
Abstract
Skeletal muscle health and function are important determinants of systemic metabolic homeostasis and organism-wide responses, including disease outcome. While it is well known that exercise protects the central nervous system (CNS) from aging and disease, only recently this has been found to depend on the endocrine capacity of skeletal muscle. Here, we review muscle-secreted growth factors and cytokines (myokines), metabolites (myometabolites), and other unconventional signals (e.g. bioactive lipid species, enzymes, and exosomes) that mediate muscle-brain and muscle-retina communication and neuroprotection in response to exercise and associated processes, such as the muscle unfolded protein response and metabolic stress. In addition to impacting proteostasis, neurogenesis, and cognitive functions, muscle-brain signaling influences complex brain-dependent behaviors, such as depression, sleeping patterns, and biosynthesis of neurotransmitters. Moreover, myokine signaling adapts feeding behavior to meet the energy demands of skeletal muscle. Contrary to protective myokines induced by exercise and associated signaling pathways, inactivity and muscle wasting may derange myokine expression and secretion and in turn compromise CNS function. We propose that tailoring muscle-to-CNS signaling by modulating myokines and myometabolites may combat age-related neurodegeneration and brain diseases that are influenced by systemic signals.
Keywords
Muscle contraction (exercise) is one of the most effective interventions to prevent age-related diseases. These effects have been ascribed to increased nutrient utilization but also to circulating factors produced by many tissues, including skeletal muscle. Here, we review how muscle-derived signaling factors induced by exercise and associated processes mediate muscle-brain and muscle-retina communication. We highlight the impact of myokines and myometabolites on proteostasis, neurogenesis, cognitive functions, and behavior. We propose that harnessing knowledge on protective and deleterious myokines may provide new opportunities for combating age-related brain functional decline and neurodegeneration.
INTRODUCTION
Skeletal muscle is one of the most abundant tissues in the human body. In addition to its role in voluntary and involuntary movements, skeletal muscle is an important determinant of systemic metabolic homeostasis and influences organism-wide responses [1, 2] via its capacity to secrete a myriad of signaling factors in response to basal contraction, physical activity, atrophy, disease, and metabolic demands/imbalances [3, 4]. Such endocrine capacity of skeletal muscle has been found to impact many tissues and organs and increasing evidence indicates that also the central nervous system is a target of muscle-initiated signaling [3–8].
The impact of skeletal muscle on the central nervous system has been best described mostly in response to exercise [9–14]. Many studies have found that exercise decreases the risk of onset and progression of neurodegenerative diseases such as Alzheimer’s disease (AD) [15–17]. Exercise reduces age-related brain atrophy [18–21], improves brain metabolic functions and mitochondrial biogenesis [22–24], reduces oxidative stress and neuroinflammation [25–28], and improves cerebral blood flow and cognition [29–36]. For example, exercise improves memory [29, 37, 38], object recognition [39], contextual fear memory [39], and spatial navigation [40, 41]. Interestingly, exercise preserves the function not only of neurons but also of microglia by decreasing cell senescence, and this preserves cognitive functions [42].
Although the beneficial effects of exercise are well known, it has also be noted that excessive exercise might be deleterious by impairing mitochondrial function and by decreasing glucose tolerance in humans [43, 44] and this may apply also to the brain, as it was found that excessive oxidative stress caused by exhaustive exercise impairs cognitive functions in mice [45].
Aerobic exercise also preserves the function and structure of the retina from light-induced degeneration in mice [46, 47], although other types of exercise can be detrimental [48]. By acting on the visual cortex, exercise can also correct visual acuity and depth perception in rats with amblyopia [49] and similar results are also observed in humans [50].
There are multiple modes of muscle-to-brain signaling (Fig. 1), which can occur via muscle-secreted growth factors and cytokines known as myokines [51]. Although many circulating factors cannot pass the blood-brain barrier (BBB), they can influence the brain by binding to their receptors on the endothelial cells of the BBB, as observed for GDF11 [52, 53]. However, some circulating factors can pass through the BBB and therefore can signal directly to brain cells [54–56]. This is also the case for some myokines, including irisin, cathepsin B, BDNF, IL6, and FGF21 [57–63]. In addition to myokines, muscle-released metabolites (myometabolites) and other unconventional signals (such as bioactive lipid species, enzymes, and exosomes) can contribute to communication with the central nervous system (CNS; Fig. 1). In addition to endocrine signaling through the circulation, direct muscle-to-nerve connections may also provide a route for signaling from skeletal muscle to the brain [3]. In agreement with this hypothesis, fluorescent tracers injected into skeletal muscle are delivered to the spinal cord via retrograde axonal transport in motor neurons [64], suggesting that myokines may also employ this route [65, 66].
