Abstract
BACKGROUND:
Nevoid basal cell carcinoma syndrome (NBCCS) is a rare autosomal dominant disease with a complex genetic etiology. Although three causative genes (PTCH1, PTCH2, SUFU) have been identified through linkage analysis and Sanger sequencing, the genetic background of NBCCS hasn’t been fully understood.
METHODS:
We performed a whole-exome sequencing (WES) in a Han Chinese NBCCS family and two unaffected volunteers to search for its causative gene. Bioinformatic analysis was used to select candidate genes and analyze the functional networks of each candidate gene.
RESULTS:
A total of 8 single-nucleotide variants (SNVs) were detected in PTCH1, PTCH2 and SUFU in all the 5 subjects, however none of them was considered the pathogenic genetic mutation in this NBCCS family. The following filtering process identified 17 novel candidate genes (GBP3, AMPD1, ASPM, UNC5C, RBM46, HSPA1L, PNPLA1, GPR126, AP5Z1, ZFHX4, KIF24, C10orf128, COX15, GPRC5A, UGGT2, RHBDF1, RPUSD1). Among them ZFHX4 had been already identified as a new basal cell carcinoma susceptibility loci through a genome-wide association study (GWAS) and was considered the most likely pathogenic gene for this NBCCS family. The functional network analysis revealed that ZFHX4 may be involved in notch signaling pathway.
CONCLUSIONS:
Our study reported the identification of 17 novel candidate genes in a Han Chinese family through WES. ZFHX4 may be a susceptibility gene for NBCCS in Chinese population.
Keywords
Background
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare genetic disease characterized multiple basal cell carcinomas, palmar/plantar pits, jaw cysts, and bony deformities like kyphoscoliosis and frontal bossing [1]. This disease is transmitted in an autosomal dominant manner and has the characteristic feature of high penetrance and variable expressivity [2, 3]. There is a significant racial variation in the incidence of the disease worldwide and is most prevalent among Caucasians [4, 5], only a few reports has been reported in Asians so far.
