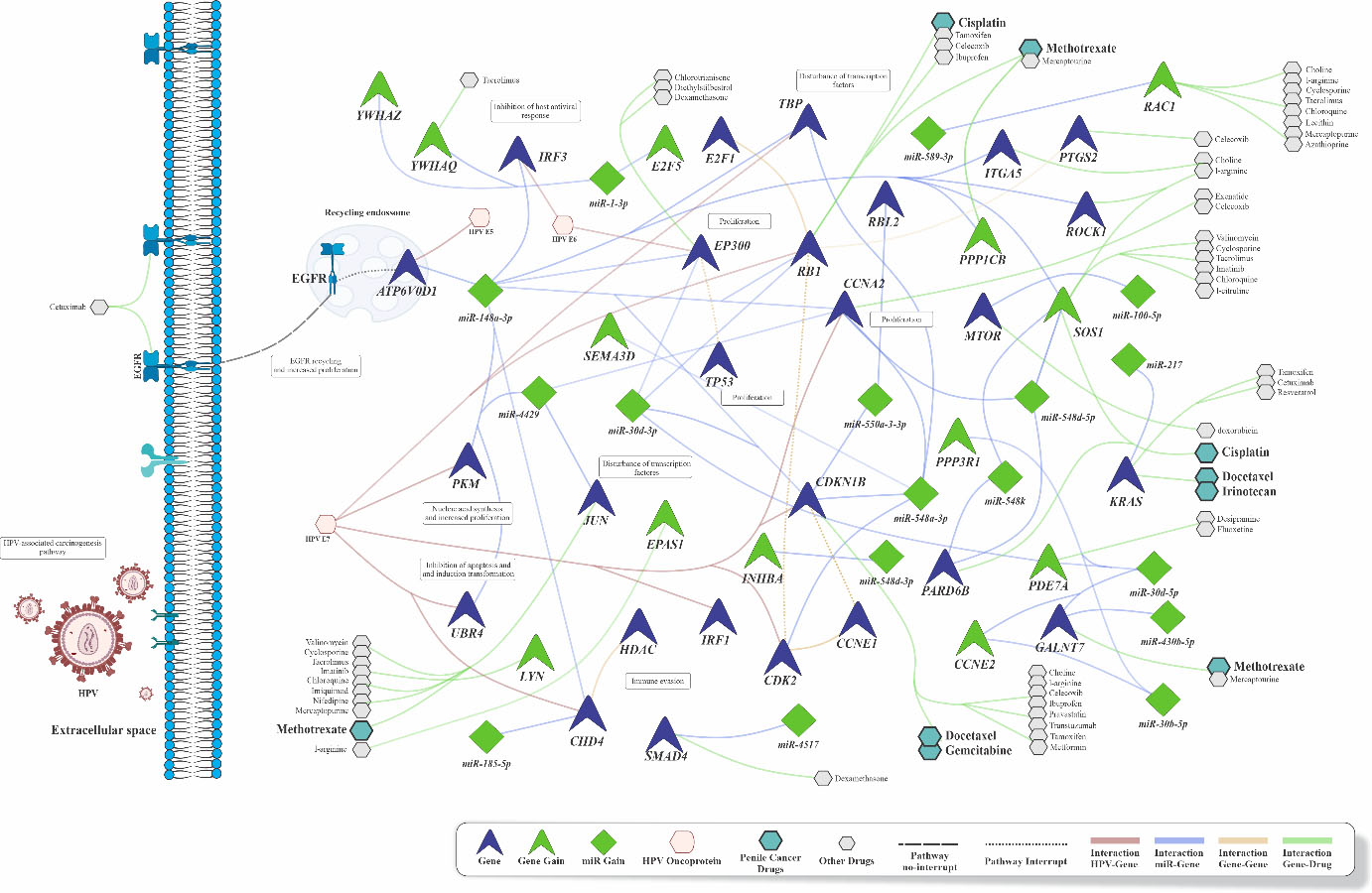
Finally, drug prediction analysis was performed using the five common pathways identified from CNA-miRNA (hippo signaling, lysine degradation, O-glycan mucin biosynthesis, prion, and proteoglycan diseases in cancer), as well as using the 13 main common pathways identified from the CNA-genes, and CNA-miRNAs analysis. Moreover, considering the high prevalence of HPV infection in the PeCa patients of this study, the HPV-mediated carcinogenesis pathway was also used for drug prediction. This analysis revealed drugs currently used to treat PeCa, such as Cisplatin, which acts on the PARD6B (regulated by miR-548k/548d-5p), and RB1 (regulated by miR-30d-3p and miR-148a-3p) genes. In addition, the MTOR (regulated by miR-100-5p) and SOS1 (regulated by miR-148a-3 and miR548k/548d-5p) pathways were found to be susceptible candidates to pharmacological inhibition by Doxorubicin and Imatinib, respectively. Interestingly, Cetuximab (an EGFR inhibitor) and Celecoxib (selective cyclooxygenase-2 inhibitor (COX2) were detected as alternative therapies for PeCa (Fig. 3 and Supplementary Table 4).
