
Abstract
BACKGROUND:
The molecular mechanisms involved in the prostate cancer and their relationship with immune cell infiltration are not fully understood. The prostate cancer patients undergoing standard androgen deprivation therapy eventually develop castration resistant prostate cancer (CRPC) for which there is no effective treatment currently available, and the hub genes involved in this process remain unclear.
OBJECTIVE:
To study prostate cancer systematically and comprehensively.
METHODS:
Differentially expressed genes (DEGs) of prostate cancer were screened in The Cancer Genome Atlas (TCGA) database. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. Connectivity Map (Cmap) software was applied to discover potential treatment drugs. A protein-protein interaction (PPI) analysis was performed to obtained the hub genes, and the relationship between hub genes and immune cell infiltration was investigated. Next, RNAseq data of hormone-sensitive prostate cancer samples and CRPC samples obtained from TCGA database was further analyzed to identify DEGs. Finally, a PPI analysis was performed to obtain the hub genes.
RESULTS:
A total of 319 DEGs were identified between prostate cancer samples and normal adjacent samples from TCGA database using comparative analysis. The KEGG pathway analysis showed significant correlations with drug metabolism, metabolism of xenobiotics by cytochrome P450, and chemical carcinogenesis. AMACR, FOLH1 and NPY, three hub genes, were found to be upregulated. FOLH1 was positively correlated with CD8
CONCLUSIONS:
AMACR, FOLH1 and NPY may be effective therapeutic targets and specific diagnostic markers for prostate cancer. AMACR, FOLH1, and NPY are also closely associated with immune cell infiltration in prostate cancer. Moreover, aminoglutethimide and resveratrol were found to be the promising drugs for treating prostate cancer. The progression of hormone-sensitive prostate cancer to CRPC may be related to arachidonic acid metabolism, PPAR signaling pathway, AMPK signaling pathway, and other metabolic pathways. SCD and FASN are expected to be the potential therapeutic targets for CRPC.
Introduction
Prostate cancer is the second most common cancer and the fifth leading cause of cancer death in men [1]. At present, the pathogenesis of prostate cancer, the cause of disease progression, and the molecular mechanisms behind drug resistance are not fully understood. There is sufficient epidemiological evidence to suggest a genetic predisposition to prostate cancer [2]. With the development of sequencing technology, several genetically susceptible gene variants have been discovered in inherited cancer syndrome, such as the HOXB13 gene [2, 3]. In addition, polygenic panel testing has been used in clinical practice to assess the risk of cancer [4]. However, due to the existence of complex genetic heterogeneity and the continuous development of sequencing technology, the multi-gene panel’s genes need to be continuously updated in order to improve the accuracy of genetic diagnosis. The current prostate cancer diagnosis mostly relies on the prostate-specific antigen test, which increases the detection rate and results in overtreatment [5]. Therefore, there is a need to find more accurate molecular markers that can be used to detect cases that are at risk of clinical progression and that require intervention.
Prostatectomy and/or radiation therapy are currently the most common treatments used for men with prostate cancer. Androgen deprivation therapy is another therapeutic option, but many cancers treated with androgen deprivation therapy develop to castration-resistance, requiring a next generation of endocrine therapy [6]. However, the hub genes involved in the progression of hormone-sensitive prostate cancer to CRPC remain unclear, and there is still no effective treatment for CRPC. Therefore, there is an urgent clinical need to explore the molecular mechanisms of CRPC and find more effective and promising drugs.
Other studies have found a link between chronic inflammation and prostate cancer development [7, 8]. The interaction between tumor cells and immune cells in the tumor microenvironment jointly promotes prostate cancer development and disease progression. Anti-inflammatory therapy and immune cell-targeted therapy are expected to be the new therapeutic strategies for prostate cancer [9]. Therefore, it is necessary to further explore the relationship between prostate cancer and immune cell infiltration. The purpose of this study is to systematically and comprehensively study prostate cancer using data from several authoritative databases, including the molecular mechanisms involved in prostate cancer, its relationship with immune cell infiltration, and potential therapeutic drugs that could be used for targeted therapy.
