
Abstract
BACKGROUND:
Glioma is considered to be one of the most common and lethal malignant brain tumors, accounting for 40% to 50% of brain tumors. Long non-coding RNAs (lncRNAs) have been widely proved to play an irreplaceable role in the tumorigenesis and progression. Nevertheless, the role of LINC00467 in glioblastoma remained unclear.
AIM:
The current study was aimed to explore the functional mechanism of LINC00467 in glioblastoma.
METHODS:
The expression of LINC00467/miR-339-3p/IP6K2 glioblastoma tissues and cells was evaluated by RT-qPCR. The protein expression of genes (cleaved PARP, PARP, cleaved caspase 3, caspase 3, Bax, Bcl-2 and IP6K2) was measured by western blot assay. Then role of LINC00467 was demonstrated by EdU, colony formation, flow cytometry and TUNEL assays. The relationship between miR-339-3p and LINC00467/IP6K2 was validated by RNA pull down and luciferase reporter assays.
RESULTS:
The expression of LINC00467 was upregulated in glioblastoma tissues and cells. LINC00467 knockdown suppressed cell proliferation but activated cell apoptosis. Further, LINC00467 high expression was associated with shorter overall survival rate in glioblastoma patients. Further, LINC00467 could bind with miR-339-3p, and IP6K2 was targeted by miR-339-3p. IP6K2 expression was regulated by LINC00467/miR-339-3p in a ceRNA pattern. Moreover, LINC00467 could regulate the development of glioblastoma via miR-339-3p/IP6K2 axis.
CONCLUSIONS:
LINC00467 knockdown repressed cell proliferation but stimulated cell apoptosis in glioblastoma via miR-339-3p/IP6K2 axis, which may enlighten to find a novel therapeutic tactic for glioblastoma patients.
Introduction
Gliomas, referring to the tumors derived from neuroepitheles, were the most common intracranial malignancies, which account for 40% to 50% of brain tumors [1]. Surgery section, radiotherapy, chemotherapy and immunotherapy were proved to be effective for brain lower grade glioma [2, 3]. However, glioblastoma, classified into advanced stage of glioma tumor, attributed to a dismal prognosis of glioblastoma patient with low overall survival rate [4, 5]. Therefore, better understanding and further exploration of the molecular mechanism related to tumorigenesis and progression was critical for the treatment of glioblastoma.
Long non-coding RNAs (lncRNAs) belong to a category of non-coding RNAs (ncRNAs) with longer than 200 nucleotides and lack the ability to encode proteins [6, 7]. Further, the dysregulation of lncRNAs was generally associated with the initiation or development of tumors, including glioblastoma [8]. For illustration, lncRNA GAS5 downregulation increased GDF15 expression to promote ovarian cancer cell proliferation [9]; silencing of lncRNA RHPN1-AS1 suppressed the EMT (epithelial-to-mesenchymal transition) process to inhibit breast cancer progression [10]; lncRNA NEAT1 regulated by the EGFR pathway facilitates glioblastoma progression via Wnt/
Moreover, the common molecular regulatory mechanism of lncRNAs, competitive endogenous RNA (ceRNA), in tumors has been intensively reported. For example, lncRNA RP4 serves as a ceRNA via sponging miR-7-5p in colorectal cancer [15] and lncRNA NEAT1 modulates the progression of papillary thyroid cancer via regulation of miR-129-5p/KLK7 expression [16]. About glioblastoma, there are also some reports about ceRNA regulation mechanism, like lncRNA AC016405.3 exerted tumor-suppressor function through regulating TET2 by sponging miR-19a-5p in glioblastoma [17], lncRNA LINC01446 contributes to glioblastoma development by targeting miR-489-3p/TPT1 axis [18] and lncRNA AC003092.1 activates temozolomide chemosensitivity by the modulation of miR-195/TFPI-2 signaling in glioblastoma [19]. In addition, lncRNA could regulate gene expression in several ways including mRNA splicing, chromatin modification and post-transcriptional modulation [20, 21, 22]. In the meantime, the ceRNA regulation pattern of LINC00467 in cancers has been illustrated of lately, such as colorectal cancer [23], lung adenocarcinoma [24] and cervical cancer [25]. . Moreover, proliferation is the common cellular activity in glioblastoma [26, 27]. Cell proliferation and apoptosis became our researching focus in glioblastoma. Accordingly, we aimed to confirm whether LINC00467 regulated the cell proliferation and apoptosis of glioblastoma via ceRNA mechanism.