Discussion
PeCa is a rare disease in developed countries and, despite the recent increase in the number of molecular studies performed in these tumours [25, 26, 27, 28, 29], their molecular basis is still not well understood. In this study we evaluated the role of miRNAs in the molecular pathways associated with HPV-positive tumours, considering the high frequency (63.3%) of high-risk HPV genotypes (HPV16) in the PeCa patients. This is an unusual high frequency compared to the literature, which have described the HPV16 genotype as the most prevalent in PeCa, although in frequencies less than 50% [3, 27].
The aCGH analysis revealed 89.5% of the altered cytobands are mapped in HPV integration sites in the host genome. These findings support that HPV integration sites are generally located on or nearby regions of fragile sites, and that CNAs in these regions can be potentially triggered by the virus insertion [30]. It is also relevant to point out, that all the 15 cytobands that showed an association with clinical and histopathological parameters are regions of HPV integration sites. For example, the 14q12 region showed an association with the HPV16 genotype and multiple infections. CNAs on chromosomes 2p, 9p, 11q and 15q were associated to early stages of the tumour, while CNAs on 4p, 7p, 7q, 14q and 22q to intermediate tumour stages. These findings, together with our previous study [4], reinforce that CNAs play an important role in HPV-related PeCa, and can indicate candidate genes within these affected regions, that can be used as molecular markers for these patients.
Herein, we identified potential molecular markers affected by CNAs using prediction analysis to determine the interaction of genes and miRNAs mapped at these regions with CNAs and their corresponding gene targets. To our knowledge this is the first study conducted in PeCa patients, where this integration approach was performed. This integrative analysis, followed by functional enrichment pathways analysis, using both tarbase and microT-CDs algorithms, revealed five common pathways: Hippo signaling, lysine degradation, mucin type O-Glycan biosynthesis, prion diseases, and proteoglycans in cancer. Hippo signaling, which is a pathway associated with cell proliferation and survival [31]. The miRNAs that were mapped in the CNAs, miR-548d-5p, miR-548k, miR-548n, and miR-4517, were previously demonstrated to regulate several genes involved in these cellular process [32, 33, 34, 35]. Interestingly, this pathway presents a feedback relationship with the TGF-B signaling pathway [36], in which the downstream genes BMPR2 and SMAD4 are involved. These genes are also targets of miR-548d-5p (BMPR2) and miR-4517 (BMPR2 and SMAD4), which loss was previously associated with aggressive tumour phenotypes, such as tumours with larger sizes, metastasis development, and chemotherapy resistance [37, 38]. Therefore, we suggest that the negative regulation of these genes by miR-548d-5p, and miR-4517 can be one of the molecular mechanisms relevant to the penile tumorigenesis.