Materials and methods
The differentially expressed genes (DEGs) in prostate cancer were searched based on The Cancer Genome Atlas (TCGA) database. TCGA covers 33 cancer types and has been widely used in cancer research. Gene Expression Profiling Interactive Analysis (GEPIA) is a powerful online analysis tool [10] that can directly obtain DEGs, survival rates, and clinically relevant data of different types of cancer in TCGA. In this study, DEGs were obtained from prostate cancer tumor samples and normal adjacent samples using the GEPIA tool. The Limma package was used in the analysis. The analysis parameters were set to
The Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were used to explore the molecular and biological mechanisms of DEGs in the development and progression of prostate cancer. MetaScape is a widely used web tool (
Next, the hub genes involved in prostate cancer development were searched. Based on DEGs and the STRING database [15], a protein-protein interaction (PPI) network of prostate cancer was constructed. The hub genes that play a crucial role in PPI networks were then analyzed with the Cytoscape software [16]. Subsequently, GEPIA was used to analyze the expression level of hub genes in TCGA. The GEPIA was used to further explore the distribution of the hub genes in other tumors in the TCGA database. The Cell Marker database [17] was used to find the distribution of hub genes in prostate cancer tissues.
TIMER [18, 19] a web server that integrates comprehensive resources and analyzes immune cell infiltration in different cancer types and correct for tumor purity, was employed to investigate the relationship between genes of interest and survival prognosis in prostate cancer patients. The relationship between these hub genes and the proportion of infiltrating immune cells was further analyzed.