In present research, we elucidated that LINC00467 knockdown suppressed cell proliferation but activated cell apoptosis in glioblastoma via miR-339-3p/IP6K2 axis, which might inspire people to find out a novel therapeutic marker for glioblastoma treatment.
Materials and methods
Tissue samples
The glioblastoma samples (
Cell lines
The glioblastoma cell lines (U87, U251, A172 and U373) and normal human astrocytes (NHA) were bought from the Chinese Academy of Science Cell Bank (Shanghai, China). Cells were cultivated in the Dulbecco’s modified Eagle medium (DMEM, Gibco, New York, NY, USA) with 10% fetal bovine serum (FBS). The cell lines were incubated in a humid atmosphere at 37
Cell transfection
The short hairpin RNA (shRNA) targeting LINC00467 or IP6K2 (sh-LINC00467#1/2 or sh-IP6K2#1/2) with negative control (sh-NC) was applied to knockdown LINC00467 or IP6K2 expression. MiR-339-3p mimics/inhibitor with negative control (NC mimics/inhibitor) was used to overexpress/knockdown miR-339-3p. The above-mentioned vectors were purchased from GenePharma (Shanghai, China) and transfected into A172 and U373 cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) under the manufacturer’s instruction.
Real-time reverse-transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted from glioblastoma tissues or cells by using TRIzol solution (Invitrogen). Reverse transcription was performed in PrimeScript™RT Master Mix (TaKaRa, Dalian, China). For miRNA, the reverse transcription of miRNAs was carried out with TIANScript M-MLV (Tiangen, Beijing, China). RT-qPCR was operated on a LightCycler 480 instrument (Roche, Basel, Switzerland) using SYBR Premix Ex Taq II (TaKaRa). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U2 served as the internal control. The relative levels of genes were calculated by using the 2
5-ethynyl-2’-deoxyuridine (EdU) assay
The proliferation abilities of A172 and U373 cells transfected with sh-LINC00467#1 or sh-NC were evaluated by EdU assay (Ribo, Guangzhou, China). Briefly, the EdU solution (Invitrogen) was added to the culture medium (1000:1). The cells in proliferating phase were tabbed with EdU for 2 hours. Subsequently, the cells were purified with 0.5 g/mL of PBS for three times. Afterwards, DAPI (Invitrogen) nuclei counterstained the purified cells for 10 minutes at normal temperature in the dark place. The DAPI-marked cells were then rinsed with PBS for three times. Finally, the ratio of the fluorescent positive cells to total cells was calculated as proliferation rate.
Colony formation assay
A172 and U373 cells (totally 500) were seeded in a six-well plate and cultured in a DMEM with 10% FBS. The medium would be changed every 3 days. The cells were fixed with methanol and stained with 0.1% crystal violet (Sigma, San Francisco, CA, USA) after 2 weeks. The number of colonies was calculated.
Terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) assay
According to the protocol of TUNEL assay, A172 and U373 cell apoptosis was detected in the In Situ Cell Death Detection kit (Roche). Apoptosis was evaluated by counting the positive cells as well as the total number of cells at five random fields. Cell nuclei were positive if they were labelled with FITC (green), whereas 4, 6-diamidino-2-phenylindole (DAPI) staining indicated the cell nucleus under the florescence microscope. The positive staining cells were counted using image-Pro Plus 6.0 software.
Cell apoptosis analysis
Transfected cells were collected and resuspended with phosphate-buffered saline (PBS). Afterward, the density of cells was modulated to 1
Nuclear-cytoplasmic fractionation
The location of LINC00467 in cytoplasm or nucleus was segregated with the application of a PARIS kit (Life Technologies, MA, USA). Briefly, A172 and U373 cells were collected and lysed on ice. After centrifugation, the supernatant was harvested. GAPDH and U2 were utilized as the cytoplasm and nucleus controls, respectively. The extracted RNAs were tested by RT-qPCR.
Fluorescence in situ hybridization (FISH)
Subcellular localization of LINC00467 was determined utilizing a FISH Kit (Roche, Basel, Switzerland). Transfected A172 and U373 cells were added with paraformaldehyde for fixation. Subsequently, a LINC00467 probe (Sigma-Aldrich) hybridization solution incubated in digoxigenin was added into plate. An antagonistic LINC00467 probe was produced and deemed as NC. Hoechst (Sigma-Aldrich) stained nucleus for 10 min. Last, a laser confocal scanning microscopy (Olympus) was used for obtaining the fluorescence images.