The role of miRNAs in the lysine’s degradation pathway is not completely understood. MiR-548d-3p, mapped at 8q24.13 and affected in our cases by gain of copy number, is predicted to regulate several gene targets involved in this pathway, such as KMT2 A, KMT2D, SUV420H1, WHSC1L1, and SETD7. The altered expression of these genes has been described in several cancers [39, 40, 41, 42, 43], however the mechanisms that lead to their expression alterations is not clear.
The third pathway affected by the miRNAs located in regions with CNAs of this study, is the biosynthesis of mucin’s type O-glycan. O-glycans-type mucins are constituents of the main compounds of mucous, forming physical-chemical barriers [44]. The O-glycosylation process involve several genes from the GALNT or GalNac-T (N-acetyl-galactosaminyltransferases) family, which abnormal expression have been associated with cancer progression [44, 45]. In our study, the GALNT1, GALNT2, GALNT3 and GANLT7 genes were identified as potential targets for miR-30b-5p and miR-30d-5p, mapped at 8q24.22 region, affected by gain of copy number. Interestingly, Gaziel-Sovran et al. [46] observed an increase in the metastatic potential in melanoma by the repression of GALNT1 and GALNT7 due to the high expression of miR-30b/30d. In gastric carcinoma, negative regulation of GALNT2 is associated with more advanced stages of the tumour, lymph node metastasis and reduced disease-free survival [45]. It was also observed that the downregulation of GALNT2 modifies the O-glycosylation of EGFR and affects the EGFR-AKT signaling [44]. Additionally, the lower expression of GALNT3 in pancreatic cancer has been associated with altered glycosylation of the ErbB family receptors, and with increased aggressiveness [47]. Altogether, these findings support the role of miR-30b/30d in the O-glycan mucin pathway via regulation of the GALNT gene family, and their association with aggressive tumour types.
Our analysis also revealed that miR-20d-3d and miR-148a-3p target the PRNP gene. This gene is involved in Prion diseases, where it regulates pathways of oxidative stress, proliferation, adhesion, and cell survival. Although most of the studies on PRNP are focused on the nervous system, recent evidence indicates that this gene also regulates these process in cancer cells [48]. The role of miRNAs in the regulation of PRNP is still uncertain, since the physiological functions of the PrP
Considering that in this study all the tumours were HPV positive, we also investigated the HPV-associated carcinogenesis pathway. The tarbase algorithm revealed that both miRNAs and HPV oncoproteins can negatively regulate genes related to proliferation, cell cycle and apoptosis. For example, although endosomal acidification is required for the initial stages of HPV infection [52], inhibition of acidification by the HPV oncoprotein E5 has already been reported [53, 54, 55]. Moreover, the ATP6V0D1 gene has been identified as a target for miR-148a-3p. This gene encodes one of the subunits of the vacuolar-ATPase enzyme and is responsible for endosomal acidification and consequent degradation of some cellular components, such as the EGFR [56]. Thus, miR-148a-3p and HPV-E5 may both inhibit the endosomal acidification and, consequently, increase the availability of EGFR. The involvement of EGFR in PeCa was previously reported in other studies, including our own [4], where EGFR was observed with gain of copy number and gene overexpression.
Other genes that are usually targeted by HPV oncoproteins have also been identified as targeted by miRNAs in the HPV-mediated carcinogenesis pathway. In the cases of this study, miR-148a-3p and miR-30d-3p are mapped at regions with gains (at 7p15.2 and 8q24.22, respectively), which were among the miRNAs predicted to regulate RB1 and CDKN1B, both genes involved in cell cycle and apoptosis [57]. Previously, we demonstrated the reduced expression of RB1 in PeCa and suggested the occurrence of other mechanisms for RB repression than the canonical HPV/TP53/RB1 signaling pathway [4]. Therefore, based on this present study, upregulation of these miRNAs’ expression, could be another mechanism that leads to the negative regulation of RB1 in HPV
Additional integrated analysis considering the 78 genes affected by CNAs and regulated by CNA-miRNAs highlighted the involvement of other genes (CCNE2, E2F5, EPAS1, INHBA, LYN, PDE7A, PPP1CB, PPP3R1, RAC1, SEMA3D, SOS1, YWHAQ, and YWHAZ) in the previously discussed pathways, such as P53 signaling, hippo and TGF-
The conventional treatment for PeCa patients is usually based on 5-Fluorouracil, Doxorubicin, Cisplatin, Ifosfamide, Methotrexate, and Paclitaxel, mostly in combination with radiotherapy. This treatment regimen is effective, however, patients’ tumours eventually become resistant, which can lead to disease recurrence and death [6, 9].
