
Abstract
BACKGROUND:
Genetic and epigenetic dysregulation of Wnt signaling pathway is widely linked up with abnormal proliferation and/or epithelial-to-mesenchymal transition, in different cancer cell types.
OBJECTIVE:
In the present research, we have tested whether promoter DNA methylation of a set of Wnt/non-Wnt genes such as [cadherin-2 (CDH2)], “present in circulation”, could serve as “bone-marrow biopsy surrogate” and help in diagnosing the status, sub-type or treatment outcome in pediatric acute lymphoblastic leukemia (ALL) patients.
METHODS:
Promoter DNA methylation was quantified in the bisulfite modified blood from the pediatric ALL patients (
RESULTS:
The observed methylation index, sensitivity and specificity of selected molecular markers (viz., SALL1, WNT5
CONCLUSIONS:
Whilst the reported metadata provides useful insight into the plausible involvement of epigenetic glitches in leukemogensis, our findings strengthen the remarkable functional consequences of dysregulated Wnt signaling genes in the hematological malignancies besides offering a novel panel of epigenetic marks.
Keywords
Introduction
With 450,000 new cases and over 320,000 deaths, leukemia (a hematological malignancy) has been placed alongside the top-ranked cancers, worldwide [1, 2]. Amongst the subtypes of leukemia, acute lymphoblastic leukemia (ALL) is most frequently recorded in infants (onset at an age
Despite remarkable advancements made in the fields of human biology and pathophysiology, there are still several ‘grey areas’ in our understanding of the underlying, concealed molecular perturbations that trigger and essentially drive the genesis- and/or progression of hematological malignancies, including ALL [5, 6, 7, 8, 9]. Conventionally, the genetic abnormalities, copy number variations and the chromosomal rearrangements are regarded as the key drivers contributing towards malignant transformation of lymphoid progenitors into the ‘leukemic’ cells, in the B- and/or the T-cell lineages. However, accumulating evidences suggest that epigenetic alterations such as hypo-/hyper-methylation of promoter DNA, histone modifications, acetylations, etc., also trigger the leukemogensis by activating the oncogenes and/or silencing the tumor suppressor genes [7, 8, 9, 10, 11, 12]. Studies have shown that in diverse cancers, promoter DNA and/or CpG islands display raised methylation levels contrasting an overall reduction in the methylation frequencies at the genome-wide scale. Whereas, promoter DNA hypermethylation of certain tumor suppressor genes has been linked to cancer chemotherapeutic resistance; the treatment with methylation inhibitors (e.g., 5
Owing to the potential involvement of epigenetic glitches in leukemic gene regulations, we here tested the hypothesis whether promoter DNA methylation of a “set of genes” present in circulation, could help in diagnosing the status, sub-type or treatment outcome in pediatric ALL patients, either alone or in combination with available stream of tests. To assess and explore the potential gene signatures with functional CpG site methylation, extensive data mining was performed; interestingly, the search came up with a set of genes that mostly linked up at the interface of Wnt canonical/non-canonical signaling pathways along with a distantly linked non-Wnt gene CDH2 [a modulator of cell migration, proliferation, and stem cell differentiation]. The findings of our wet-lab study are interesting and provide useful insight into the plausible involvement of aberrant promoter DNA methylation in derailing an otherwise finely-tuned normal Wnt signaling cascade, besides offering a panel of gene signatures for potential non-invasive, liquid biopsy-based diagnosis of ALL.