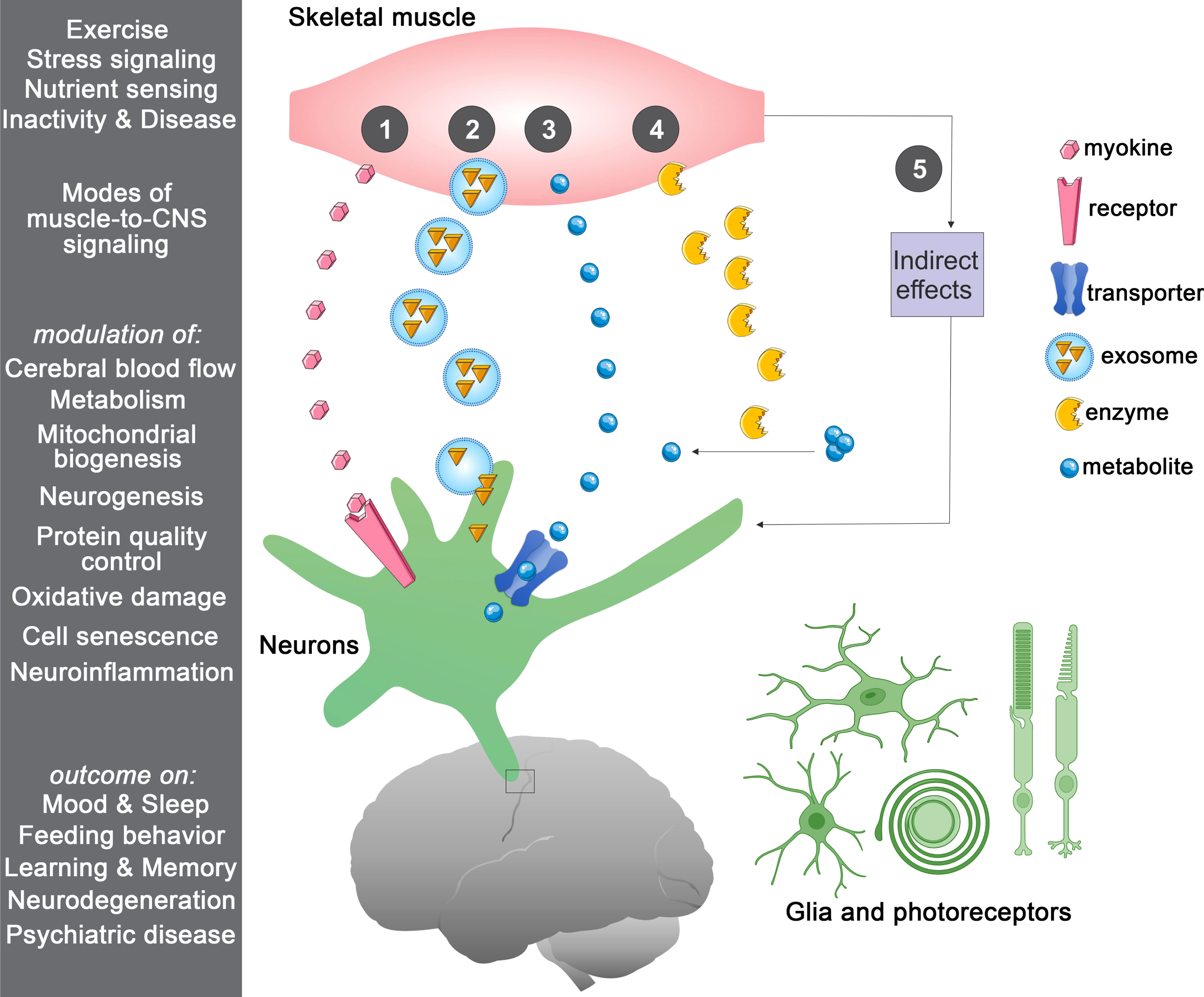
Multiple routes of muscle-to-CNS signaling. In response to a variety of stimuli, skeletal muscle can communicate in a number of ways with the central nervous system (CNS), including the following: (1) by secreting signaling proteins (myokines) that can bind to receptors in the blood-brain barrier (BBB) and brain cells (neurons and/or glia), with the consequent induction of downstream signaling; (2) by releasing extracellular vesicles such as exosomes that contain signaling factors; (3) by releasing metabolites (myometabolites) that enter the brain through solute transporters present on the BBB and brain cells; (4) by secreting enzymes that produce signaling factors in the muscle, in the circulation, and/or in the brain; and (5) via indirect effects stemming from modulation of muscle metabolism and/or myokine signaling to other tissues distinct from the brain. Regulated processes include improvement in cerebral blood flow, brain metabolic functions, mitochondrial biogenesis, and neurogenesis whereas protective signaling reduces oxidative stress, cell senescence, and neuroinflammation. Altogether, the action of muscle-brain signaling on these cellular processes improves cognitive functions.