According to the clinical information downloaded from TCGA, the samples that received endocrine therapy and had clear indications of its therapeutic effect were screened out. The samples were then divided into CRPC and hormone-sensitive prostate cancer based on their response to endocrine therapy. Comparative analysis of RNAseq data from the two groups was performed using the Limma package (version 3.40.6) of R (version 3.6.1) to obtain DEGs. The parameter was set as
The DEGs in prostate cancer samples and adjacent normal samples were analyzed using the Limma package. The parameter was set as
Results
DEGs of prostate cancer were screened, and GO analysis and KEGG pathway enrichment analysis were performed according to the DEGs. Using the GEPIA tool, 319 DEGs of prostate cancer were screened from TCGA. Among these, 95 genes were upregulated and 224 genes were downregulated (Fig. S1). The GO analysis showed that the biological processes associated with these genes were mainly related to peroxidase activity, structural molecule activity and oxygen binding. The cell components were mainly located in the extracellular matrix and blood microparticles. The molecular functions were related to extracellular structure organization, hydrogen peroxide catabolic process and benzene-containing compound metabolic process (Fig. 1). Enrichment of KEGG pathways showed significant correlations with drug metabolism, metabolism of xenobiotics by cytochrome P450, and chemical carcinogenesis (Fig. S2).
GO analysis of the DEGs in prostate cancer. (A) Biological process. (B) Cellular components. (C) Molecular function.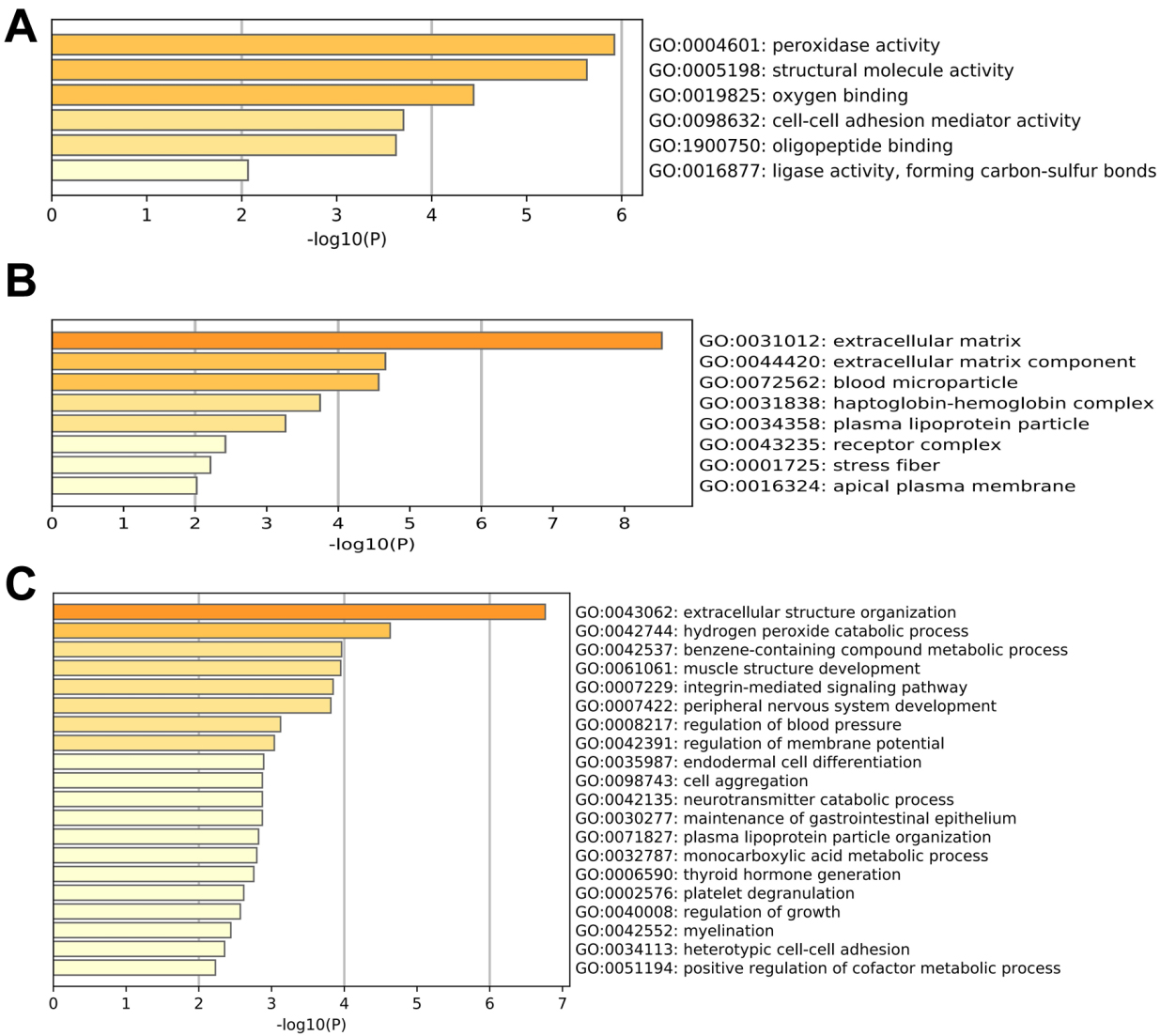
We then proceeded forward to predict potential drugs for prostate cancer treatment. According to the DEGs, 125 drug candidates were selected using the Cmap database. Among these, aminoglutethimide and resveratrol were discovered to have great potential in the drug treatment of prostate cancer. To construct a network of candidate drug targets, we explored the action targets of the top 10 drugs using the STITCH database (Fig. S3). The drugs Butacaine and Prestwick-665 did not reveal any suitable drug targets.