Western blot
A172 and U373 cells were lysed with the help of RIPA lysis buffer (Beyotime Biotechnology, China). Total protein extracts were separated by 10% SDS-PAGE and immediately transferred onto PVDF membranes (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Afterwards the membranes were incubated with primary antibodies at 4
Luciferase reporter assay
LINC00467-WT (or IP6K2-WT) and LINC00467-Mut (or IP6K2-Mut) were constructed into pmirGLO plasmids (Promega, Madison, USA). The constructed plasmids were co-transfected with miR-339-3p mimics or NC mimics into A127 and U373 cells for 48 hours. Luciferase reporter assay system (Promega, Madison WI, USA) was adopted to examine the relative luciferase activities.
RNA pull down
MiR-339-3p-WT and miR-339-3p-Mut were transcribed employing Transcript Aid T7 High Yield Transcription Kit (ThermoFisher Scientific, USA). Biotinylated miR-339-3p-Wt, miR-339-3p-Mut and negative control (Bio-NC) were co-cultivated with cell lysate (A127 and U373) at 4
Statistical analysis
All data were presented as the means
Result
LINC00467 knockdown inhibited cell proliferation but encouraged cell apoptosis in glioblastoma
Recently, LINC00467 has been reported to exacerbate lung adenocarcinoma progression [13, 14], which aroused our interest. To figure out the role of LINC00467 in glioblastoma, a series of experiments were implemented. As shown in Fig. 1A, data from GEPIA (
LINC00467 knockdown inhibited cell proliferation but encouraged cell apoptosis in glioblastoma. (A) Data from GEPIA showed the up-regulation of LINC00467 in GBM tissues. (B and C) RT-qPCR assay tested LINC00467 levels in glioblastoma tissues (or cells) and normal tissues (or