Materials and methods
Study population and sample collection
20 subjects (15 pre-treatment ALL
Selection of marker set – systematic data mining
In order to select the candidate genes, whose promoter DNA methylation is associated with ALL, we conducted a systematic review of the literature published during 1997–2019. A total of 337 research articles were retrieved through searching different databases, using keywords such as “Pediatric Acute Lymphoblastic Leukemia”, “Biomarker”, “Diagnostic”, “Hypermethylation”, and “Promoter”. After excluding 56 duplicate records, 281 studies were screened to identify the eligible studies for analysis. From these, 241 studies were further excluded including the 46 review articles, 50 book chapters, 17 encyclopedia records, 56
(A) PRISMA flow diagram detailing the literature search and inclusion and exclusion criteria for studies. (B) Venn-diagram showing different sample types used in the reported literature. (C) Selection of candidate gene signatures; the frequency of promoter methylation (%) has been plotted against the number of articles reporting their hypermethylation. The genes displaying methylation frequency 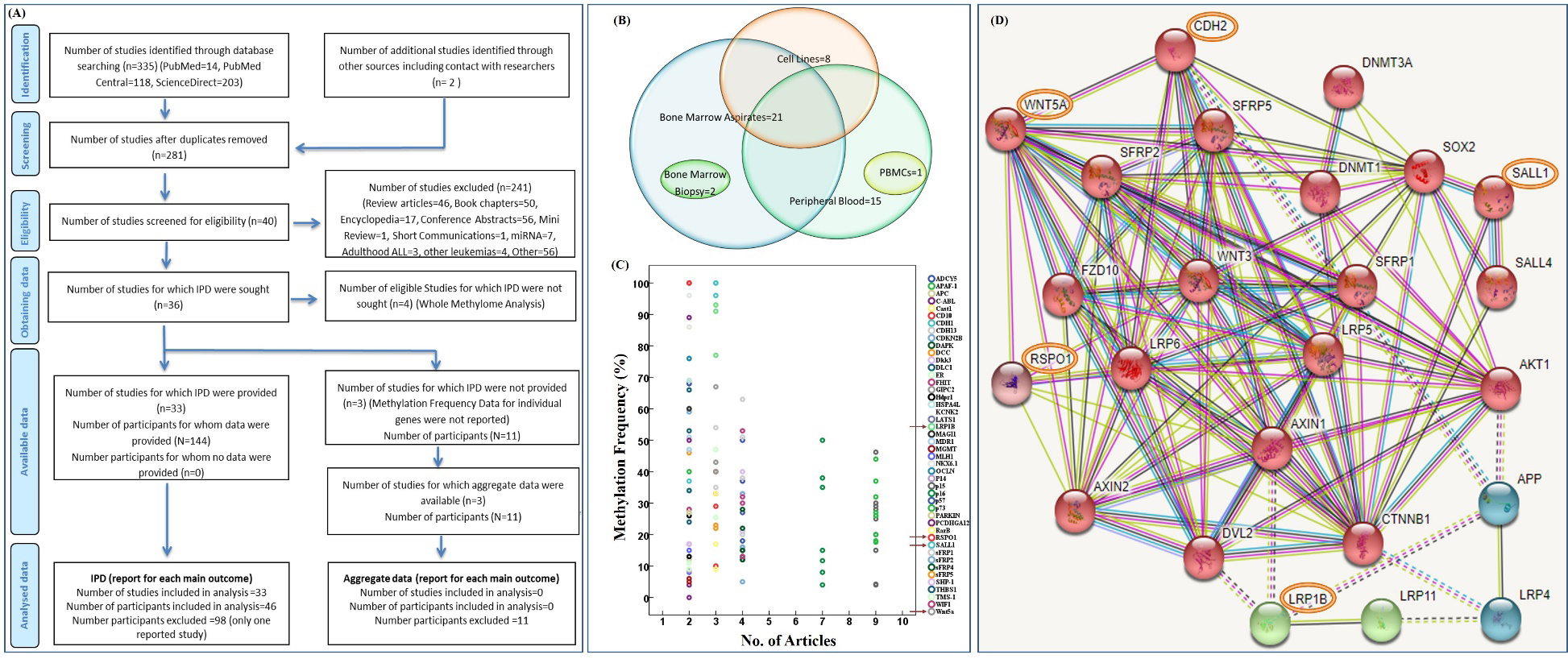
conference abstracts, 1 mini-review, 1 short communication, 7 studies investigating the role of miRNAs, 3 addressing adult ALL, 4 analyzing the promoter hypermethylation in other leukemia types and 46 monitoring the genetic/genomic/proteomic biomarkers (Fig. 1A). Forty studies were thereafter screened for eligibility; however, individual patient data (IPD) could be sought only for 36 studies. Amongst these, 3 were further excluded as lacking % methylation frequency for individual genes thus only 33 articles could be considered for final analysis.