PPI analysis of the hub genes in the regulatory network in prostate cancer.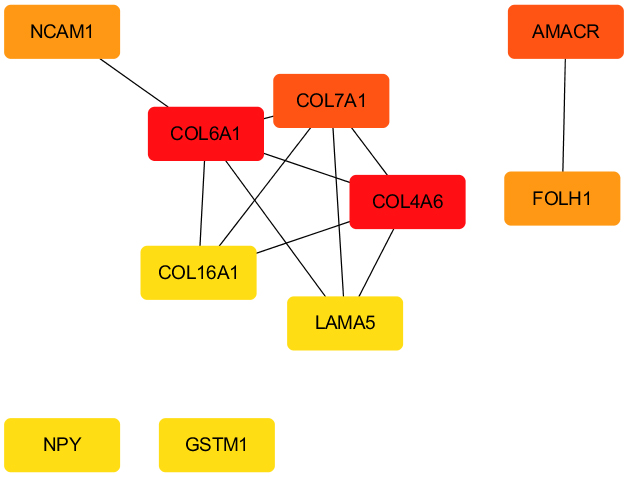
Cytoscape was used to screen ten hub genes (COL6A1, COL4A6, AMACR, COL7A1, NCAM1, POLH1, LAMA5, GSTM1, COL16A1, and NPY) in the PPI regulatory network (Fig. 2). We found seven downregulated genes and three genes that were upregulated in prostate cancer tumor samples (Fig. 3). The three upregulated genes (FOLH1, AMACR, and NPY) could be used as potential diagnostic and therapeutic targets. The 10-year survival rate of AMACR overexpression in prostate cancer patients was observed to have prognostic value (Fig. S4). To assess the distribution of these three hub genes in prostate cancer tissue cells, we searched the distribution of these genes in the Cell Marker database. We observed that FOLH1 expression is associated with peripheral blood circulating tumor cells, and AMACR is expressed in prostate cancer stem cells. We further explored the distribution of these three hub genes in other tumors in the TCGA database. We found that AMACR is highly expressed in renal papillary cell carcinoma and prostate cancer (Fig. S5). FOLH1 is highly specifically expressed in prostate cancer (Fig. S5), and NPY is highly expressed in pheochromocytoma, paraganglioma, and prostate cancer (Fig. S5). In summary, in the three selected hub genes, FOLH1 has the highest specificity as a diagnostic marker for prostate cancer.
Differential expression of hub genes in prostate cancer. 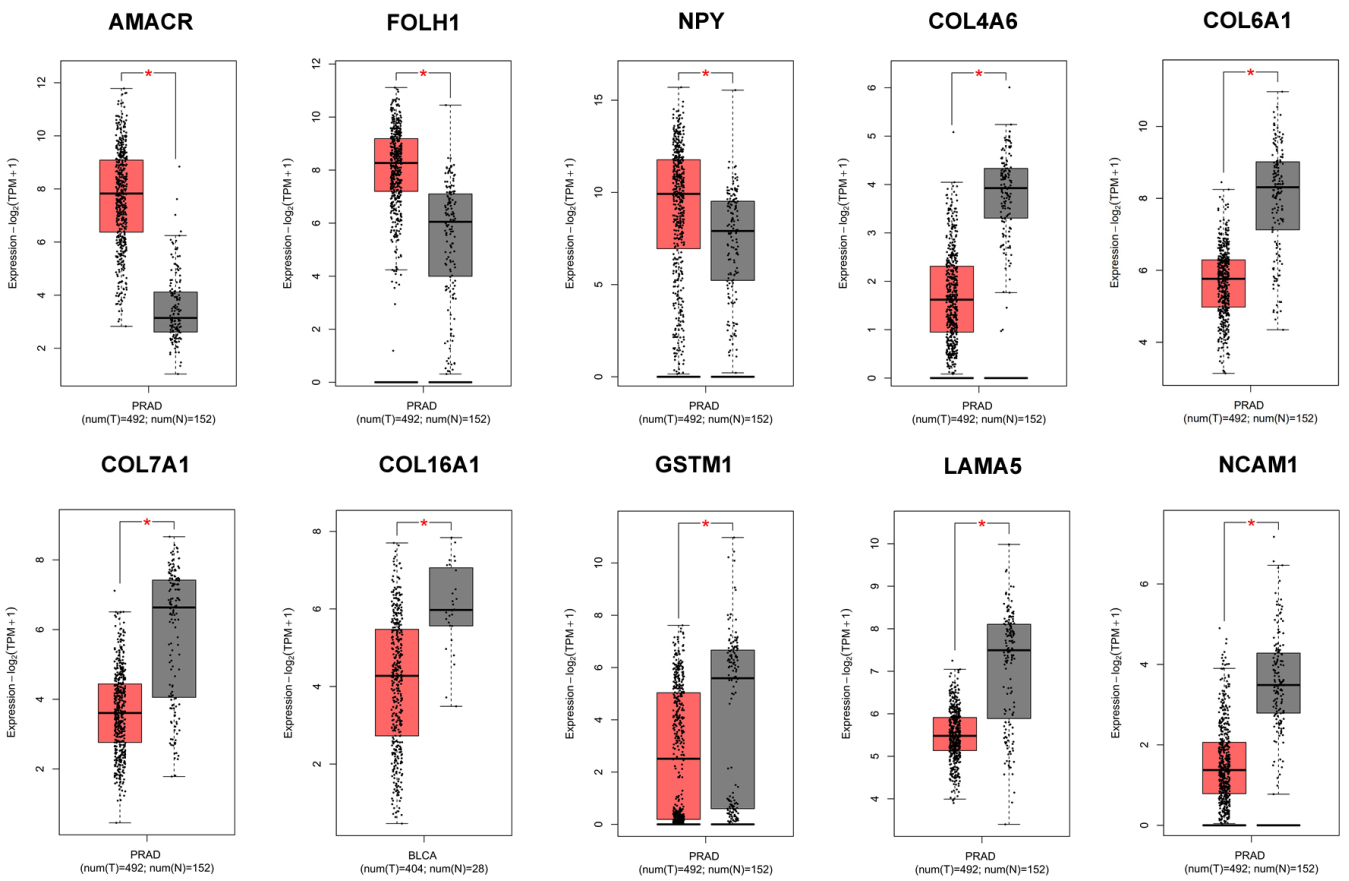