cells). (D) The knockdown efficacy of LINC00467 in A172 and U373 cells was evaluated by RT-qPCR assay. (E and F) The proliferative ability of sh-LINC00467#1 transfected A172 and U373 cells was assessed by EdU and colony formation assays. (G and H) Flow cytometry and TUNEL assays were operated to measure apoptosis in sh-LINC00467 transfected A172 and U373 cells. (I) The expression of apoptosis-related proteins (cleaved PARP, cleaved caspase 3 and Bax) was evaluated by western blot assay when downregulating LINC00467 in A172 and U373 cells. GAPDH served as internal control. 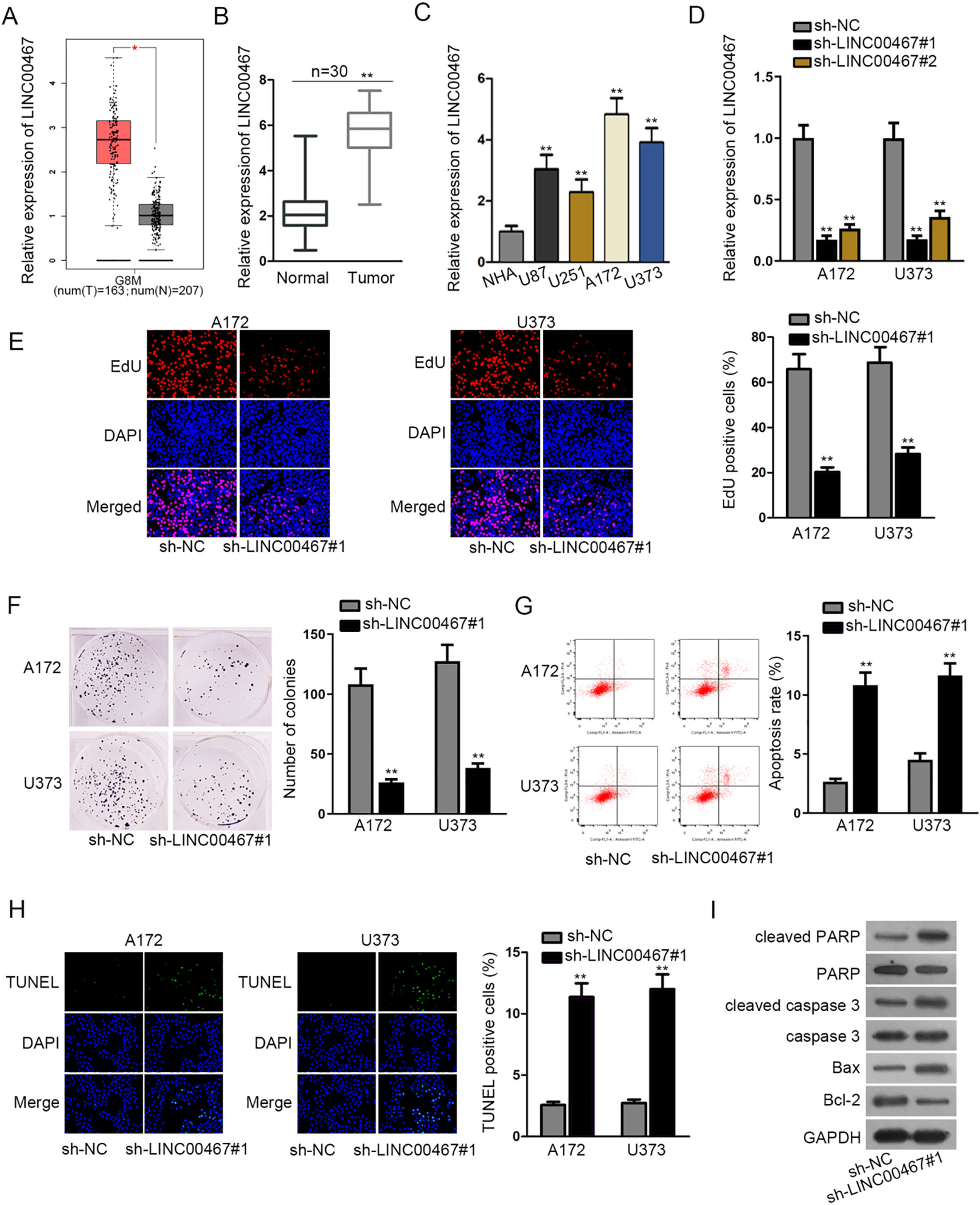
LINC00467 bound with miR-339-3p in glioblastoma. (A and B) Nuclear-cytoplasmic fractionation and FISH assays were used to locate LINC00467. (C) The expression of 7 miRNAs that may bind with LINC00467was examined by RT-qPCR assay in glioblastoma tissues and the matched normal tissues. Pearson correlation analysis demonstrated the correlation between miR-339-3p and LINC00467 in tissue samples (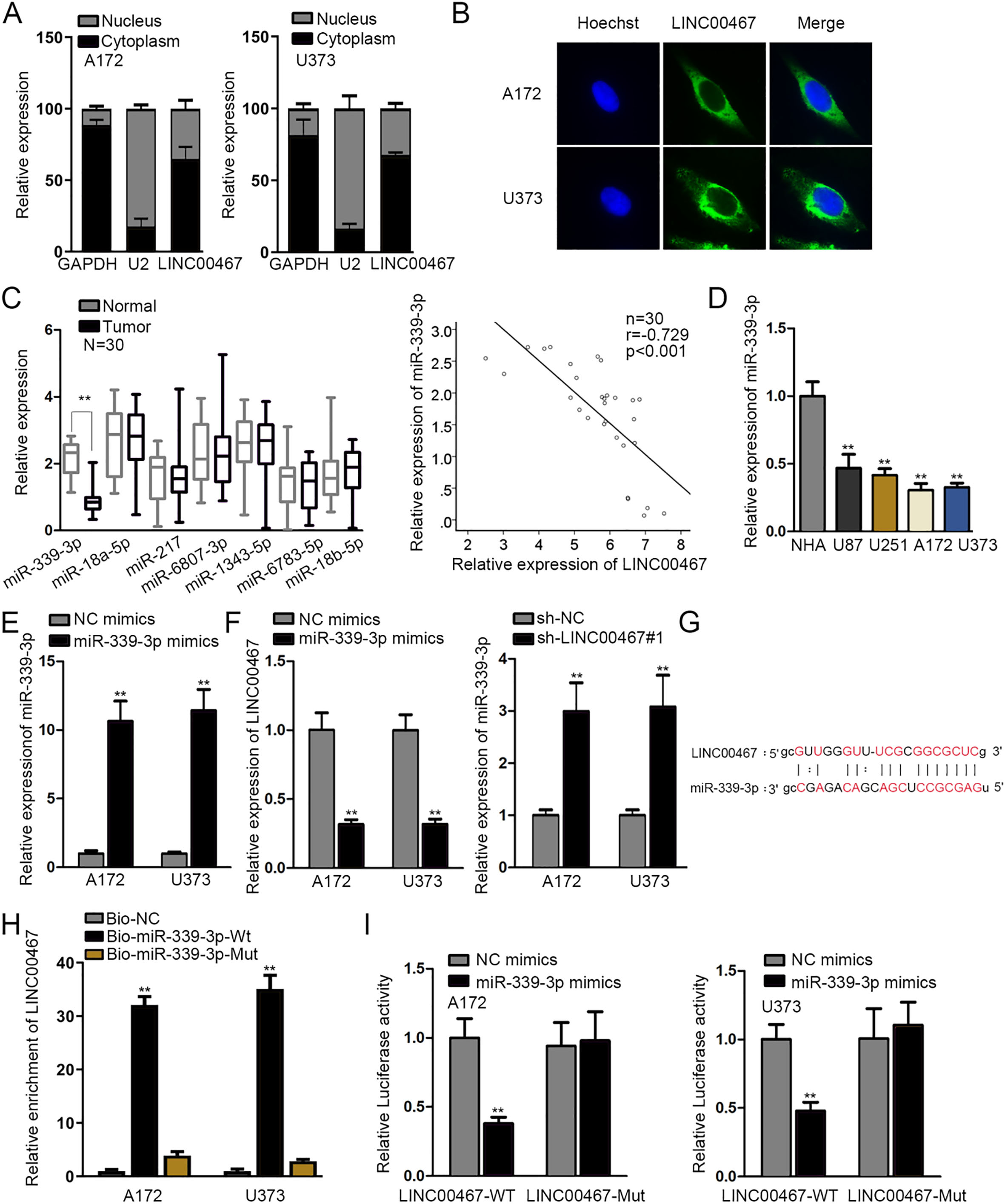
To confirm whether LINC00467 exerted regulatory function in glioblastoma through modulating miRNA via serving as ceRNA, firstly, nuclear-cytoplasmic fractionation and FISH assays detected the nuclear and cytoplasmic distribution of LINC00467. We found that LINC00467 mainly located in cytoplasm of A172 and U373 cells, suggesting that LINC00467 could function as a ceRNA to regulate gene expression at post-transcriptional level (Fig. 2A and B). Then, 7 miRNAs harboring the binding site with LINC00467 were screened out (conditions: medium stringency of CLIP Data; 2 cancer types of Pan-Cancer) via starBase (