Clinical phenotype of the patient. (a) basal cell carcinomas (BCCs) around the eyelid. (b) a large BCC on the right temporal. (c) small asymmetric palmar-plantar pits on the right palm. (d) BCCs in the mouth. (e) dermal cysts on the left arm. (f) small asymmetric palmar-plantar pits on the left sole. (g) mandible cysts. (h) amellar calcification of the falx cerebri.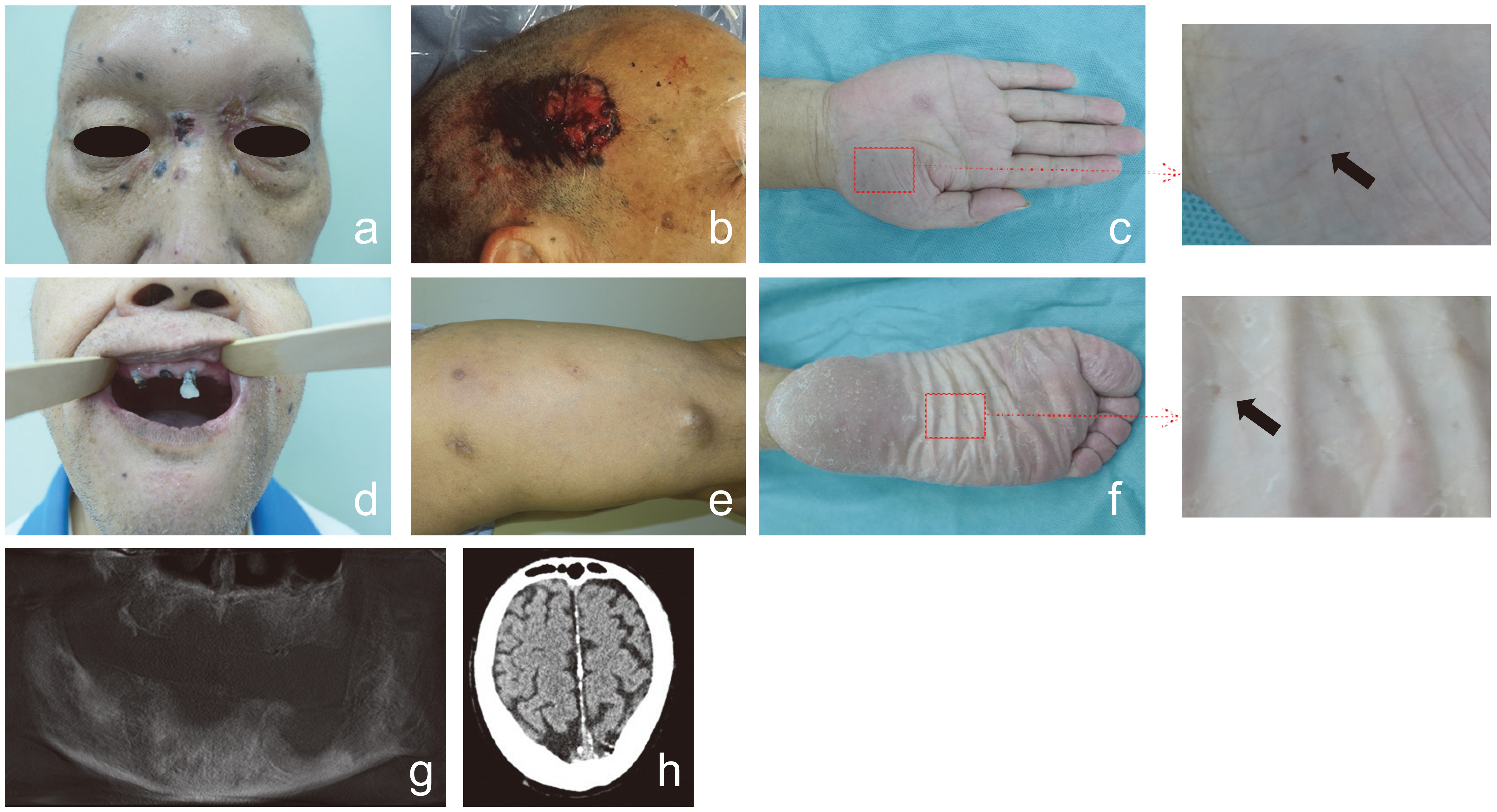
Clinical diagnosis of NBCCS relies on specific criteria, gene mutation analysis may confirm the diagnosis, genetic counseling is mandatory. NBCCS has a complex genetic etiology, linkage analysis and Sanger sequencing have identified three susceptible genes so far, including PTCH-1, PTCH-2 and SUFU [6, 7, 8], which are all involved in the Hedgehog (Hh) signaling pathway. However, mutations in theses genes may not account for all the NBCCS cases and there might be additional unidentified genetic defects responsible for this disease.
The next generation sequencing technologies, including whole genome and whole exome sequencing (WES) have been successfully applied in genetics research, especially in human diseases [9, 10]. This technique is a cost-effective one and especially suitable for the study of Mendel genetic disease: the candidate genes for monogenic disorders can be identified by exome sequencing of only a small number of individuals [11]. In this report, we applied WES in a Han Chinese NBCCS family and two unaffected volunteers to discover causative genes for this disease in this particular family.
Case presentation
A 70-year-old man was referred to our department due to a large ulcerated lesion located on the right temporal. The lesion was insidious in onset and gradually increased in size. Our physical examination showed that there were multiple, discrete, pigmented, well defined, nontender mobile plaques and nodules of varying sizes present all over the body, especially around the eyelids and in the mouth; small asymmetric palmar-plantar pits could be found on his right palm and left sole; dermal cysts were found on his left thigh and left arm. In further examination, lamellar calcification of the falx cerebri was detected through brain CT scan; several mandible cysts were found through oral CT scan (Fig. 1). There was no bifid, fused or markedly splayed ribs found in chest X ray.