Sequence of the primer pairs used for qMSP
M: methylated primer; U: unmethylated primer; IC: internal control primers.
The genomic location of CpG islands and binding position of methylated and unmethylated primer sets in the promoter regions of the study genes.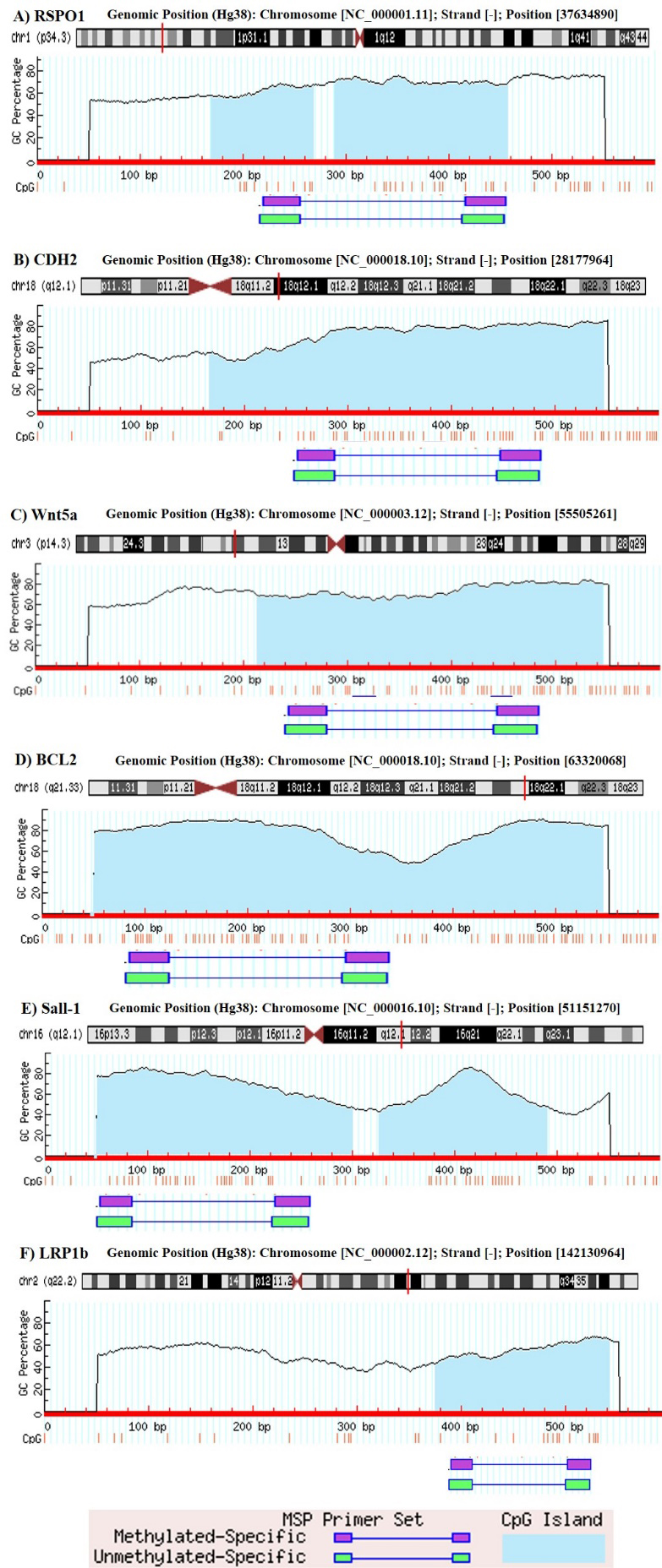
From the 33 studies, we sorted out 46 candidate genes based on the fact that these were reported in at least two studies. Most of the reported data was involving bone marrow aspirates (21 articles) and blood samples (15 articles), along with eight studies employing cell lines, two involving bone marrow biopsies and only one with mononuclear cell samples (Fig. 1B). To narrow down the selection of gene signatures, we applied another filter i.e., genes with reported methylation frequency of
Genomic DNA extraction from the whole blood and its bisulfite modification was performed using EZ DNA Methylation-Direct
Primer designing and real-time methylation specific PCR (qMSP)
The promoter sequences of all candidate genes were retrieved from the eukaryotic promoter database (EPD). Using MethPrimer v.2.0. software, pairs of methylated- and unmethylated primers were designed for each of the selected set of genes (Table 1). This tool initially performs in-silico bisulphite conversion of the sequence and then designs two sets of primers, one for the converted sequence and other for the original sequence to distinguish methylated and the unmethylated DNA sequences. The genomic location of CpG islands and binding position of methylated and unmethylated primer sets in the promoter regions is presented in Fig. 2.