IP6K2 was targeted by miR-339-3p in glioblastoma. (A) Venn diagram showed that IP6K2 possessed the binding ability with miR-339-3p by miRmap, microT and pictar databases. (B and C) The expression of IP6K2 in tumor tissues, tumor cells and corresponding matched groups was detected by RT-qPCR assay. Pearson correlation analysis demonstrated the correlation between IP6K2 and miR-339-3p/LINC00467 in tissue samples (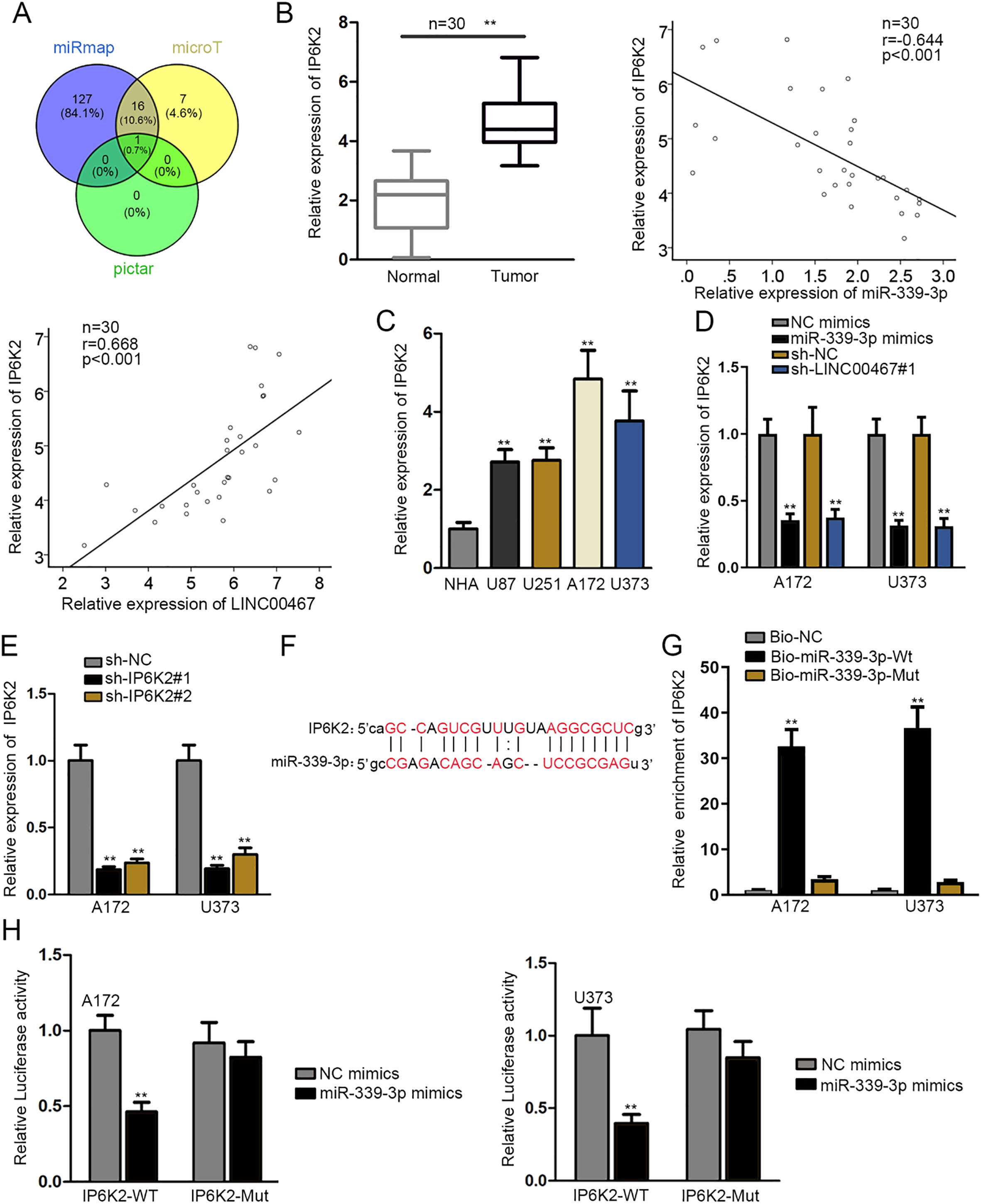
To complete the ceRNA regulation mechanism of LINC00467, seeking the target gene of miR-339-3p was necessary, and bioinformatics analysis was applied. As shown in Venn diagram, Inositol hexakisphosphate kinase 2 (IP6K2) was screened out (Fig. 3A). To confirm our hypothesis that IP6K2 acted the target gene of miR-339-3p, firstly, RT-qPCR assay was operated and the result demonstrated that IP6K2 was upregulated in glioblastoma tissues and cells compared with normal tissues and cells. In addition, IP6K2 was negatively correlated with miR-339-3p, but positively correlated with LINC00467 in tissue samples (Fig. 3B-C). Moreover, the regulation function of LINC00467 and miR-339-3p on IP6K2 was investigated by RT-qPCR. And results showed that miR-339-3p overexpression or LINC00467 depletion decreased the expression of IP6K2 (Fig. 3D). Then, we detected that IP6K2 level plummeted after the transfection of sh-IP6K2#1 or sh-IP6K2#2, and the former was picked for following exploration because of its better knockdown efficacy (Fig. 3E). In addition, both LINC00467 and miR-339-3p displayed no differential expression in the context of IP6K2 knockdown compare with NC group (Supplementary Fig. 2C). Subsequently, the binding sequences between miR-339-3p and IP6K2 were predicted by starBase (Fig. 