Flow chart of the whole study.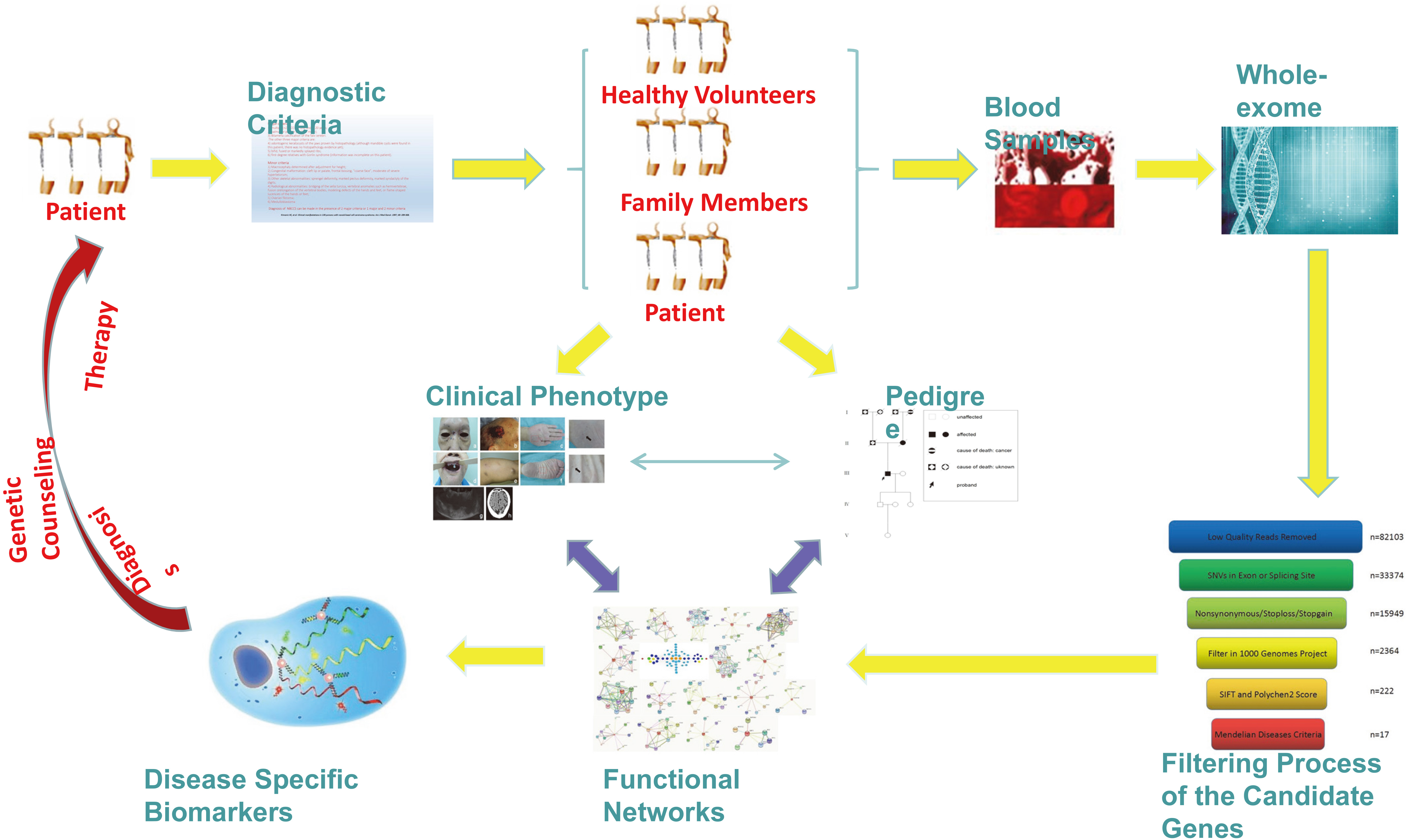
The patient received a scalp lesion removal operation 50 years ago, he took another large hip lesion removal and skin graft surgery 6 years ago, histopathology in both times were suggestive of basal cell carcinoma (BCC). He had a history of cataract in both eyes.
On family history, both of the patient’s parents deceased for decades. His mother had a similar manifestation of “black skin nodules over the body” just like himself. His maternal grandmother died of abdominal cancer, he failed to provide more information on the cause of death of his father, maternal grandfather, paternal grandfather and paternal grandmother. This patient had a daughter, a son and a granddaughter, none of them carried the clinical phenotype of NBCCS (Fig. 2).