The relationship between hub genes (AMACR, FOLH1 and NPY) and the proportion of infiltrated immune cells in prostate cancer.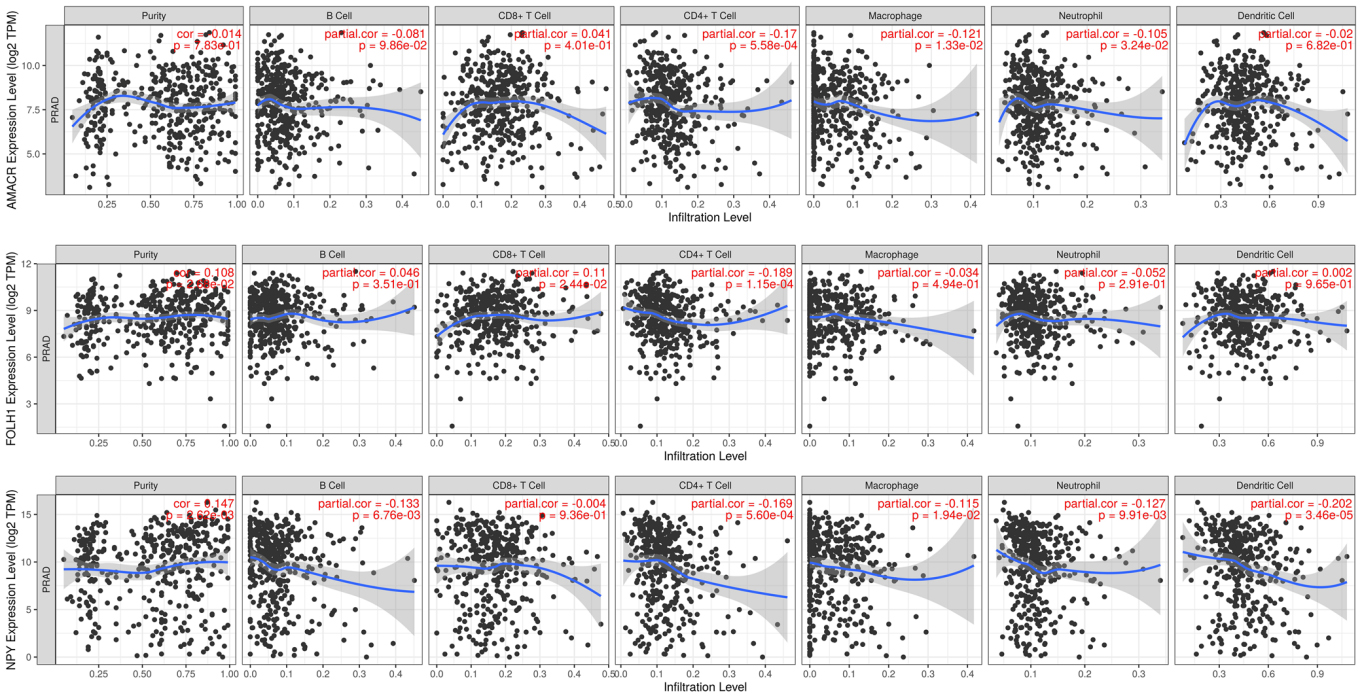
We then analyzed the relationship between hub genes and immune cell infiltration. We found that FOLH1 was positively correlated with CD8
In order to further obtain the hub genes that promote the progression of hormone-sensitive prostate cancer to CRPC, the TCGA database was used to further screen out hormone-sensitive prostate cancer samples and CRPC samples for further analysis. 5 pairs of hormone-sensitive prostate cancer samples and CRPC samples were selected from the TCGA database (Table S1). A total of 426 DEGs were selected through further comparative analysis of RNAseq data from hormone-sensitive prostate cancer and CRPC (Fig. 5). Enrichment of KEGG pathway showed that there are significant correlation with arachidonic acid metabolism, PPAR signaling pathway, AMPK signaling pathway, and metabolic pathways (Fig. 6). The top 10 hub genes (PPARG, SREBF1, SCD, HMGCR, FASN, PTGS2, HMGCS2, SREBF2, FDFT1, and INSIG1) were obtained from the PPI network combined with analysis using the Cytoscape software platform (Fig. 7). Among these, SCD and FASN were upregulated in prostate cancer samples, and are expected to be potential therapeutic targets for CRPC (Fig. S8).
Volcano plot shows DEGs in hormone-sensitive prostate cancer and CRPC.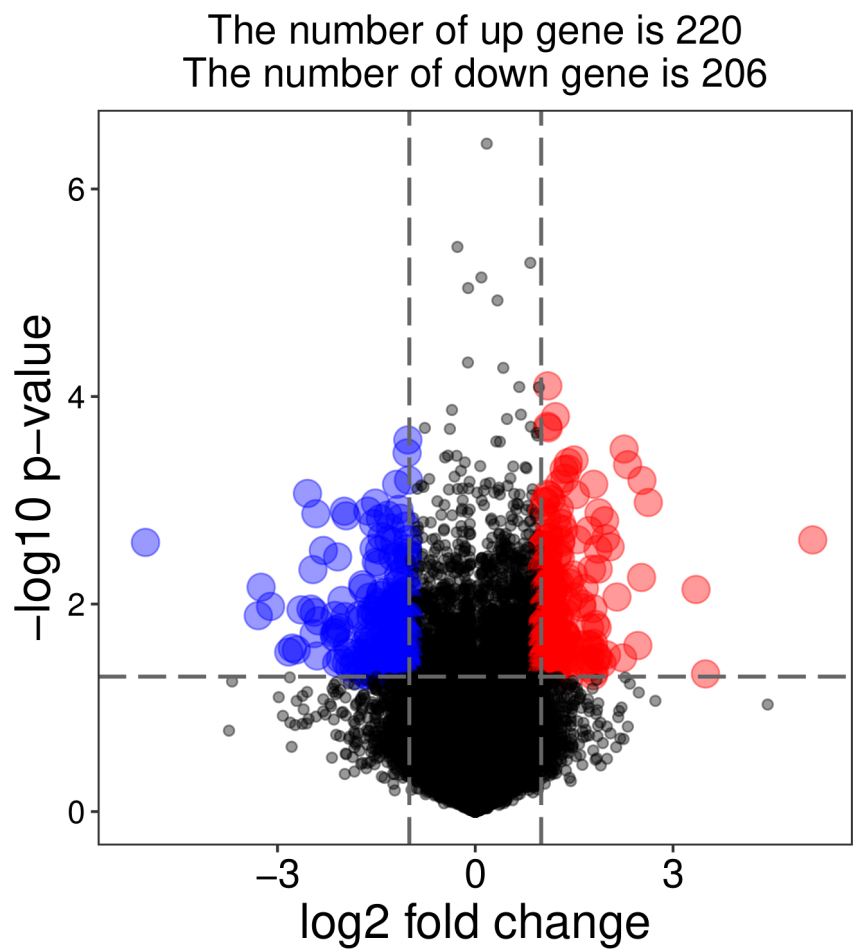
The KEGG enrichment pathway analysis of DEGs in CRPC.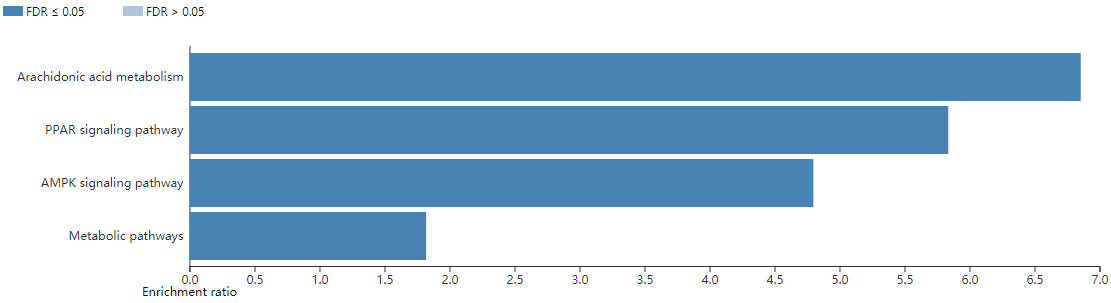