3F). RNA pull down assay uncovered that IP6K2 was enriched in Bio-miR-339-3p-Wt of A172 and U373 cells, implying the binding possibility between miR-339-3p and IP6K2 in glioblastoma cells (Fig. 3G). Eventually, data from luciferase reporter assay manifested that the luciferase activity of pmirGLO-IP6K2-WT was decreased by miR-339-3p mimics whilst pmirGLO-IP6K2-Mut displayed no subtle alteration, validating directly the binding relation between miR-339-3p and IP6K2 (Fig. 3H). To sum up, miR-339-3p targeted and inhibited IP6K2 negatively, and LINC00467 interacted with miR-339-3p to upregulate the expression of IP6K2 in glioblastoma.
LINC00467 boosted glioblastoma development by upregulating IP6K2 or inhibiting miR-339-3p
Rescue assays were adopted to prove whether LINC00467/miR-339-3p/IP6K2 axis boosted glioblastoma development. At first, IP6K2 overexpression remedied the inhibitory effect of LINC00467 suppression on both mRNA and protein expression of IP6K2 (Fig. 4A and B). Besides, the satisfactory knockdown efficiency of miR-339-3p was achieved by miR-339-3p inhibitor (Supplementary Fig. 2D). Furthermore, EdU and colony formation assays measured that the inhibition of cell proliferation resulting from LINC00467 knockdown was restored by transfection of pcDNA3.1/IP6K2 or miR-339-3p inhibitor (Fig. 4C and D; Supplementary Fig. 2E and F). Last but not least, flow cytometry and TUNEL assays revealed that the promoting impact on cell apoptosis caused by LINC00467 deficiency was abrogated by IP6K2 amplification or miR-339-3p downregulation (Fig. 4E and F; Supplementary Fig. 2G and H). All in all, LINC00467 boosted glioblastoma development by upregulating IP6K2 or inhibiting miR-339-3p.