All qMSP reactions were performed in BioRad CFX-96 Real-Time PCR System. The reaction mixture (25
MI (%)
Bisulfite amplicon sequencing
Bisulfite amplicon sequencing (BAS) of all five genes from two randomly selected paired ALL samples (pre- and post-treatment) was performed to validate the results obtained from the qMSP analysis. The bisulfite converted DNA was subjected to PCR amplification using primers listed in Table 1. The PCR products were purified using PureLink Quick Gel Extraction kit and sequence analyzed using the standard automated Sanger sequencing (Macrogen, Korea).
Statistical analysis
The Origin and SPSS statistics ver.21 software, were used for statistical analyses. Methylation frequencies in the patient- and the control groups were compared using Fisher’s exact test; while Spearman non-parametric test was applied to assess correlation. The diagnostic and prognostic significance of the selected set of gene(s) was predicted using Receiver Operator Characteristics (ROC) and Kaplan-Meier survival analyses, respectively.
Results
Promoter DNA methylation analysis
The clinicopathological features of pediatric ALL patients (both discovery- and validation-phase) are listed in Suppl. Table A. The patients’ group was dominated by pre-B ALL cases, median age ranged between 2.5 months to 15 years with male to female ratio being over 2:1. Only four patients had a family history of ALL; 7 cases were Philadelphia positive, 4 had CNS involvement, 21 showed hyperdiploidy while 48 presented normal karyotype. Hypodiploidy (an unfavorable risk feature associated with poor patient outcome), which according to WHO is manifested by 1–2% pediatric ALLs only [7, 8 and references therein], was found in around 20% cases (17/86); out of these 4% were baby girls while 16% were boys. Diagnosis of all the cases was based on the standard morphological, cytochemical and immunophenotypic criteria, as per the WHO classification guidelines (Suppl. Fig. S1).