Here, we review emerging paradigms for skeletal muscle signaling to the CNS and the resulting impact on brain health and disease.
MUSCLE-TO-BRAIN SIGNALING MEDIATED BY EXERCISE-INDUCED MYOKINES
To dissect how exercise influences brain function and neurodegeneration, many studies have examined the role of myokines induced by signaling pathways and transcription factors modulated by exercise. Here, we review exercise-induced myokines responsible for muscle-brain signaling.
PGC1α is a key mediator of exercise-induced local and systemic adaptations [12, 67, 68] and therefore extensive research efforts have examined the role of PGC1α-modulated myokines, some of which were found to act on the central nervous system [5, 7]. The myokine irisin derives from proteolytic cleavage of its transmembrane precursor FNDC5 (Fibronectin type III domain-containing protein 5) and contributes to the systemic benefits of exercise by promoting browning of the adipose tissue [69], and also by signaling to the brain in mice [58]. Specifically, irisin is upregulated in response to exercise in skeletal muscle and hippocampus and in turn induces expression of brain-derived neurotrophic factor (BDNF) [58], which is key for the effects of exercise on neurogenesis and cognition [70]. However, irisin derived from peripheral tissues is not merely a nuance but is rather important for neuroprotection, as demonstrated by the fact that it can cross the BBB and that adenovirus-mediated expression of irisin in the liver increases expression of BDNF and other neuroprotective genes in the mouse hippocampus [58, 71, 72]. Moreover, irisin protects from neuronal injury associated with cerebral ischemia via Akt and Erk signaling [73] and also improves both the cognitive deficits and neuropathology in AD mouse models [74–76]. Also in this context of AD-linked neurodegeneration, it was found that circulating irisin has important roles, in addition to irisin produced by the brain. Specifically, circulating FNDC5/irisin rescues memory impairment in AD mouse models, whereas blockade of either peripheral or brain FNDC5/irisin impedes the amelioration of synaptic plasticity and memory by physical exercise [74–76]. These neuroprotective actions have been ascribed to the capacity of irisin to trigger cAMP/PKA/CREB signaling as well as to the transcriptional induction of BDNF and of other neuroprotective genes in the mouse hippocampus [76].
Another PGC1α-regulated myokine, neurturin [77], is a member of the glial cell line-derived neurotrophic factor (GDNF) family, which broadly regulates the survival and function of motor neurons. Neuturin acts as a paracrine myokine that induces a shift in myofiber type and motor neurons towards slow identity, hence coordinating their functional properties [78]. Additionally, neurturin enhances mitochondrial function, capillary density, fatty acid oxidation, essentially recapitulating the effects of PGC1α over-expression in mouse skeletal muscles [78]. Although neurturin is effective on motor neurons, it remains undetermined whether this myokine can also act in an endocrine manner and signal to the brain.
In addition to PGC1α-mediated responses, a key component of exercise is the depletion of energy stores and the consequent activation of the AMP-activated kinase (AMPK), which senses AMP/ADP concentrations. Via the administration of the AMPK agonist 5-aminoimidazole-4-carboxamide riboside (AICAR), Kobilo et al. found that beneficial effects of AICAR on memory and motor functions are age- and dose-dependent [79]: a 3-day administration of AICAR was sufficient to improve cognition in young wild-type mice but prolonged treatment over 14 days was required to achieve cognitive benefits in old mice [79]. However, since the BBB is impermeable to AICAR, the impact on cognition was ascribed not to the effects of AICAR on the brain but rather on its action on peripheral tissues. To ascertain if these effects arise from the action of AICAR on skeletal muscle, the authors utilized muscle-specific AMPK-DN (AMPK dominant-negative) mice and found that the cognitive and motor benefits of AICAR were lost in the absence of AMPK signaling in muscle [79]. Altogether, this study indicates that the metabolic adaptations and/or myokine/myometabolite signaling induced by AMPK in skeletal muscle may contribute to preserve cognitive function during aging.
Subsequent proteomic analyses identified an unusual muscle-secreted factor, the protease cathepsin B (CTSB), which was enriched in the conditioned medium of skeletal muscle cells treated with AICAR and in the human, mouse, and monkey plasma upon exercise [57]. Administration of recombinant CTSB increased expression of brain-derived neurotrophic factor (BDNF) and doublecortin in cultures of hippocampal progenitor cells [57]. Moreover, exercise did not promote adult hippocampal neurogenesis and spatial memory function in CTSB knockout mice [57], further confirming the importance of CTSB for exercise-induced neuroprotection [57, 80, 81].