PPI analysis of the hub genes in the regulatory network in CRPC.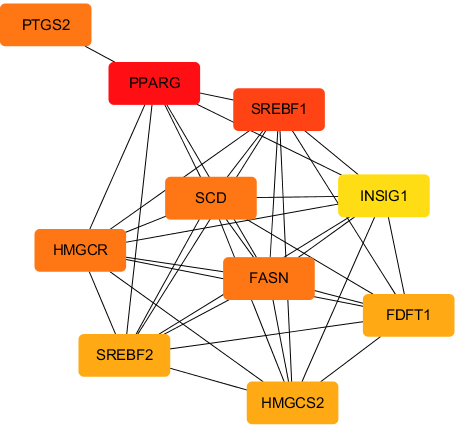
This study first screened DEGs obtained from the TCGA database using the GEPIA tool to gain insight into the molecular mechanisms involved in prostate cancer. We found 95 up-regulated genes and 224 down-regulated genes in prostate cancer samples. We performed GO analysis and KEGG pathway enrichment analysis on these DEGs. The GO analysis showed that the biological processes involved were related to peroxidase activity, structural molecule activity and oxygen binding. Some studies have shown that peroxidase activity plays a vital role in maintaining the oxidative capacity of prostate cancer cells [20]. Other studies have observed higher peroxidase activity in breast cancer samples than in normal samples [21]. In addition, studies have shown that increased expression of genes associated with oxygen binding function was strongly associated with breast cancer metastasis [22]. Further KEGG enrichment analysis of DEGs was performed using the WebGestalt analysis tool, showing that the involved pathways were mainly related to drug metabolism, metabolism of xenobiotics by cytochrome P450, and chemical genesis. In tumors, the metabolism of chemotherapy drugs directly affects the effective dose and the maintenance time of drugs in vivo. Another enzyme associated with the first stage of drug metabolism is cytochrome P450 [23]. In addition, studies have shown that chemokines and cytokines in tumor microenvironment can downregulate the enzymic activity of cytochrome P450 enzyme and further affect the efficacy of drugs [24]. In summary, the main biological pathways involved in prostate cancer are related to drug metabolism, which may indicate the low efficacy of current drugs for prostate cancer. Further studies on drug metabolism in prostate cancer should be carried out to offer novel approaches for chemotherapy of prostate cancer.
After obtaining DEGs, the Cmap database was further used to screen potential therapeutic agents for prostate cancer therapy. A total of 125 candidate drugs for prostate cancer were screened out. We focused on the top 10 drugs based on the most significant scores. The STITCH database was used to analyze the action targets and build the target network of the top 10 drugs. We further reviewed the relevant literature and concluded that aminoglutethimide and resveratrol have a broad prospect for the treatment of prostate cancer. Some scholars have suggested that aminoglutethimide has great application potential as a second-line treatment drug for androgen-dependent prostate cancer with great application prospect [25]. In addition, multiple studies have shown that resveratrol can lead to apoptosis and inhibit the proliferation and metastasis of prostate cancer cells [26]. Currently, there are many studies on aminoglutethimide and resveratrol, with the further development of other drugs, the candidate drugs mentioned above may be developed into new drugs with promising application prospects.