The calculated promoter methylation index (% MI) of SALL1, LRP1b, WNT5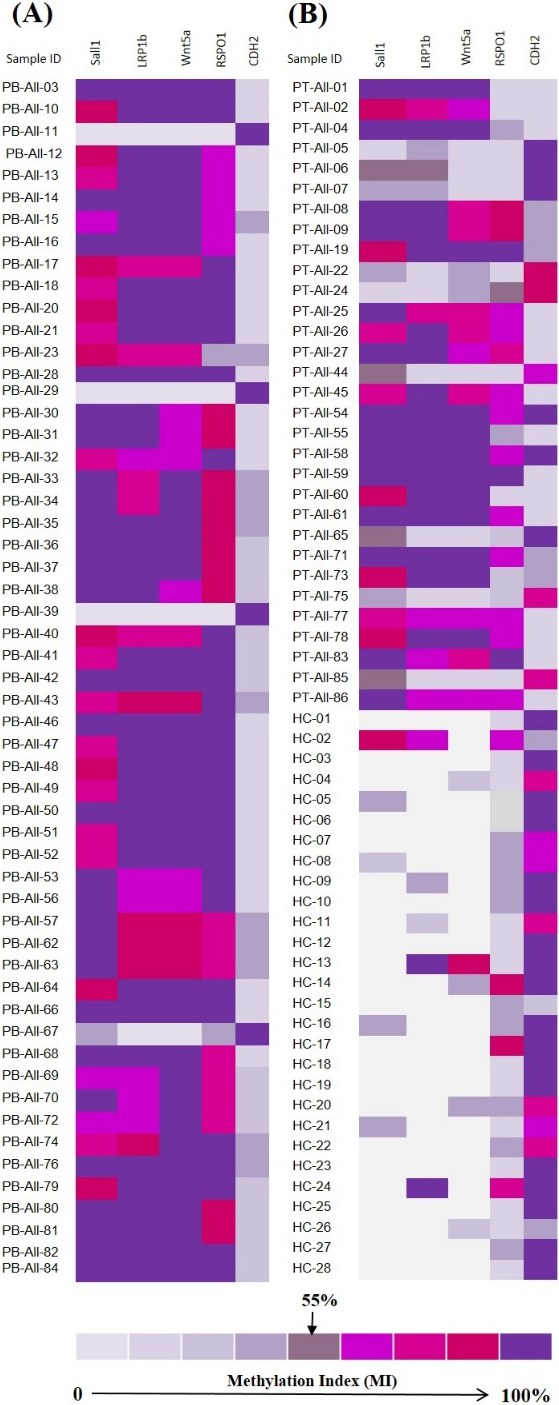
The promoter methylation/demethylation pattern of five selected candidate genes was analyzed in the pre- and post-treatment ALL patients and the results were compared with the control group (Fig. 3). The individual methylation frequencies of SALL1, LRP1b, WNT5
Methylation profile of ALL patients and cancer-free control
SD: Standard deviation.
Correlation of clinical features with promoter methylation index of each gene
Representative receiver operating characteristics (ROC) of the informative sets of genes for the detection of ALL
TP: True Positive; TN: True Negative; FP: False Positive; FN: False Negative.
Receiver operating characteristic (ROC) curves depicting prognostic significance of SALL1, LRP1b, WNT5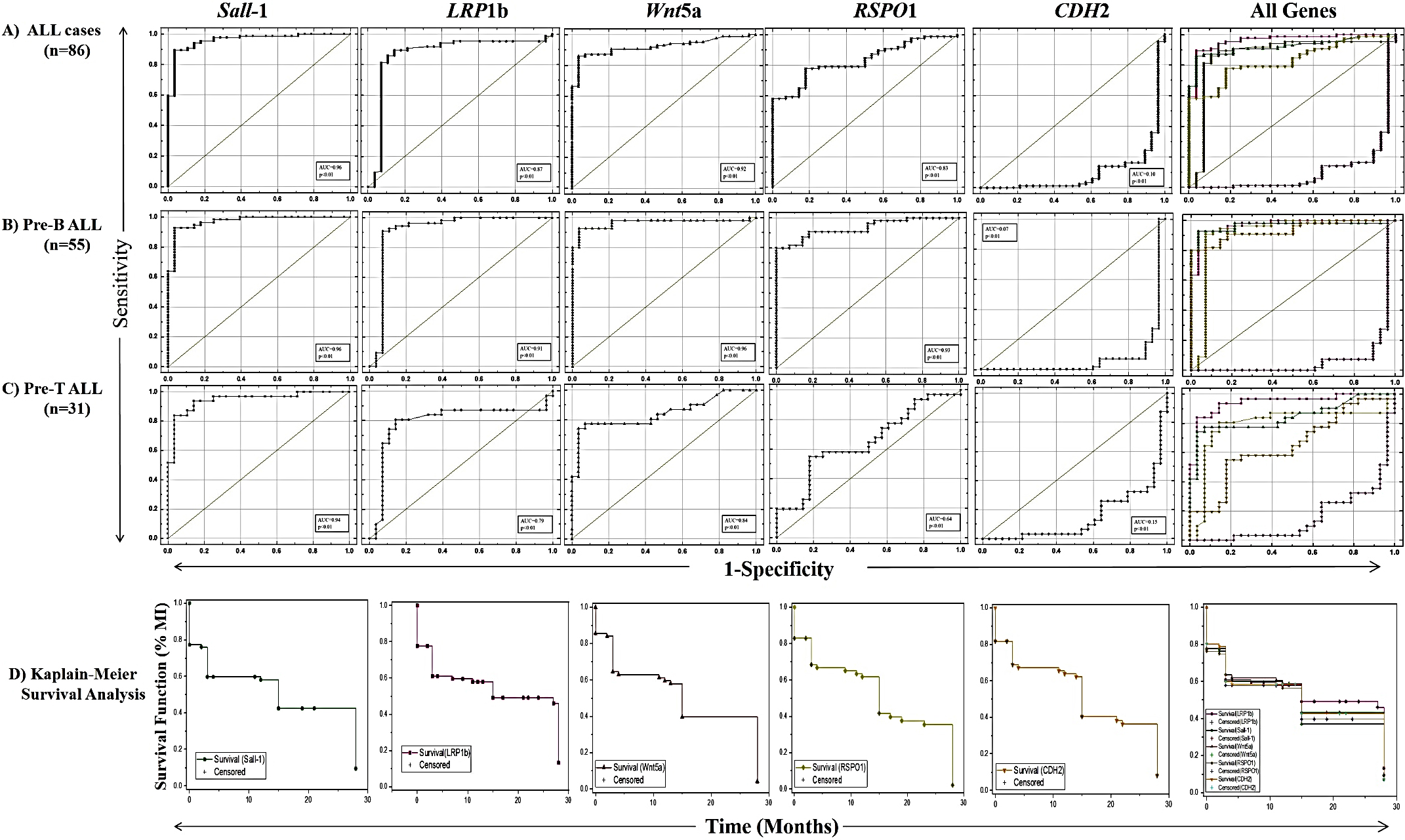
Further, the methylation levels were assessed in the post-treatment patients. Notwithstanding the fact that analysis was performed with a limited number of paired pre- and post-treatment blood samples (
Spearman correlation coefficient was calculated to evaluate the relationship between the % MI value of each candidate marker gene and the clinicopathological features (Table 3). With a few exceptions, almost all features showed a very strong, statistically significant correlation with % MI (highlighted).