After his administration, the patient was diagnosed NBCCS. He received a lesion removal and skin graft operation and recovered well after the surgery.
We harvested the peripheral blood from the NBCCS patient (sample 1), his unaffected daughter (sample 2), his unaffected son (sample 3), an unaffected female volunteer (sample 4) and an unaffected male volunteer (sample 5).
By using Qiagen DNA kit, genomic DNA was extracted from peripheral blood. IlluminaHiSeq 2500 in a paired end 2
We aligned the paired-end reads to the reference human genome (hg19) using the third-party software BWA (Burrows – Wheeler Alignment, version 5.9). The average mapping ratio was as high as 99%. The Flagstat tool was utilized to assess the mapping information. Next, we analyzed the distribution of each sample’s reads in the target region and the enrichment of reads in the genome. SNVs (single nucleotide variations) were then processed using the GATK UnifiedGenotyper (GenomeAnalysisTK-3.1-1). Finally, we annotated the mutations using ANNOVAR software (GenomeAnalysisTK-3.1-1).
Identification of candidate genes
SNVs in PTCH1, PTCH2 and SUFU in all the samples were firstly analyzed. Since there was no positive finding, novel NBCCS pathogenic genes were identified through the following filtering process: low quality reads were firstly removed; SNVs located out of exon region and splicing site were then removed; synonymous SNVs and common SNVs (mutation frequence
Summary of original exome sequencing data
Summary of original exome sequencing data
Exonic SNVs in PTCH1, PTCH2 and SUFU
Chr: Chromosome ID. Pos: mutation position in each chromosome. Ref: reference base. Alt: altered base. Alts: altered bases, if there is more than one altered base. S1–S5: sample1-sample 5. 1000 G: allele frequencies in 1000 genomes project database.
For the remaining 17 genes, a protein-protein interaction networks analysis (
Results