LINC00467 boosted glioblastoma development by enhancing upregulating IP6K2 or inhibiting miR-339-3p. (A and B) The mRNA and protein levels of IP6K2 were respectively detected by RT-qPCR and western blot assays. (C and D) Cell proliferation in differently transfected groups was examined by EdU and colony formation assays. (E and F) The measurement of cell apoptosis under diverse transfecting conditions was carried out in flow cytometry and TUNEL assays. 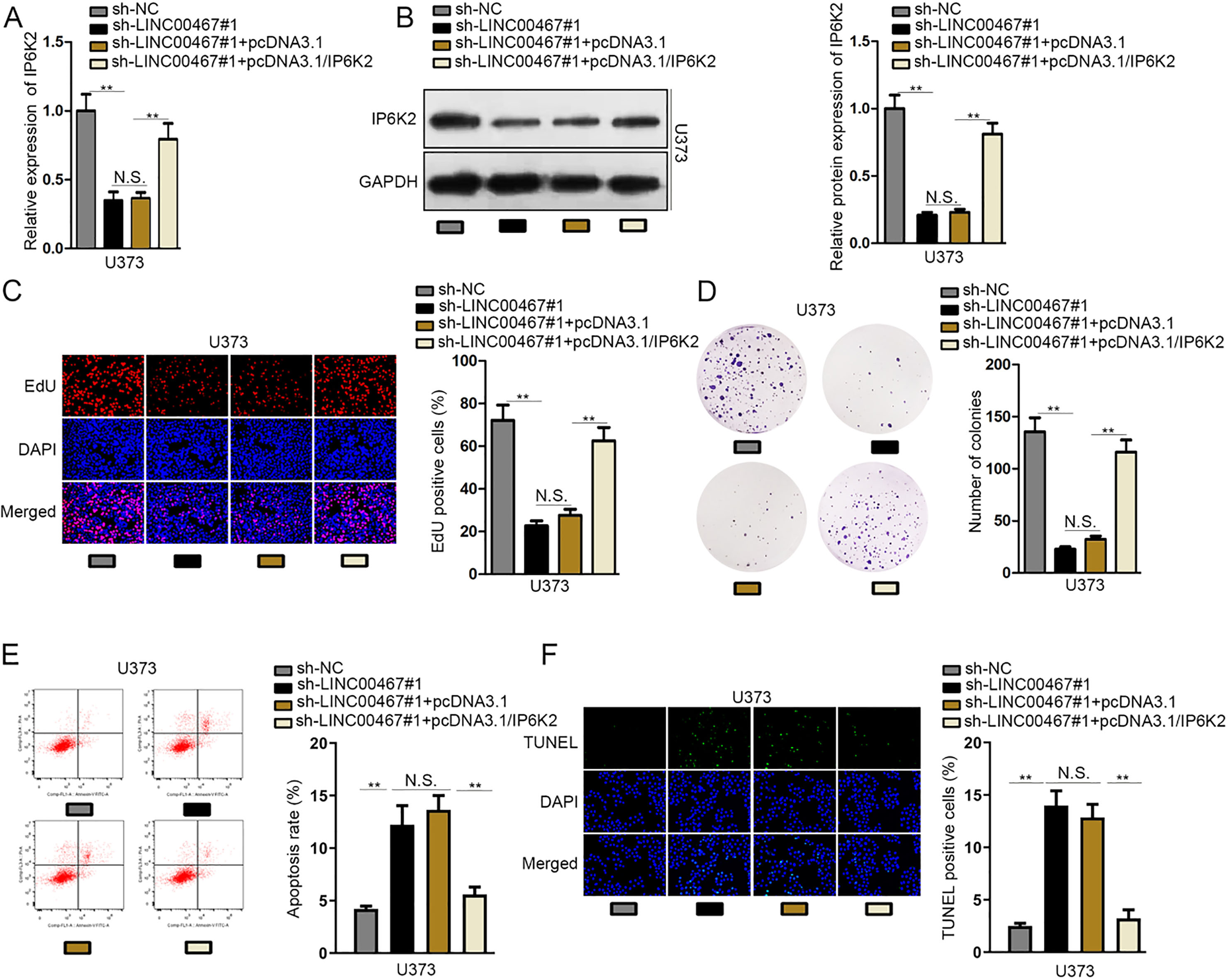
LncRNAs has been widely reported to be involved in various cellular processes such as proliferation, differentiation, migration and apoptosis [29, 30]. Furthermore, the abnormal expression of lncRNA was closely associated with the regulation of a series of diseases and cancers, like cervical cancer [31] and osteoarthritis [32]. In glioblastoma, lncNRA MALAT1 was validated to regulate chemoresistance to temozolomide [33], lncRNA LINC00470 to mediate cell autophagy [34] and lncRNA LINC01446 to promote cell proliferation and invasion [18]. Although several lncRNAs has been identified in glioma, the specific role of LINC00467 was unclear in glioma. Besides, it has reported the LINC00467 facilitates proliferation, invasion and metastasis in colorectal cancer [35], and promotes cell proliferation and stemness in lung adenocarcinoma [36]. In this research, we found that LINC00467 was upregulated in GBM (glioblastoma multiforme) tissues and glioblastoma tissues as well as cell lines according to GEPIA public database and RT-qPCR analysis. In addition, LINC00467 exhibited remarkably higher expression in glioblastoma patients in advanced stage and it upregulation was closely related to the lower survival rate of glioblastoma patients. Functionally, LINC00467 downregulation repressed cell proliferation but promoted cell apoptosis in glioma, indicating the oncogenic role of LINC00467 in glioma.