The sensitivity and specificity of the candidate genes, when worked out using ROC analysis, yielded an area under the curve (AUC) of 0.96, 0.92, 0.87 and 0.83 for SALL1, WNT5
Curated pathway depicting the mechanism of Wnt signaling in the normal (A, left half) and the leukemic lymphoblasts (B, right half). Detailed mechanism in normal cells is summarized under the ‘Discussion’ section. In case of leukemia, the promoters regulating the transcription of SALL1, LRP1b, WNT5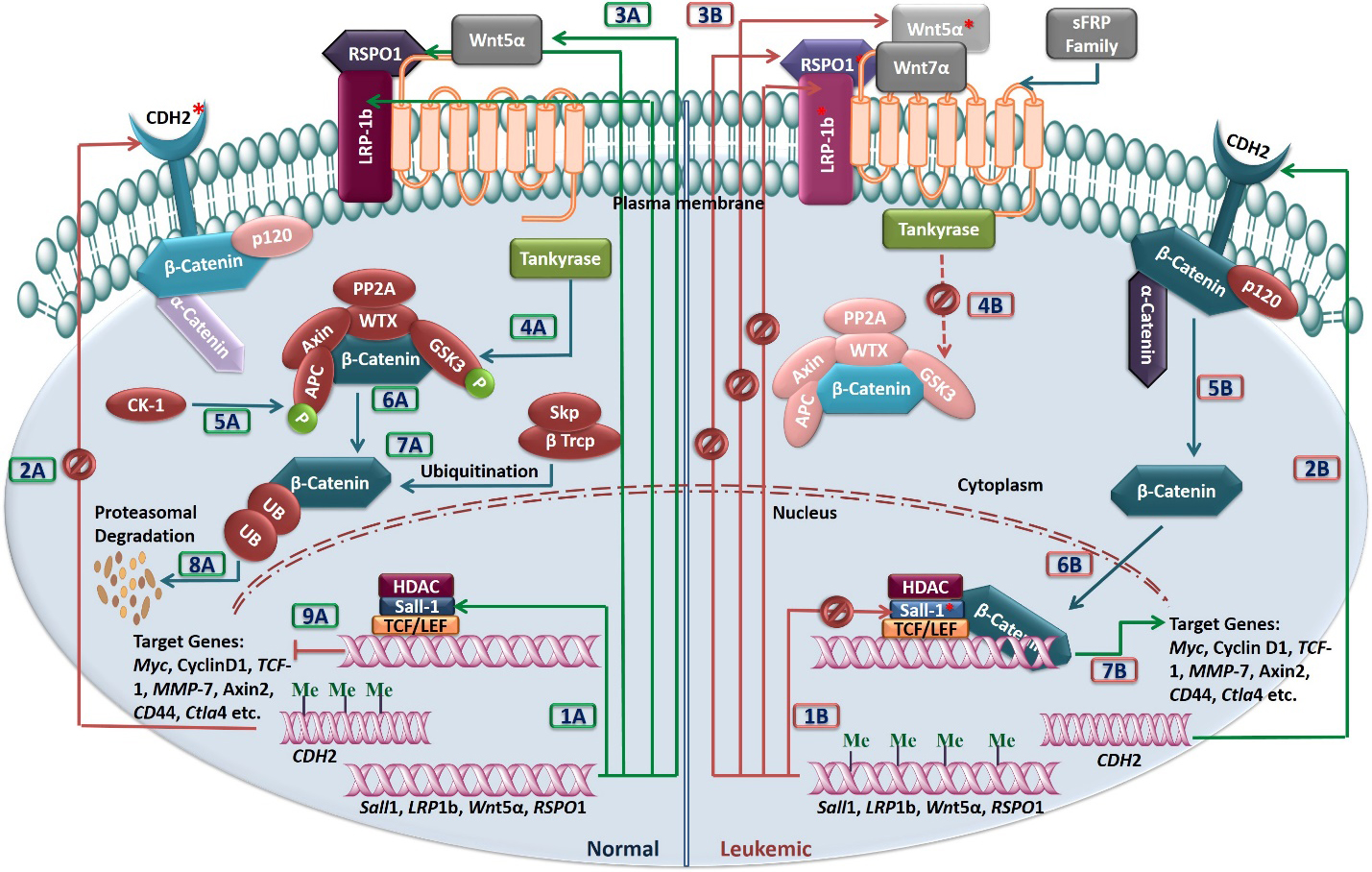
Wingless type (Wnt) signaling cascade is amongst the key signaling pathways that play important role in the normal cell growth, proliferation and development of many organ systems; and hence its dysregulation is believed to be linked up with abnormal cellular proliferation and/or epithelial-to-mesenchymal transition (EMT), in different solid tumors [20, 21, 22, 23]. Recent studies have documented that impaired regulation of Wnt proteins and their antagonists also have adverse impact on the thymocytes proliferation, lymphocytes differentiation, and/or the self-renewal of hematopoietic stem cells [[24, 25, 26] and references therein].
