
Abstract
BACKGROUND:
EGFR-mutant lung cancer inevitably develops resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs).
OBJECTIVE:
To investigate the clinical relevance of microRNAs (miRNAs) in TKI therapy response and resistance.
METHODS:
We performed a miRNA PCR array analysis and used The Cancer Genome Atlas (TCGA) database to identify potential miRNAs related to EGFR TKIs resistance. We then correlated miRNA expression in 70 surgical and 50 malignant pleural effusion specimens with patient outcomes in those with non-small cell lung carcinoma. Molecular manipulation was performed in EGFR mutant lung cancer cells to assess the effect of miR-200c-3p on cell migratory ability and EGFR-TKI sensitivity.
RESULTS:
We identified miR-200c-3p and miR-203a-3p as potential EGFR TKI resistance regulators via their modulation of epithelial-to-mesenchymal transition (EMT). MiR-200c-3p and miR-203a-3p were down-regulated in EGFR TKI-resistant cell lines. Progression-free survival (PFS) with EGFR-TKI treatment of patients with high miR-200c-3p expression, but not miR-203a-3p, in the specimens was significantly longer than that of patients with low expression. MiR-200c-3p overexpression inhibited the EMT process in EGFR TKI resistance cell lines and promoted cell death. MiR-200c-3p silencing in EGFR TKI sensitive cell lines increased drug resistance.
CONCLUSION:
MiR-200c-3p plays a role in sensitivity to EGFR TKIs via modulating EMT process.
Keywords
Abbreviations
Background
The epidermal growth factor receptor (EGFR) has been implicated in the development and progression of non-small-cell lung cancer (NSCLC). Several EGFR-targeting small molecule tyrosine kinase inhibitors (TKIs) are used in the treatment of NSCLC. Although EGFR TKIs produce a high clinical response rate in EGFR-mutant NSCLCs, tumor relapse is inevitable owing to several well-known mechanisms of acquired resistance, including the presence of secondary EGFR mutations (such as the T790M mutation), parallel activation of other signalling pathways (e.g., MET amplification), phenotypic transformation, and other genetic alterations [1]. However, many of the underlying mechanisms of acquired resistance remain unexplained. As such, deeper investigations into predicting EGFR TKI responses and resistance mechanisms are required.
MicroRNAs (miRNAs) are small non-protein-coding RNAs approximately 19–24 nucleotides in size that can act as endogenous RNA interference, thus negatively regulating gene expression at the post-transcriptional level. Thousands of miRNAs have been identified in various organisms via random cloning, sequencing, or computer-based prediction models. The human genome may encode over 1000 miRNAs [2], and their scope and diversity have recently begun to be investigated more thoroughly. MiRNAs have been shown to participate in the control of many fundamental cellular and physiological processes such as cell development, differentiation, growth, and survival [3]. Increasing evidence shows that miRNAs are linked to cancer and play critical roles in cellular transformation and carcinogenesis, acting either as oncogenes or tumor suppressors [4, 5, 6].
Recent studies show that the tumor microenvironment stimulates a miRNA-based process that induces resistance to anti-EGFR therapy and drives lung tumor cells to undergo epithelial-to-mesenchymal transition (EMT), invasion, and metastasis [7]. EMT is a biological process whereby epithelial cells lose their recognizable characteristics and acquire a mesenchymal-like phenotype [8], and was shown to be partially responsible for acquired resistance to EGFR TKIs in patients with NSCLC [9]. Differential profiling of miRNAs indicates that they modulate cellular plasticity through the suppression of EMT inducers such as Slug, Twist, and Zbe1/2, as well as of EMT repressors [10].
We therefore hypothesized that differential expression of EMT-associated miRNAs contributes to EGFR TKI resistance. To that end, we aimed to identify specific EMT-associated miRNAs that correlated with the response to EGFR TKI therapy in those with lung adenocarcinoma using The Cancer Genome Atlas (TCGA) database. We also aimed to clarify the function of these microRNAs in NSCLC and their association with the efficacy of EGFR TKIs and resistance to them.
Materials and methods
Cell lines
EGFR TKI-sensitive cell lines- PC9 and HCC287, and gefitinib-resistant PC9/gef cells that harbor a deletion in exon 19 of EGFR were kindly provided by Dr. James Chih-Hsin Yang (Graduate Institute of Oncology, Cancer Research Center, National Taiwan University) [9]. HCC4006 was purchased from the American Type Culture Collection (Manassas, VA, USA). Gefitinib-resistant HCC827/gef and erlotinib-resistant HCC4006/ER cells were generated from HCC827 or HCC4006 cells using stepwise escalation of concentrations of the EGFR TKIs gefitinib and erlotinib, respectively, up to 10
TaqMan
human microRNA array
The TaqMan
Correlation analysis of miRNA expression and EMT
The matched mRNA and miRNA expression datasets from lung adenocarcinoma (
Patient population and specimens
To evaluate the relationship between miRNA expression and the response to EGFR TKI therapy, we retrospectively investigated EGFR-mutant NSCLC patients with surgical specimens or malignant pleural effusion samples available for miRNA expression analysis. The first group included patients with NSCLC harbouring EGFR mutations who underwent surgery and from whom surgical specimens were therefore obtained. Lung cancer tissues obtained at surgery were immediately snap frozen in liquid nitrogen and stored until use. To analyse EGFR TKI resistance mechanisms, we only selected patients who received the EGFR TKIs gefitinib, erlotinib, or afatinib as their first line therapy after lung cancer relapse. RNA was extracted from fresh-frozen surgical specimens, whereupon miRNAs expression was analyzed. The other group comprised newly diagnosed EGFR-mutant lung cancer patients with malignant pleural effusions; such effusion samples were collected from patients who regularly required thoracentesis. The pleural fluids were acquired aseptically in vacuum bottles by thoracentesis in the ultrasonography examination room of the National Taiwan University Hospital, and the presence of adenocarcinoma was confirmed by a pathologist. The primary cancer cells in the malignant pleural effusions were isolated and then cultured using a modified version of a previously reported protocol [14]. Media were replaced every 2–3 days, and cells were harvested after 10 days. Total RNA was extracted, and the miRNA expression level was determined using qRT-PCR. These patients underwent regular computed tomography at 12-week intervals to assess tumour response. Data on clinical characteristics and the durations of EGFR TKI treatment were also obtained from the medical records. The collection of human tissue samples and clinical data was approved by the Institutional Review Board of National Taiwan University Hospital.