Summary of whole exome sequencing
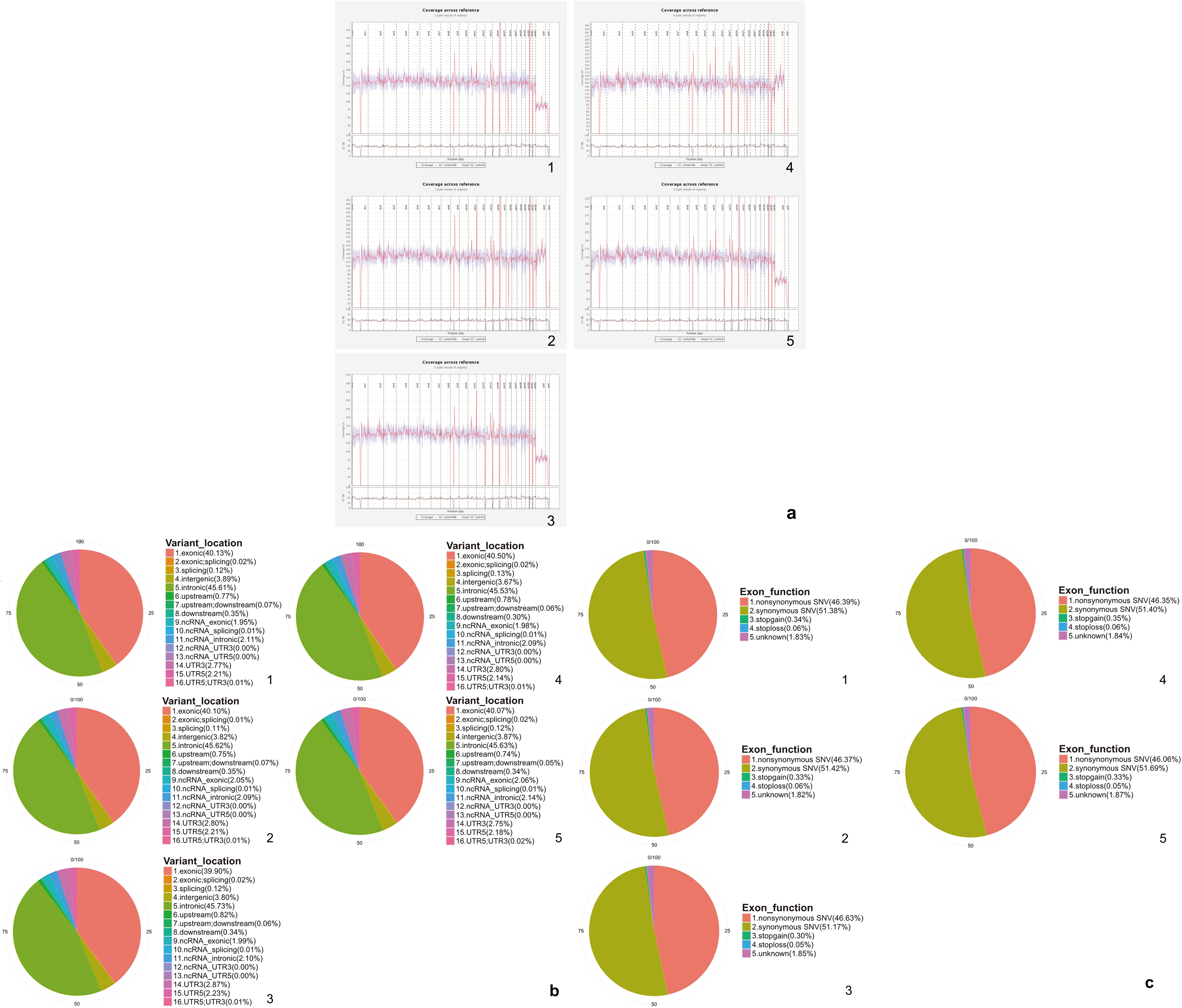
Overall, we obtained more than 87% uniquely mapped reads and more than 99% good matched reads. In each sample over 84% reads were on the target region. In total about 98.2% of the targeted bases were covered sufficiently to pass the quality assessment and were aligned to the human reference genome (hg19) with the BWA. The average sequencing depth in the exome region of the case was approximately more than 120 X (Table 1); the average sequencing depth in each autosome was approximately more than 120 X and the mean GC content was about 50% (Supplement Fig. 1a). The target SNV distribution and target SNV exonic function were presented in Supplement Fig. 1b and c.
In summary, we obtained a high quality WES data which could provide reliable information of the disease.
Comparison SNVs with PTCH1, PTCH2 and SUFU
In exon region, 5 SNVs were detected in PTCH1, 2 SNVs were detected in PTCH2 and 1 SNV was detected in SUFU respectively. However in the total 8 SNVs, 6 SNVs were synonymous variations, only 2 SNVs in PTCH1 gene were nonsynonymous variations. Between these 2 nonsynonymous SNVs, only 1 SNV was detected in the affected patient; however this SNV was a common variation, the mutation frequency in 1000 genomes project was 0.4, moreover both of his children carried this mutation (Table 2).
In summary, we detected no pathogenic SNVs in the three known suspected genes in this family.
Identification of candidate genes
Through the initial filtering process, 222 mutated genes were considered potentially hazardous in all the five subjects (Fig. 2). Considering the autosomal dominant inheritance pattern, we were finally able to narrow down to 17 genes as the candidate genes in this patient: GBP3, AMPD1, ASPM, UNC5C, RBM46, HSPA1L, PNPLA1, GPR126, AP5Z1, ZFHX4, KIF24, C10orf128, COX15, GPRC5A, UGGT2, RHBDF1, RPUSD1 (Table 3).