Through the PPI network, we searched hub genes of prostate cancer, including COL6A1, COL4A6, AMACR, COL7A1, NCAM1, POLH1, LAMA5, GSTM1, COL16A1 and NPY. The upregulated genes FOLH1, AMACR and NPY might serve as potential diagnostic and therapeutic targets for prostate cancer. We further explored the distribution of these three hub genes in other tumors using the TCGA database and found that FOLH1 was specifically expressed in prostate cancer. Therefore, FOLH1 is a relatively specific molecular marker for prostate cancer. At present, there are few research reports on FOLH1, and further studies focusing on this gene are needed. AMACR is highly expressed in kidney and prostate cancers. There are many reports suggested that AMACR can be used as a diagnostic marker for prostate cancer [27, 28]. We further observed that AMACR has prognostic predictive value for the 10-year survival rate of patients with prostate cancer. We searched the cell marker database to further assess the distribution of the three hub genes in prostate cancer cells. FOLH1 was observed and considered as a marker of peripheral blood circulating tumor cells, and AMACR was found to be a marker of prostate cancer stem cells. With the advent of single-cell sequencing technology, tumor research can be performed at a single-cell resolution. This technology allows targeting research and treatment to individual cells. Therefore, our future studies will aim to conduct single-cell studies related to prostate cancer, to further assess prostate cancer cells’ molecular biological mechanisms at the single-cell level.
Recent studies have shown that immune cells play an essential role in tumor occurrence and progression [29, 30, 31]. At present, there are few reports about immune cells and prostate cancer. Immune cells in the prostate cancer tumor microenvironment play various roles during different disease stages [32]. Therefore, in our present study, the relationship between prostate cancer hub genes and common immune cell infiltration was analyzed. We found that FOLH1 expression was positively correlated with CD8
CRPC is a challenging topic in prostate cancer research. There is an urgent need to find the hub genes that dominate the progression of hormone-sensitive prostate cancer to CRPC and to determine the effective therapeutic targets for CRPC. We found that the progression of hormone-sensitive prostate cancer to CRPC may be related to arachidonic acid metabolism, PPAR signaling pathway, AMPK signaling pathway and other metabolic pathways. A recent study has shown that the mechanism of arachidonic acid-induced steroid generation is related to the activation of androgen receptors in CRPC progression; therefore, the targeting of fatty acid pathways in CRPC can be attempted for treatment [36]. Zeng et al. found that prostate leucine zipper regulates autophagy through the AMPK pathway in prostate cancer [37]. We also found that SCD and FASN are potential therapeutic targets for CRPC. Dłubek et al., demonstrated that fatty acids are a potential target for the treatment of prostate cancer and may inhibit tumor growth by reducing the synthesis of fatty acids in prostate cancer [38].
To summarize, we first identified hub genes of prostate cancer by using PPI network. These hub genes may be key targets for targeted therapy in prostate cancer. We propose FOLH1 as a specific marker for the diagnosis of prostate cancer. Our study is the first to reveal 125 candidate drugs for prostate cancer and suggest that aminoglutethimide and resveratrol are the most promising drugs for this disease. We also correlated these hub genes with immune cell infiltration. We found that the progression of hormone-sensitive prostate cancer to CRPC may be related to arachidonic acid metabolism, PPAR signaling pathway, AMPK signaling pathway, and other metabolic pathways. SCD and FASN are expected to be potential therapeutic targets for CRPC.
Footnotes
Acknowledgments
This research is funded by the following research projects: 1. Project of the Ministry of Science and Technology, Project No. 2017YFC0908000; 2. National Natural Science Foundation of China, Project No.81770759; 3. Guangxi Innovation Driven Development, Project No. AA18118016.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-200939.