RNA extraction and qRT-PCR for miRNAs and mRNA
Cell-derived total RNA was extracted using the TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) and reversely transcribed into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems) as previously described [15]. Quantitative PCR was performed using an ABI PRISM 7900 HT system using a standard protocol; RNU6B and TBP (TATA-binding protein) were used as an internal miRNA and mRNA control, respectively. Relative miRNA expression levels were calculated using the 2
Over-expression and silence of miR-200c-3p
For over-expressing miR-200c-3p or silencing miR-200c-3p, cells were transiently transfected with a miR-200c-3p mimic or miR-200c-3p inhibitor, respectively, using the Lipofectamine RNAiMax reagent (ThermoFisher Scientific). The miR-200c-3p specific-mimic (#MC11714) and miR-200c-3p inhibitor (#MH11714) were obtained from ThermoFisher Scientific.
Western blot analysis
Proteins were extracted from cells using an RIPA buffer (Cell Signaling Technology, CST). The extracted proteins were quantified, resolved in 7% SDS-polyacrylamide gel, and then transferred to a PVDF membrane. The membranes were incubated with the indicated primary antibodies (E-cadherin [BD Biosciences, #60181], N-cadherin [CST, #13116], zeb1 [CST, #3396], or
Migration assay
Transwells (BD Biosciences; Franklin Lakes, NJ, USA) were used for migration assay. Indicated cells were seeded into the upper chamber supplied with serum-free medium, and medium in the lower chamber supplied 5% FBS was used as chemoattractant. After 18 hours of incubation, cells remaining on the top of the insert were removed and then the inserts were fixed with 70% ethanol and stained with 0.5% crystal violet. Total migratory cells on the lower surface of the insert were counted at 200
Cytotoxicity and apoptosis assays
Apoptosis was evaluated by using an Annexin-V-FITC detection kit (BD Biosciences) according to the manufacturer’s instructions. Briefly, cells were stained with fluorescein isothiocyanate (FITC)-labelled Annexin-V and propidium iodide (PI) for 15 minutes in the dark, and analyses were performed using a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
Expression levels of miRNAs were dichotomized into 2 groups of ‘high’ and ‘low’ expression level. The Student’s
Identification and characterization of microRNAs (miRNAs). MiR-200c-3p and miR-203a-3p, which are negatively correlated with the mesenchymal-associated gene signature, are downregulated in cancer cells with acquired resistance to the epidermal growth factor receptor tyrosine kinase inhibitors gefitinib and erlotinib. (A) Flow chart of microRNA (miRNA) expression analysis using the TaqMan Human MiRNA PCR Array. (B) The heat map shows the expression correlation of Epithelial-to-mesenchymal transition (EMT)-related genes and miRNAs using The Cancer Genome Atlas database. Nine miRNAs are negatively correlated with mesenchymal-associated genes; whereas 27 miRNAs are positively correlated with mesenchymal-associated genes. (C) A dot plot showing the correlation between the expression of the downregulated microRNAs in gefitinib-resistant HCC827/gef cells and the EMT score. The plotted values are Pearson’s correlation coefficients. (D) Quantitative real-time PCR was conducted to measure the expression levels of miR-200c-3p and miR-203a-3p in the indicated cell lines. The expression of microRNAs was normalized to RNU6B expression. Quantitative real-time PCR data are presented as the mean 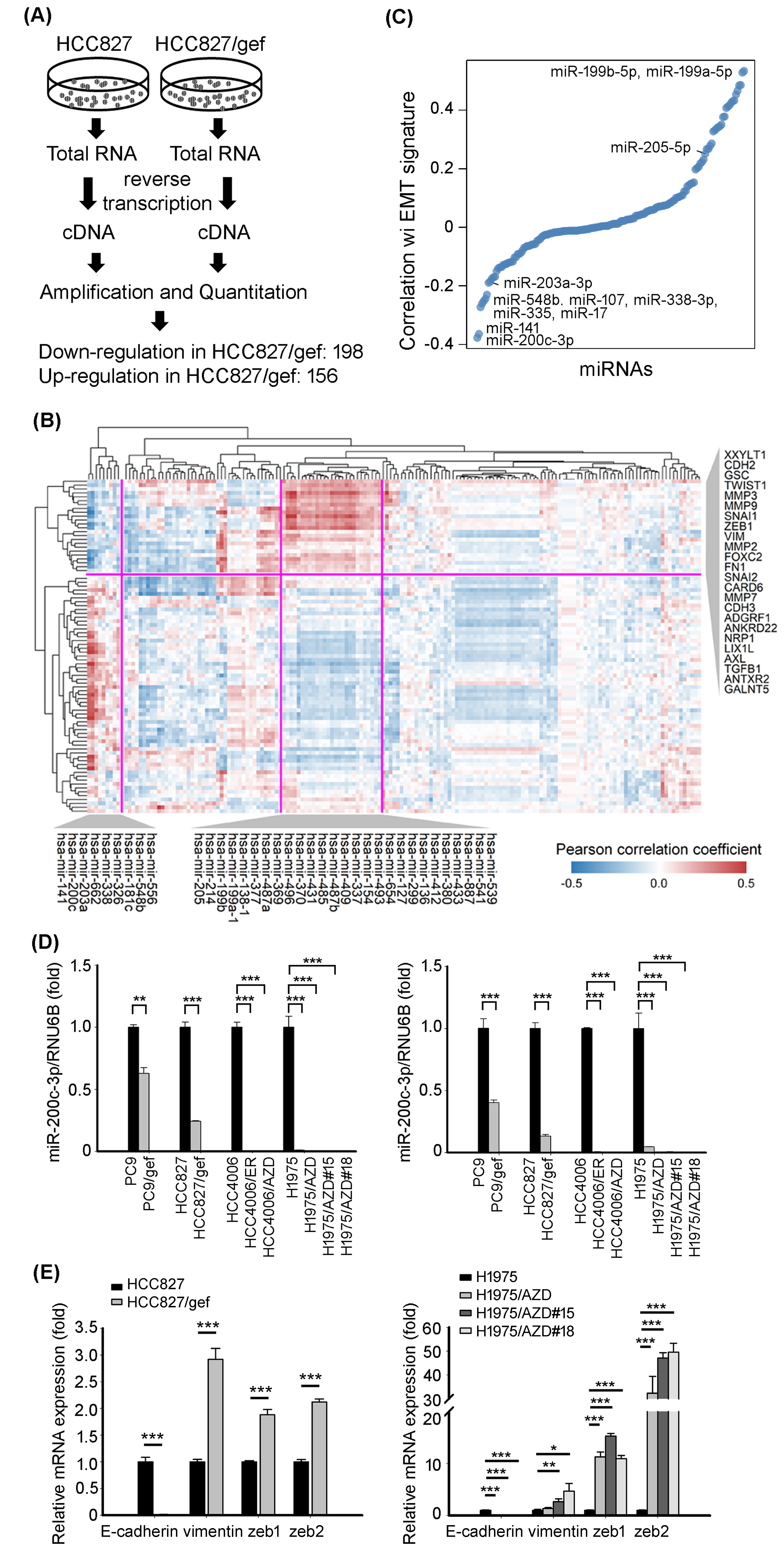
Clinicopathologic characteristics of lung adenocarcinoma patients with high vs. low miR-200c-3p expression
The cut-off point for high vs. low expression was the median value.