MicroRNAs (miRNAs) were another class of ncRNAs with less than 22 nucleotides [37]. Likewise, miRNAs also have been proved to function as regulators in diverse biological processes [38]. Mechanistically, miRNAs could interact with lncRNAs to release the downstream targets [39]. In our exploration, 4 miRNAs were found to interact with LINC00467 possibly after prediction and screening by applying bioinformatics analysis. However, only miR-339-3p expression was significantly downregulated in glioma tissues. Additionally, miR-339-3p serves as a tumor-inhibitor in melanoma [40] and colorectal cancer [41]. Previous literature has studied that lncRNA and miRNA interact with each other in various ways, and they can also form ceRNA patterns. Of note, ceRNA is not a requirement for that lncRNA and miRNA interact with each other [42, 43]. In this study, miR-339-3p overexpression could suppress luciferase activity LINC00467 promoter, implying that miR-339-3p possibly regulate the transcription level of LINC00467. Therefore, on one hand, LINC00467 expression could be impaired by miR-339-3p overexpression. On the other hand, miR-339-3p expression was increased by LINC00467 silencing. And this regulatory function of lncRNAs to miRNAs has been widely researched in cancers [44, 45, 46]. Then, the binding capacity between LINC00467 and miR-339-3p was validated. Further, Inositol hexakisphosphate kinase 2 (IP6K2) was selected as the potential target gene of miR-339-3p according to bioinformatics analysis and the filter of RT-qPCR. And previous studies have demonstrated that IP6K2 promotes cell migration and invasion in tumor [47]. Afterwards, we confirmed that IP6K2 was negatively or positively modulated by miR-339-3p or LINC00467 in glioblastoma cells. In the development of glioblastoma, the ceRNA regulation pattern has been researched. For example, lncRNA TRG-AS1 accelerates proliferation of glioblastoma cells via competitively binding with miR-877-5p to release SUZ12 expression [48]. The miR155HG/miR-185/ANXA2 loop facilitates tumor growth and progression in glioblastoma [49]. Therefore, we also verified whether LINC00467/miR-339-3p/IP6K2 axis facilitated glioblastoma development. Finally, rescue assays illustrated that IP6K2 overexpression or miR-339-3p suppression partially counteracted the hindering effects of LINC00467 knockdown on cell proliferation or the activating effects of that on cell apoptosis. Furthermore, the treatment function of molecular targets in tumors has received an increasing attention. For illustration, lncCAMTA1 may be an efficient prognosis factor and an underlying therapy target for hepatocellular carcinoma [50]. LncRNA MAPKAPK5-AS1 is identified as a fresh prognostic biomarker and possible therapeutic target for colorectal cancer [51]. Our data revealed the prognostic significance of LINC00467 by Kaplan-Meier analysis, suggesting LINC00467 as a potential prognostic marker in glioblastoma, but clinical application of our findings in glioblastoma treatment waits to be further explored.
All in all, for the first time, our present investigation manifested that LINC00467 knockdown repressed cell proliferation but stimulated cell apoptosis in glioblastoma via miR-339-3p/IP6K2 axis, which maybe inspirational for finding a novel therapeutic target to glioblastoma. However, this was the initial exploration of LINC00467 in glioblastoma and other regulatory mechanism remained to be explored.
Footnotes
Acknowledgments
We appreciate all people involved in this study.
Conflict of interest
The authors declare that there are no competing interests in this study.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-190939.