List of the disease specific candidate genes
List of the disease specific candidate genes
Protein-protein interaction networks between the candidate genes and Hedgehog (Hh) pathway.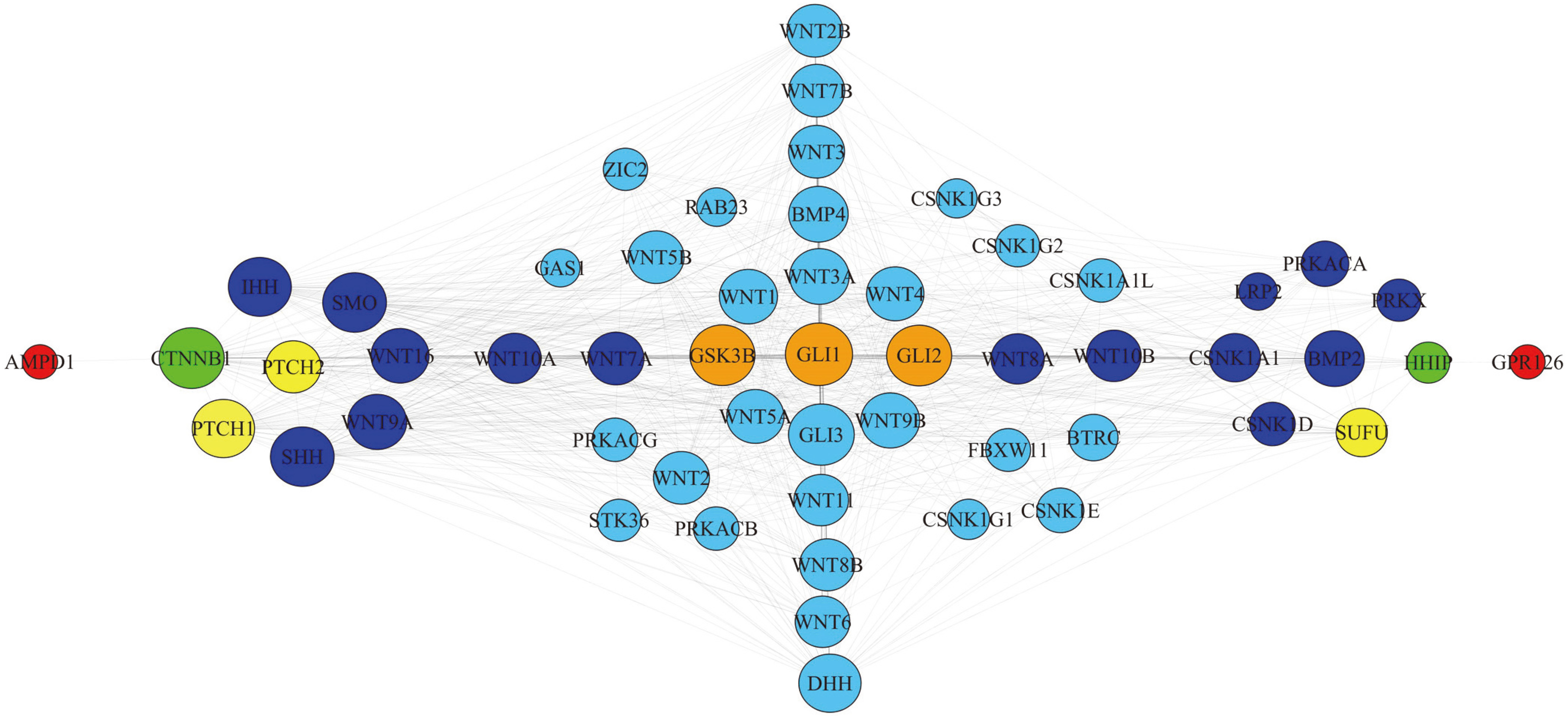
The protein-protein interaction networks analysis revealed that 2 genes (AMPD1, GPR126) were involved indirectly in the Hh pathway: AMPD1 is linked to PTCH1 through CTNNB1, GPR126 is linked to PTCH2 and SUFU through HHIP. However none of the 17 candidate genes were directly involved in Hh pathway based on available research data (Fig. 3).
Functional networks of ZFHX4
Since ZFHX4 was discovered as a new basal cell carcinoma susceptibility loci in a genome-wide association study (GWAS), we specifically focused on the functional networks of this gene. The predicted functional partner of ZFHX4 include CHD4, C1QBP, NOTCH1, NOTCH2, NOTCH3, NOTCH4, ZEB1, TMEM14A, PIAS1, HIVEP3 (Fig. 4). These findings revealed a possible linkage between ZFHX4 and notch signaling pathway.