Clinicopathological characteristics of lung adenocarcinoma patients with high vs. low miR-203a-3p expression
The cut-off point for high vs. low expression was the median value.
MiR-200c-3p and MiR-203a-3p are potential regulators of EGFR TKI resistance
To determine the potential miRNAs involved in the mechanism of resistance to EGFR TKIs, we analyzed the expression profile of miRNAs in a pair of TKI-sensitive (HCC827) and TKI-resistant (HCC827/gef) cell lines to screen for the miRNAs that potentially regulate the EGFR TKI resistance process. Based on the expression ratio criteria of
The EGFR TKI-resistant cells (HCC827/gef, HCC4006/ER, HCC4006/AZD, and H1975/AZD) became spindle-shaped as compared with EGFR TKI-sensitive cells (HCC827, HCC4006, and H1975) (Fig. S1). We previously demonstrated that EMT were responsible for EGFR TKI resistance [9]. We further surveyed the matched mRNA and miRNA expression of lung adenocarcinoma specimens using public TCGA database to identify the EMT-associated miRNAs. An integrated analysis was conducted and revealed the correlation between EMT-related genes and miRNAs down-regulated in HCC827/gef cells (Fig. 1B). A cluster containing 9 miRNAs whose expression levels were negatively correlated with known mesenchymal markers, such as VIM, SNAI1, and Slug (SNAI2), were identified; in contrast, a cluster comprised of 27 identified miRNAs were positively correlated with these mesenchymal-associated genes (Fig. 1B). In addition, 6 (miR-200c-3p, miR-141, miR-548b, miR-338-3p, miR-335, and miR-203a-3p) out of 9 miRNAs negatively correlation with mesenchymal-associated genes were also negatively correlated with EMT signature (Fig. 1C). The two miRNAs, miR-200c-3p and miR-203a-3p, have the larger expression changes between TKI-resistant and -sensitive cells than the others and became the focus of this study.
The differential expression of miR-200c-3p and miR-203a-3p was verified by qRT-PCR assays in 4 pairs of EGFR TKI-sensitive (PC9, HCC827, HCC4006, and H1975) and EGFR TKI-resistant (PC9/gef, HCC827/ gef, HCC4006/ER, HCC4006/AZD, and H1975/AZD) cells. We found that the expression levels of both miR-200c-3p and miR-203a-3p were down-regulated in EGFR TKI-resistant cell lines (Fig. 1D). We also examined resistance-associated mutations including EGFR T790M, EGFR C797S, and MET amplification in those EGFR TKI-resistant cell lines. However, acquired resistance-associated EGFR mutations or MET amplification were not detected in EGFR TKI-resistant cell lines. We suggested that the change of miR-200c-3p was not associated with the acquisition of resistant mutations.
MiR-200c-3p and miR-203a-3p have been identified as a key element in driving EMT process [16, 17]. Further evaluation of EMT-associated genes was performed. Consistent with observation in lung adenocarcinoma specimens from TCGA database, lower miR-200c-3p- and miR-203a-3p-expressing cells are characterized of lower epithelial-associated gene (E-cadherin) expression and higher mesenchymal-associated gene (vimentin, zeb1, and zeb2) expressions (Fig. 1E). Furthermore, the miR-200c-3p level of TKI-resistant cells transfected with miR-200c-3p mimic is far more than the level in the TKI-sensitive cells, which resulting in phenotypic changes from mesenchymal-like into epithelial-like in TKI-resistant cells. By contrast, miR-200c-3p levels in miR-200c-3p inhibitor-transfected TKI sensitive cells are as low as they are in TKI-resistant cells, in which leading miR-200c-3p inhibitor-transfected TKI sensitive cells undergoe phenotype change from epithelial-like into mesenchymal-like shapes (Fig. S2). These results indicated that expression levels of miR-200c-3p was associated with the transition of phenotype and predicted EGFR TKI resistance.