In the present study, the genes selected to assess the promoter DNA methylation status in pediatric ALL vs non-cancer controls (i.e., SALL1, WNT5
While respectively acting as a ligand for the members of leucine-rich repeat-containing G-protein coupled receptors (LGR) 4/5/6 and the frizzled family of seven transmembrane receptors, the R-spondin 1 (RSPO1) and the Wnt family protein type 5A (WNT5A) modulate/activate the canonical and non-canonical Wnt pathways [24, 30, 31]. Likewise, the large-sized transmembrane protein low-density lipoprotein receptor-related protein 1B (LRP1B; genomic sequence spanning 1.9 Mb, coding sequence extending 16.5 Kb, total number of exons 91), is a member of LDL-receptors family that performs a myriad of functions at the cellular-, extracellular-, and the cell membrane level. Unlike LRP5/LRP6, the LRP1B is not directly linked with Wnt pathway (Fig. 1D); its unique role in embryonic development and as a signal modulator, however, is evident from several recent reports [32]. Furthermore, mutations (deletion/point), copy number variations, and aberrant expression of LRP1B, reported in different malignancies such as gastric-, lung-, breast-, oral-, esophageal-, neuronal-, renal-, and ovarian carcinomas [32, 33], also make it a strong candidate for analysis in hematological malignancies.