Functional networks of ZFHX4 gene.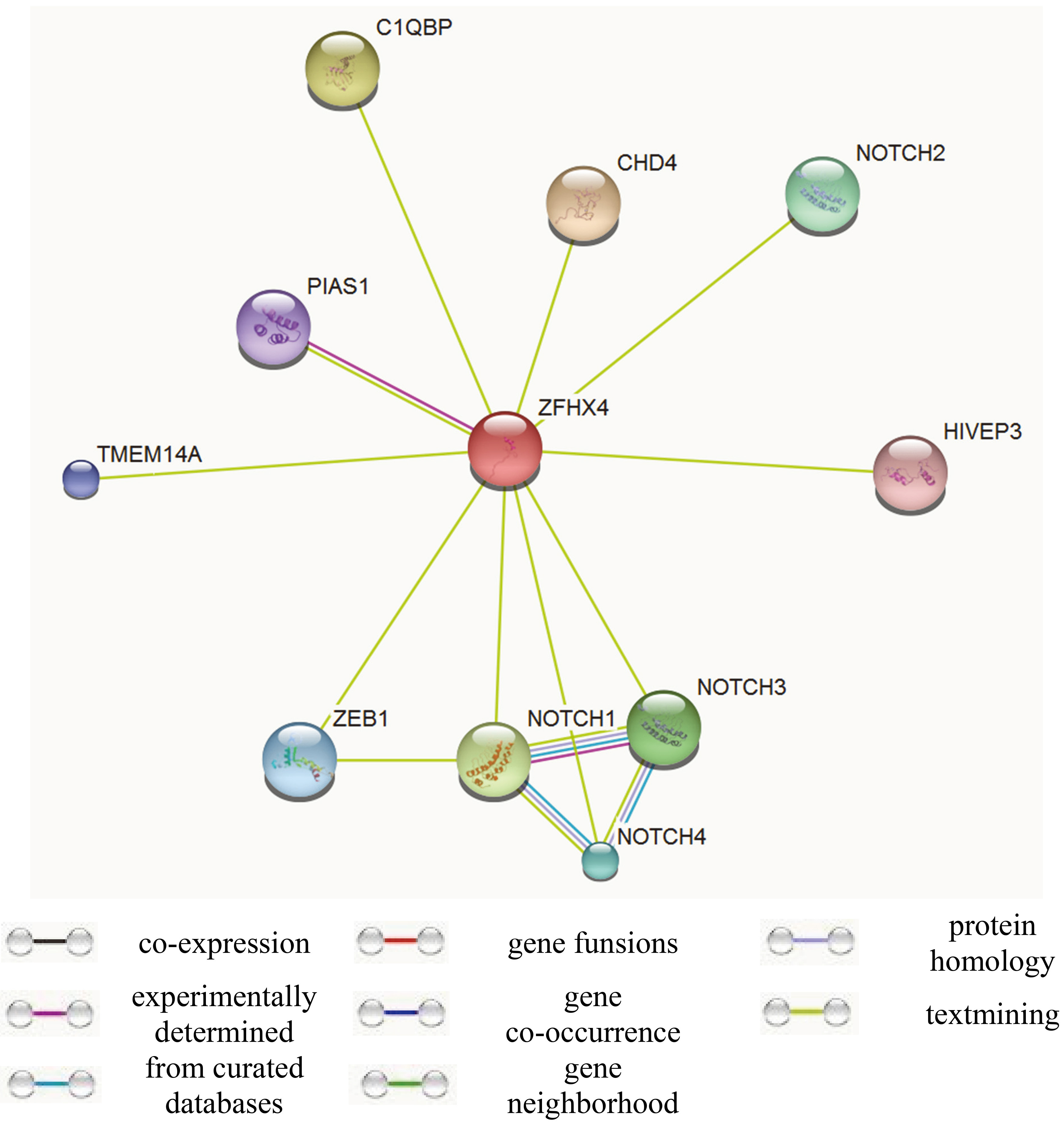
In this study a WES was performed in a Han Chinese NBCCS family and two unaffected volunteers to validate the three known pathogenic genes and to identify other potential pathogenic genes for this disease. A total of 17 candidate genes were found and most of their gene functions haven’t been systematic studied yet.