The expression levels of miR-200c-3p and miR-203a-3p in clinical specimens and patient characteristics
We measured the expression of miR-200c-3p and miR-203a-3p in the surgical specimens of the first group; a total of 70 patients were included in this analysis. Tumor relapse was noted during the post-operative follow-up period, whereupon EGFR TKIs were initiated as the first-line systemic anti-cancer therapy. The second group (malignant pleural effusion) comprised 50 patient samples. These patients were all initially diagnosed with stage IV lung adenocarcinoma and received EGFR TKIs as first-line treatment. All these patients had common EGFR mutations, including L858R and exon 19 deletion. Most of the patients had a partial response to the EGFR TKI therapy initially and tumor progression developed later. The detailed clinical characteristics of these patients are shown in Supplementary Table S2. There were no differences in age, sex, disease stage, tumor status, lymph node metastasis, and type of EGFR mutations between the high vs. low expression groups for either of the miRNAs. The detailed results of the association between clinicopathological features and expression levels of miR-200c-3p and miR-203a-3p are shown in Tables 1 and 2, respectively.
Multivariate Cox analysis of risk factors for disease progression in patients with surgical specimens
Multivariate Cox analysis of risk factors for disease progression in patients with surgical specimens
CI, confidence interval.
Survival in patients from whom surgical samples were examined. Kaplan-Meier survival curves of (A) progression-free survival (PFS) and (B) overall survival (OS) after epidermal growth factor receptor tyrosine kinase inhibitor treatment in lung adenocarcinoma patients with miR-200c-3p high-expression (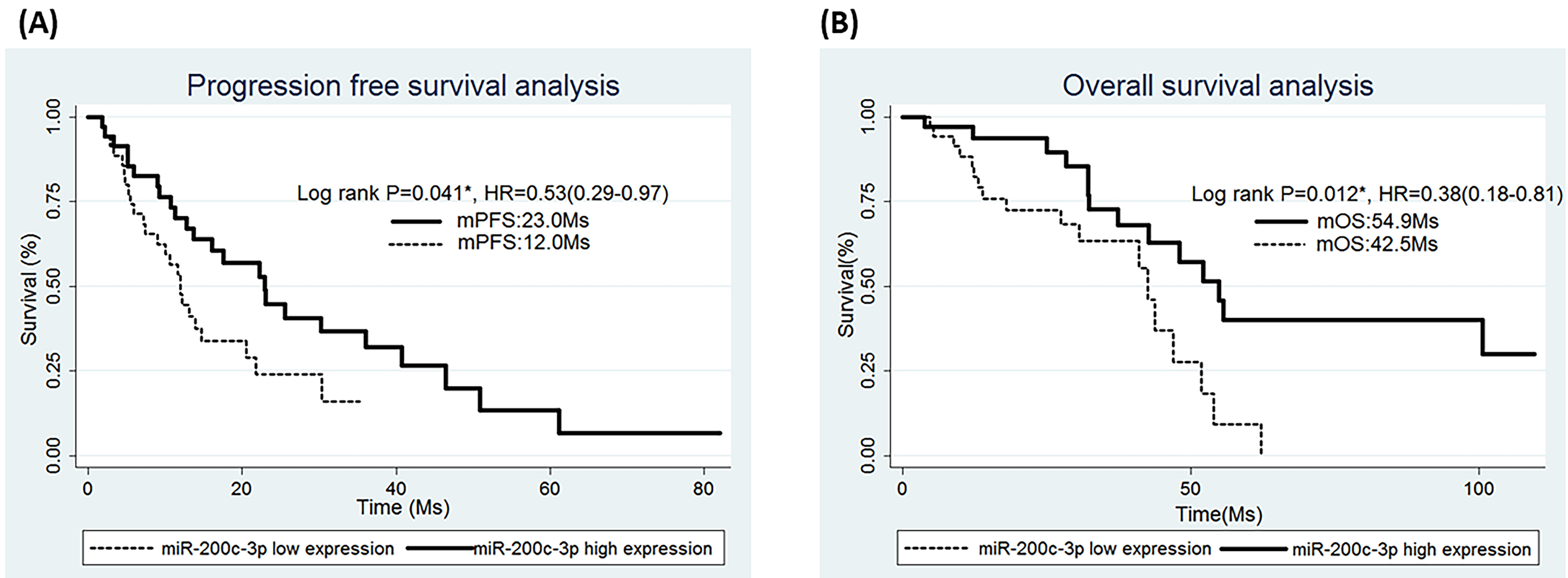
Survival in patients from whom pleural effusion samples were examined. Kaplan-Meier survival curves for (A) progression-free survival (PFS) after epidermal growth factor receptor tyrosine kinase inhibitor treatment in lung adenocarcinoma patients and (B) overall survival (OS) with miR-200c-3p high-expression (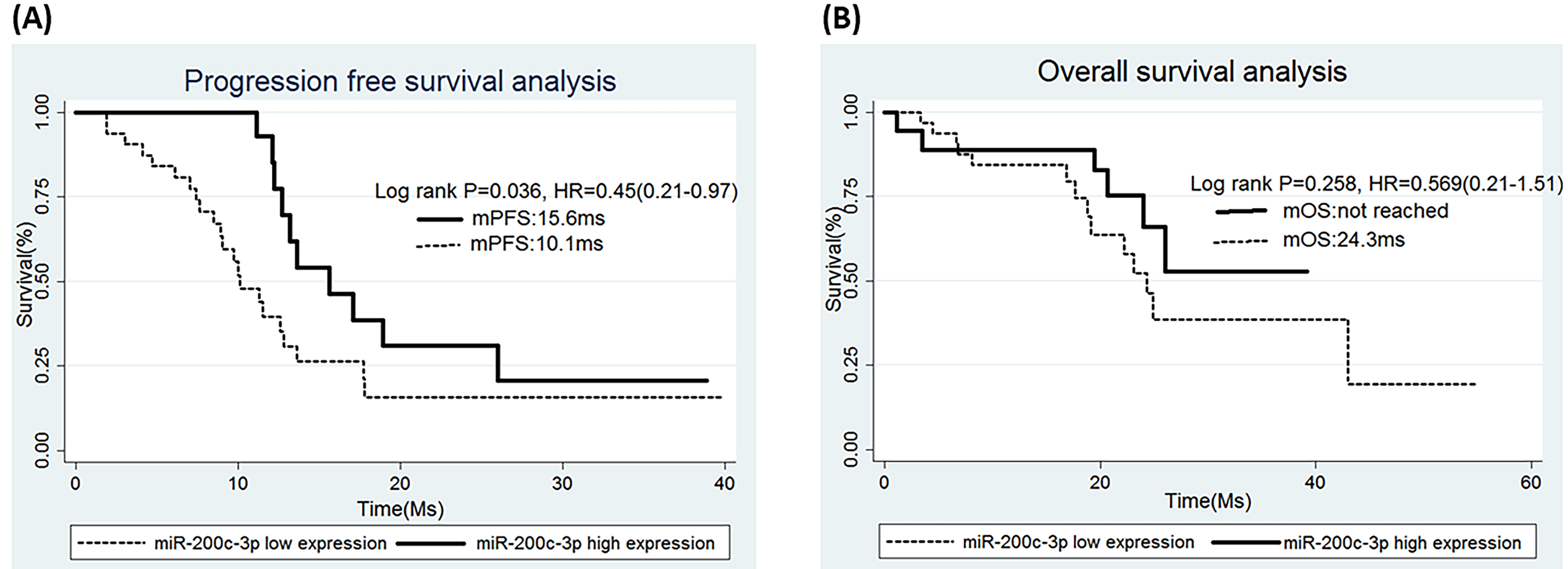
The PFS under EGFR TKIs in patients with high miR-200c-3p expression levels was significantly longer than in those with low expression levels (23.0 vs. 12.0 months; hazard ratio [HR]
High miR-200c-3p expression in malignant pleural effusions was associated with longer survival under EGFR TKI therapy
As with the patients in whom tumor samples were examined, high miR-200c-3p expression in malignant pleural effusions was associated with longer survival under EGFR TKIs based on the data from 50 patients in the second (pleural effusion) group. There were no differences in the clinical characteristics of patients with high vs. low miR-200c-3p expression (Supplementary Table S3). Longer PFS with EGFR TKI treatment was also observed in patients with high miR-200c-3p expression (15.6 vs. 10.1 months; HR
Multivariate Cox analysis of risk factors for disease progression in patients with malignant pleural effusions
Multivariate Cox analysis of risk factors for disease progression in patients with malignant pleural effusions
CI, confidence interval.