The drug prediction analysis conducted in our study, based on the main pathways described above indicated several molecular markers which inhibitory drugs could be repurposed for the treatment of HPV-related PeCa. The EGFR gene emerged as one of these known druggable targets, which we consider one of the most promising markers that can be used for PeCa treatment. This is based on our findings showing its gain of copy number and higher expression in most of the PeCa cases analyzed [4], and its involvement in the HPV-mediated carcinogenesis pathway, where its expression is affected by the endosomal acidification triggered by miR-148a-3p and the HPV-E5 oncoprotein. Thus, Cetuximab, a recombinant monoclonal antibody directed to EGFR, could be considered an alternative therapy for patients with PeCa that express this receptor. Interestingly, a recent study [58] has demonstrated that the growth of one PeCa-derived cell line overexpressing EGFR, was inhibited by EGFR inhibitors, corroborating with our findings. In addition, combined with chemotherapy, Cetuximab is the first line therapy for metastatic colorectal and squamous cell carcinomas [59, 60, 61], where it has been shown satisfactory responses [62, 63].
In addition to EGFR inhibitors, the drug prediction analysis performed revealed the Celecoxib, a selective inhibitor of COX2. Interestingly, we previously reported in the PeCa cases of this study, the association of the overexpression of EGFR and COX2 [4], suggesting that their inhibitors could be used for PeCa treatment. Furthermore, in vitro studies have demonstrated the efficacy of the combination of COX2 inhibitors with other drugs in cancer cells. For example, Mishra et al. [64] demonstrated that N-acetyl-cysteine and Celecoxib inhibit the initiation of cutaneous tumours. Velmurugan et al. [65] showed that the combined treatment of Celecoxib and Calyculin-A inhibited the epithelial-mesenchymal transition in oral cancer cells. Other studies showed that treatment with Celecoxib prevents Doxorubicin-induced drug resistance in canine and mouse lymphoma cell lines [66]. These findings reinforce that COX2 and EGFR are promising druggable targets and could be considered as alternative therapeutic strategies to treat PeCa patients.
Conclusion
In conclusion, our analysis showed that CNAs present a significant impact on the regulation of miRNAs and their corresponding gene targets. The genes, miRNAs, and mRNA targets mapped in chromosome regions with CNAs, can affect signaling pathways with relevance to the HPV-mediated carcinogenesis, and point out to druggable targets that could be potentially “repurposed” to treat PeCa patients with HPV infection.
Author contributions
Conception: J.S. and S.P.
Interpretation or analysis of data: J.S., L.C. and S.P.
Preparation of the manuscript: J.M., L.C. and S.P.
Revision for important intellectual content: J.M., L.N., R.C. A.D., A.K., R.M., A.P.S., L.C. and S.P.
Supplementary data
The supplementary files are available to download from
sj-pdf-1-cbm-10.3233_CBM-210035.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-210035.pdf
Footnotes
Acknowledgments
The authors would like to thank the personnel from Tissue Culture and Cytogenetics Laboratory of Institute of Evandro Chagas (Belém, PA, Brazil) for performing the scanning of the array slides. The authors thank Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES) for providing scholarship for J.S (Master Postgraduate Program in Health Science - Federal University of Maranhão, Brazil).
Conflict of interest
The authors declare that they have no competing interest.