Our last selected gene, CDH2, encodes cadherin 2 protein (CDH2, also known as N-cadherin), a calcium-dependent transmembrane glycoprotein involved in the intercellular- and the cell-extracellular matrix adhesions. Unlike the co-family member CDH1 (E-cadherin), the CDH2 is either absent or meekly expressed in the normal epithelium. However, loss of CDH1 with concomitant aberrant overexpression of CDH2 has been described in several epithelial- and non-epithelial/hematological malignancies [24, 33, 34]. This “cadherin-switch” is believed to have crucial role in promoting the EMT associated events i.e., cancer cells’ migration (distant metastasis), localized invasion and tumor aggressiveness through the modulation, augmentation and/or polarization of important signaling pathways such as Wnt/
Studies have shown that the CpG islands in promoters regulating the transcription of SALL1, WNT5
In the present study, more than 85% B-ALL patients showed promoter DNA “hypermethylation” of otherwise “unmethylated” genes with parallel demethylation of CDH2 (Tables 2 and 3; Fig. 2). Reversal to the ‘normal promoter methylation’ patterns (
While early-stage, cost-effective diagnosis is a critical component of success in the cancer management [39, 40] and a way to lessen the burden of complex, life-threatening disease(s); being an early event in leukemogenesis, the epigenetically dysregulated Wnt signaling pathway genes can provide important clues and thus facilitate early-on diagnosis of ALL in children [2, 24, 41, 42, 43, 44]. Whereas the findings of this research are encouraging in terms of panel sensitivity and specificity in liquid biopsies, and hence expected to have great impact on the ALL diagnosis and routine monitoring of treatment- or disease recurrence, the results need to be assessed in light of their limitations. For instance, in validation phase, we could enroll only a moderate (yet statistically significant) number of pediatric ALL patients and age-matched controls due to the reluctance of many families to give blood samples of their infants/children; difficulty in keeping track of the patients (coming from remote and distant areas of the countries) for the follow-up analyses was another great challenge. Furthermore, while designing the methylated/un-methylated primer pairs of the selected genes, we ignored scanning of the entire CpG island/shores. Since different sites in the CpG islands/promoter region differently impact the transcriptional activation and repression, undertaking ‘global methylome’ analysis instead of a site-specific analysis, may help in filling up the research gaps, still left behind. Lastly, to make this non-invasive liquid biopsy approach a bit more cost-effective in clinical oncology, we have used high-throughput qMSP technique instead of the next generation bisulfite targeted sequencing (NGS). qMSP have shown consistent results with up-to-the-mark accuracy level. The results were validated using BAS (Suppl. Fig. S3), nonetheless, NGS for an even better and complete coverage of the epigenomic alterations, may be considered, in future investigations.
In summary, our results demonstrate that the promoter regions of SALL1, WNT5
Disclosure of Interest
Authors of this manuscript have no competing interests to declare.
Funding
This study was partially supported by grants from University of the Punjab and the Higher Education Commission, Pakistan.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-200814.
sj-docx-1-cbm-10.3233_CBM-200814.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-200814.docx
Footnotes
Acknowledgments
Authors extend special thanks to all blood sample donors, both pediatric ALL patients and healthy volunteers as well as their families, who made this study possible. We are also thankful to the doctors (especially Dr. Samina) and paramedical staff of Children Hospital Lahore, who not only helped us in sample collection but also convincing the families to participate in this study.