MiR-200c-3p expression tended to decrease after the tumor acquired resistance to EGFR-TKI therapy. There was a trend that the miR-200c expression level declined after developing resistance to EGFR-TKIs. It did not reach a statistical significance (paired student’s 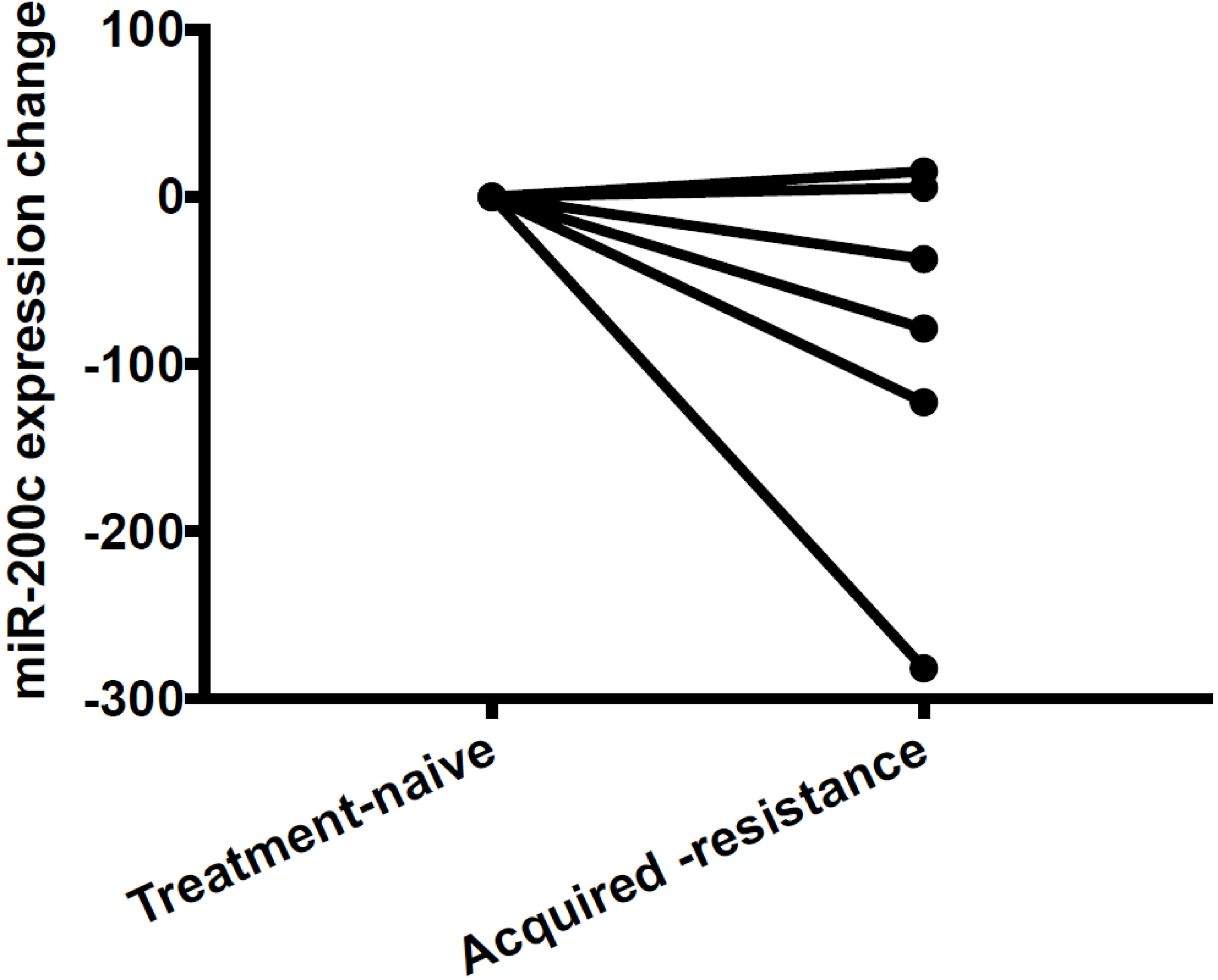
There were a total of 6 patients in our study cohort who had a pleural effusion sample collected both before EGFR TKI treatment and after EGFR TKI-resistance (Fig. 4) There was a trend that the miR-200c-3p expression level declined after developing resistance to EGFR TKIs (paired student’s
MiR-200c-3p suppressed lung cancer cell migration. (A) Quantitative real-time PCR was conducted to measure the expression levels of miR-200c-3p in the indicated cell lines. (B) Transwell migration assays were employed to detect cell migratory ability in H1975/AZD#18 and HCC4006/AZD cells after transfection miR-200c-3p mimic or miR-control mimic (miR-Ctrl). (C) The protein levels of E-cadherin, N-cadherin, and zeb1 were detected in miR-200c-3p-transfectant and miR-Ctrl-transfectant.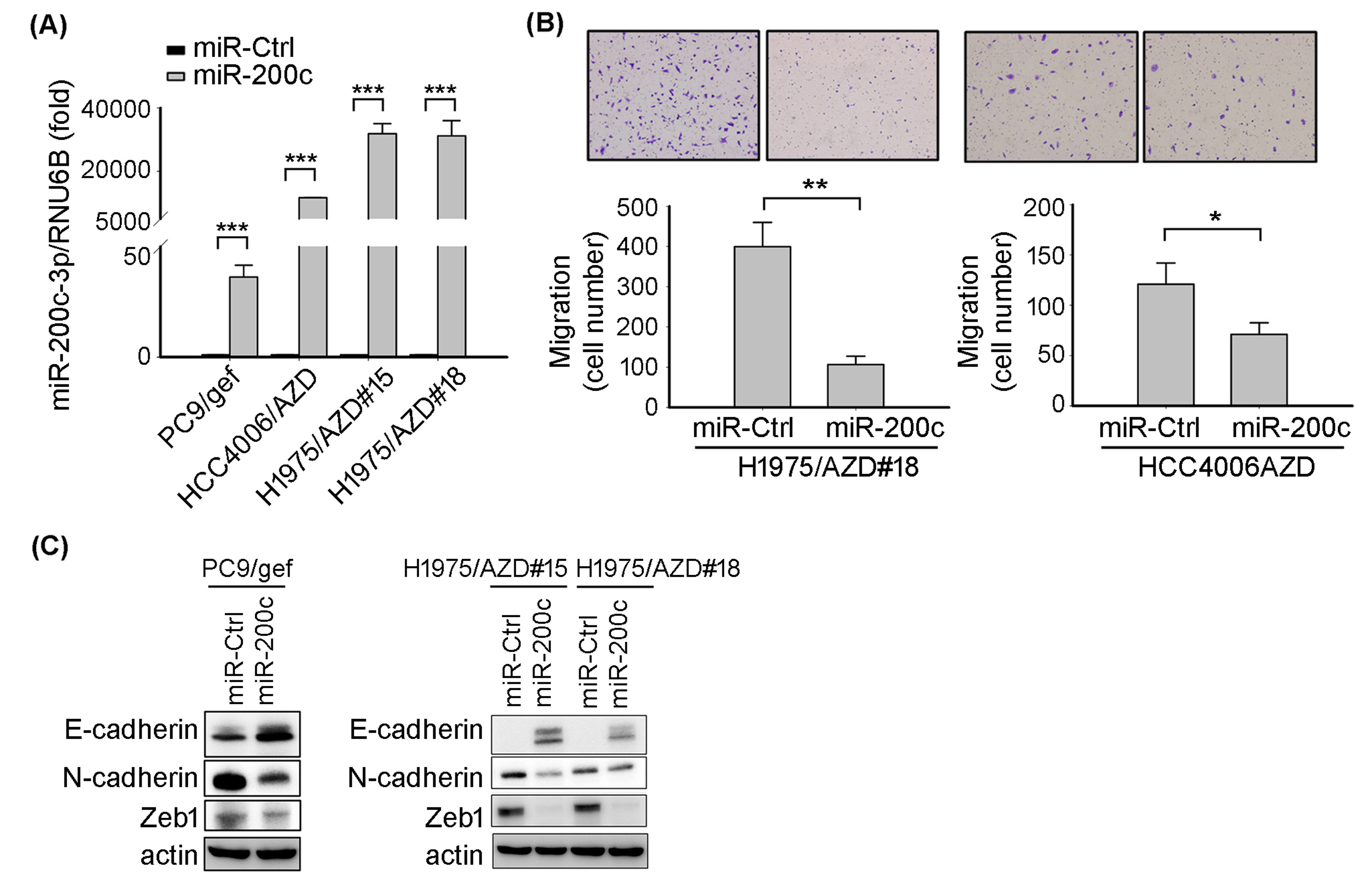
We performed transfection of miR-200c-3p mimic into EGFR TKI-resistant cancer cells to investigate whether EGFR TKI-resistant cancer cells undergo a miR-200c-3p-driven mesenchymal-epithelial transition change (Fig. 5A). The migratory ability of miR-200c-overexpressing transfectants was significantly inhibited as compared with miR-Ctrl-transfected controls (Fig. 5B). Consistent with the observation, miR-200c-3p decreased mesenchymal-associated proteins expression (N-cadherin and Zeb1) and increased epithelial-associated protein (E-cadherin) expression (Fig. 5C).