Unlike other studies [6, 7, 8], WES was used to dissect the genetic background of this disease in our research. During our early literature review, we noticed that this technique hasn’t been used in NBCCS study yet. Traditional linkage and association genetic mapping studies have limited resolution in identifying and localizing the causal genes in the genome, because a mapping region often contains hundreds of candidate genes [12, 13]. The functional testing and validation for the most promising candidates among such large lists of genes are time consuming and challenging [11]. In recent years, high-throughput sequencing technique has become cost-effective, time saving and could provide more comprehensive genetic information in each subject. Focusing on the exome region can be an efficient strategy to dissect the Mendelian disorder such as NBCCS.
None of the SNVs in the three known causative genes (PTCH1, PTCH2, SUFU) was considered pathogenic in our study. This is probably a consequence of genetic heterogeneity. As a complex genetic disease, the diagnosis criteria of NBCCS is complicated [14] and the clinical phenotype are diverse among different patients; for example, almost 10% of the patients did not manifest basal cell carcinoma [15]. Moreover, there is a significant racial variation in the incidence of the disease worldwide, the incidence in Asians are significantly lower than in Caucasus. These implied that there might be several causative genes of NBCCS among different patients.
Hh signal pathway abnormalities is the most important reason of NBCCS [16], our further functional networks analysis revealed that only two genes – AMPD1 and GPR126 have indirect relations with the components of Hh pathway. However the relationships were weak owing to the lack of sufficient information about their gene function.
GWAS have yielded a substantial number of variants that predispose to BCC, including MC1R, ASIP, TYR, SLC45A2, OCA2, IRF4 PADI6/RCC2, RHOU, TERT, KRT5, CDKN2A/B, KLF14, TP53, TGM3, RGS22, MYCN, CASP8-ALS2CR12, ZFHX4 and GATA3 [17]. Among them, ZFHX4 was the only one found in the 17 candidate genes in our study.
ZFHX4 gene was mapped to human chromosome 8q21, it was about 180 kb long, containing 12 exons that encodes a 3599-amino acid protein carrying 4 homeodomains and 22 zinc fingers. Although little is known about the function of this gene yet, our functional networks analysis proved that this gene may be involved in notch signaling pathway. Notch pathway is linked to the development of various cancers including skin cancers [18] and the notch pathway could be inactivated by zinc via a PI3K-Akt-dependent way [19]. Medulloblastoma – another clinical manifestation in NBCCS patients, was related to NuRD complex recruitment [20]. ZFHX4 was also found to interacts with the NuRD complex and regulates the glioblastoma tumor-initiating cell state [21]. This reveals a possible link between ZFHX4 and the development of medulloblastoma. In other studies, ZFHX4 was found to be susceptibility loci for several diseases, including Congenital heart defects [22], intellectual disability and/or multiple congenital anomalies [23], Hereditary congenital ptosis [24] and Peters anomaly [25]. Further studies are needed to investigate the function of ZFHX4.
We believe that this study may increase the understanding of NBCCS genetic background, especially in Asians. ZFHX4 may have the potential to become the bio-marker of NBCCS, which may be helpful in antenatal diagnosis and genetic consulting.
There were still some limitations which need to be addressed. Firstly, in this Han Chinese NBCCS family, only one living family member was affected; although we narrow the number of candidate genes down into 17 through the filtering process, a WES on two or more affected members may lead to a even less candidate gene number. Secondly, more functional analyses were warranted to elucidate the biological plausibility of this NBCCS susceptibility gene in further research.
Conclusions
In conclusion, we reported the identification of 17 novel candidate genes in a Han Chinese family through WES. Among them ZFHX4 was the most likely pathogenic gene for the disease in this particular family. However, the exact biological mechanism of these genes involved in NBCCS pathogenesis still needs to be clarified.
Footnotes
Supplementary data
Distribution of the coverage, variant location and exonic function. (a) Distribution of the coverage for each sample. The x-axis represents the position of different chromosomes. The continuous red line represents the exon coverage range in each chromosome. The discontinous red line represents the mean Guanine-Cytosine (GC) content in each chromosome. The gray line represents the GC content in each chromosome. (b) Variant location in each sample. (c) Exonic function in each sample.