Overexpression of miR-200c-3p promotes EGFR TKI-resistant cancer cell death
We further examined the role of miR-200c-3p in EGFR TKI resistance. The miR-200c-3p mimic was transiently transfected into EGFR TKI-resistant H1975/AZD#15, H1975/AZD#18, and HCC827/gef cells. After transfection, miR-200c-3p-transfectant cancer cells and miR-Ctrl-transfectant were exposed to osimertinib, and then, we determined the percentage of apoptotic cells by calculating Annexin-V-positive cells. The results revealed that the percentage of apoptotic cells was significantly increased in miR-200c-3p-transfectant cancer cells compared to in miR-Ctrl-transfectant cancer cells (Fig. 6A and Fig. S4). We also demonstrated that the cleavage of caspase-3 and PARP were increased in miR-200c-3p-transfectant cancer cells (Fig. 6B). To further investigate whether suppression of miR-200c-3p contributes to EGFR TKI resistance, we silence miR-200c-3p with miR-200c-3p inhibitor in EGFR TKI-sensitive HCC4006 cancer cells (Fig. 6C). Transfection of miR-200c-3p inhibitor decreased gefitinib or osimertinib-induced cell death compared to that in miR-scramble transfectant cancer cells (Fig. 6D and Fig. S5). These results indicate that loss of miR-200c-3p results in EGFR TKI resistance.
MiR-200c-3p promoted cancer cell apoptosis. (A) After tranfections of miR-Ctrl (control) or miR-200c-3p mimic for 24 hours, indicated cells were treated with vehicle or osimertinib for 24 hours. Ratio of cell death was measured by counting AnnexinV-positive cells, and the percentage of cell death was presented as the mean 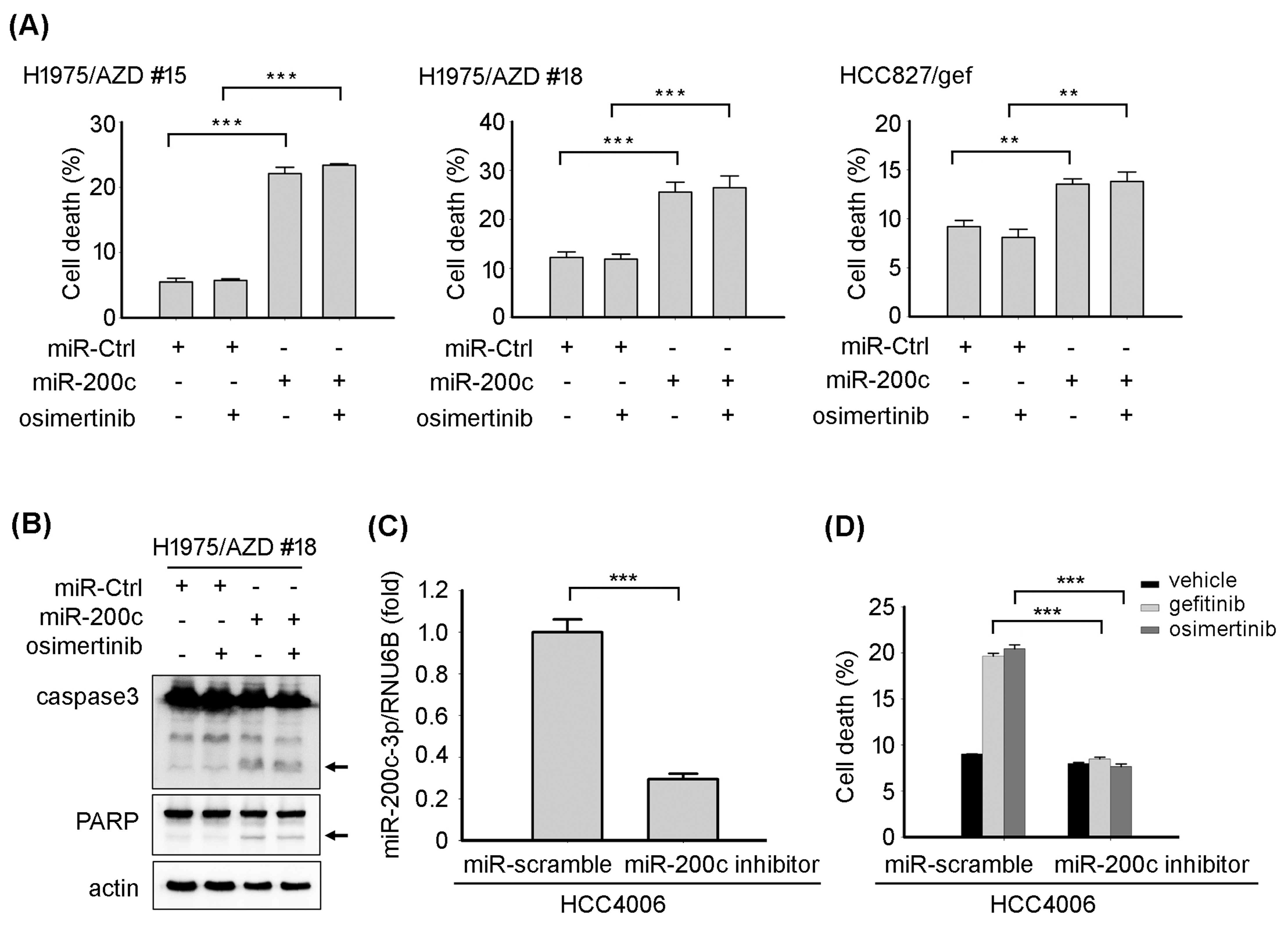
Our study demonstrated a linkage from the upstream miR200c expression to downstream EMT and phenotypic change, which finally lead to acquired resistance of EGFR TKI. We identified potential miRNAs associated with EGFR TKI resistance through bioinformatics analysis with TCGA database and miRNA expression profiles of lung cancer cell lines. We confirmed the role of miR-200c both in vitro cell lines and in vivo clinical specimens. Overexpression and silencing of miR-200c-3p in EGFR mutant lung cancer cells proved miR-200c-3p could inhibit cell migratory ability and restore EGFR-TKI sensitivity. Then, we explored the relationship between miR-200c-3p and EGFR TKI response and resistance by using clinical specimens from 2 patient groups. We provided new evidence for the potential clinical implication of miR-200c in lung cancer patient under EGFR-TKI treatment.
The miR-200 family comprises 5 members (miR-200a-3p, -200b-3p, -200c-3p, -141-3p, and -429) that are expressed as two separate genomic clusters, miR-200b-200a-429 and miR-200c-141, which are located on human chromosomes 1 and 12, respectively [18]. Of all these categories, miR-200c has been shown to play a central role in regulating EMT by directly targeting the transcription factors ZEB1 and ZEB2 and promoting the upregulation of E-cadherin [16, 19, 20, 21]. EMT is critical for tumor cell invasion and metastasis, and its occurrence correlates with poor patient prognosis [18, 22].
There are several studies focusing on the association of the expression of miR-200c and clinical outcomes in patients with NSCLC. Although comparisons of miR-200c in tumor tissues and their adjacent normal lung tissue have been inconsistent [23, 24, 25], lower miR-200c expression in patients with NSCLC was found to be significantly associated with a poor grade of differentiation and a higher degree of spreading to the lymph nodes [26]. Regarding other studies about the microRNA200c’s role as a prognostic marker for NSCLC, previous studies demonstrated that high expression of tumor miR-200c was associated with shorter OS in patients with NSCLC [23, 24, 25, 27]. This finding was not contrary to our results since our study focused on the miR-200c’ function relating to EGFR TKI’s therapeutic efficacy. On the other hand, longer OS and PFS under EGFR TKI therapy had been reported in EGFR wild-type lung cancer patients with high level of miR-200c expression as compared with low miR-200c expression subgroup [28]. However, the population in this study was different from our study which focused on lung cancer harboring EGFR mutation. Few studies provided the OS report clearly under EGFR TKI therapy based on miR-200c expression level. These inconsistent results may also be explained by a phenotypic plasticity postulated to transiently exist during EMT-related processes [29].
Since miR-200c can restore the expression of E-cadherin and inhibit EMT, it is more likely a tumor suppressor. Such a biological and pathological function of miR-200c was compatible with our observation of longer PFS of EGFR TKI treatment observed in patients whose tumors had higher levels of this miRNA. There have been several studies describing the impact of EMT on the sensitivity and resistance of lung cancer to EGFR inhibitors such as erlotinib and gefitinib [30, 31, 32, 33]. Therefore, inhibition of EMT may also be a possible explanation of the improved survival observed in patients expressing high miR-200c levels in our study. Our studies also demonstrated that overexpression miR-200c-3p in cells with acquired TKI resistance cells could inhibit migration, gain of the epithelial marker (E-cadherin) and promote cell death. Otherwise, miR-200c-3p silencing results in EGFR TKI resistance. These results provided further information about the associated between miR-200c-3p and EGFR TKI sensitivity connecting by the EMT process.
Some recent studies have also explored the relationship between miR-200c and sensitivity/resistance to EGFR TKIs in NSCLC. Li et al. demonstrated that miR-200c was downregulated in NSCLC cell lines that are resistant to EGFR TKIs, and that ectopic expression of miR-200c resulted in partial restoration of gefitinib sensitivity [28]. Zhou et al. reported that miR-200c enhanced sensitivity to gefitinib and significantly increased apoptosis through the PI3K/Akt signalling pathway via the targeting of ZEB1 in a mouse xenograft model [34]. Depletion of miR-200c may be associated with EMT as well as histone modification and may contribute to a decrease in the sensitivity to gefitinib [35]. All these findings support our conclusions of an association between PFS of patients with EGFR TKI treatment and miR-200c expression.
There were several limitations in our study. The surgical specimens used for measuring miR-200c-3p expression in the first group represented early-stage lung cancer; however, EMT and the potential effects of miR-200c-3p may be more pronounced in advanced-stage lung cancer. Surgical tumor specimens provide only snapshot in time, there is a possibility that miR-200c-3p expression could vary during the course of tumor progression. We addressed this by repeating the same analysis in the second group using malignant pleural effusions from patients with stage IV lung adenocarcinoma and obtained similar results. Furthermore, We only included tumors carrying the common mutations L858R and exon 19 deletion in our analysis for simplicity, since uncommon mutations can be regarded as confounding factors that introduce heterogeneity that in turn may influence the varying responses to EGFR inhibitors [36, 37]. A significant OS benefit of EGFR TKI treatment in patients expressing higher tumor miR-200c-3p levels was only realized in the surgical specimen group, not in the malignant pleural effusion group. Since an analysis of OS requires larger patient numbers and longer follow-up periods, and may also be confounded by subsequent therapies, differences in this parameter may be more difficult to detect than PFS. Nevertheless, the consistency in our findings as related to PFS of EGFR TKI treatment in both groups supports the linkage between miR-200c-3p and the therapeutic effects of EGFR TKIs. Finally, although we demonstrated the associated of the miR-200c-3p and TKI sensitivity mediated through EMT, we did not investigate the underlying molecular mechanisms thoroughly. The underlying biological mechanisms warrant further exploration.
Conclusion
Patients with EGFR-mutant lung adenocarcinomas that express high miR-200c-3p experience longer PFS under EGFR TKI therapy. The mechanism may involve the suppression of EMT. MiR-200c-3p may therefore be a potential predictor for EGFR TKI efficacy and play a therapeutic role in delaying EGFR TKI resistance.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-191119.
sj-doc-1-cbm-10.3233_CBM-191119.doc - Supplemental material
Supplemental material, sj-doc-1-cbm-10.3233_CBM-191119.doc
Footnotes
Acknowledgments
We would like to acknowledge the services provided by the Department of Medical Research at National Taiwan University Hospital. This project was supported by National Taiwan University Hospital Yunlin branch grant NTUHYL105.N010 (H-Y. Wang) and National Taiwan University Hospital grant 107-S3788 and 107-N3995.
Conflict of interest
Jin-Yuan Shih has received speaking honoraria from AstraZeneca, Roche, Boehringer Ingelheim, Eli Lilly, Pfizer, Novartis, Merck Sharp & Dohme, Ono Pharmaceutical, AbbVie, Chugai, and Bristol-Myers Squibb. The other authors declare no conflict of interest.