Besides being produced in the brain in response to exercise [82, 83], BDNF is also produced by contracting skeletal muscle [84, 85], where it regulates contractile strength of fast-twitch myofibers [86] and fat oxidation via AMPK activation [87]. Different cell types (including muscle satellite stem cells, myofibers, and infiltrating immune cells) have been proposed as sources for BDNF [88, 89]. Because BDNF can cross the BBB [61], muscle-derived BDNF may contribute to muscle-to-brain communication [90]. However, the capacity of peripheral BDNF to cross the BBB has been questioned [91].
Insulin growth factor I (IGF-I) has also been found to mediate muscle-to-brain signaling [90, 92]. Specifically, exercise-induced increase in circulating IGF-I was found to promote adult hippocampal neurogenesis and to improve cognition and reduce anxiety. Consistently, IGF-I mutant mice display low hippocampal neurogenesis and impaired spatial learning, and these deficits can be rescued by recombinant IGF-I systemically delivered via a subcutaneous pump but not by running [93–95].
In addition to promoting neurogenesis, myokines can also modulate other processes relevant for brain function and disease. An example of such myokines is interleukin 6 (IL-6), which is secreted by contracting skeletal muscles [96, 97] and promotes fatty acid oxidation and glucose disposal [98, 99]. Importantly, it was proposed that exercise-induced IL-6 is responsible for the long-term anti-inflammatory effects of moderate exercise [100]. However, IL-6 can induce inflammatory or conversely anti-inflammatory responses depending on the receptor engaged [101]. Although there is limited understanding on whether muscle-produced IL-6 directly signals to the CNS, IL-6 can pass the BBB [60], suggesting that muscle-derived IL-6 may indeed impact the brain. By engaging membrane-bound IL-6R receptors and consequent glycoprotein gp130 activation, IL-6 induces anti-inflammatory signaling. Alternatively, IL-6 can bind soluble IL-6R receptors generated by alternative splicing or limited proteolysis, which then interact with gp130. However, contrary to canonical signaling induced by membrane-bound IL-6R, transcellular signaling mediated by soluble IL-6R is pro-inflammatory [101]. In the brain, membrane-bound IL-6R is expressed only by the microglia whereas gp130 is expressed by all cell types, including neurons, oligodendrocytes, and astrocytes [101]. Canonical IL-6R signaling is neurotrophic and promotes regeneration whereas trans-signaling mediated by soluble IL-6R promotes inflammation and neurodegeneration [101]. At present, it remains largely undetermined whether muscle-derived IL-6 engages brain IL-6R and whether this would be skewed towards canonical versus trans-signaling. Altogether, despite being well characterized as an exercise-induced myokine, much remains to be learnt on the role of exercise-induced IL-6 in muscle-brain signaling. Likewise, other myokines structurally related to IL-6 have been characterized, including oncostatin-M and leukemia inhibiting factor (LIF), but their potential function in muscle-brain signaling remains largely unexplored.
Altogether, these studies indicate that muscle-to-brain communication is reshaped by exercise via the action of many signaling pathways and the consequent modulation of the expression and/or secretion of several myokines with neuroprotective functions [5, 7]. However, considering that the vast majority of the >600 myokines is largely uncharacterized [102, 103], it seems that our knowledge of muscle-brain signaling is in its infancy and that most likely also other myokines contribute to this intertissue communication in response to muscle contraction and other stimuli. Indeed, apart from the well-described examples of myokines reported above and known for their role in muscle-brain signaling in vivo, other myokines have been found to affect neurons in cell culture. For example, thymosin β4 is secreted by contracting myotubes and promotes neurite outgrowth in cell culture [104]. However, its role in muscle-brain signaling remains uncharacterized in vivo. In addition, there are many myokines that have protective functions on many tissues and/or that are known to be neuroprotective when expressed in the brain, as it is for apelin and its G-protein-coupled receptor APJ [105, 106]. However, it remains currently uncharted whereas muscle-produced apelin is relevant for CNS function and age-related neurodegeneration.
In addition to unraveling the roles of other myokines, future studies should determine whether such neuroprotective myokines act synergistically to preserve the brain in response to exercise and whether they regulate distinct brain functions and/or contrast hallmarks of brain aging and neurodegeneration [107]. As reviewed below, many different types of signals can be released by skeletal muscle, such as atypical myokines (e.g. enzymes) that differ from stereotypical cytokines and growth factors. Moreover, muscle-released metabolites (myometabolites) are emerging as key mediators of exercise-induced inter-tissue signaling [3, 108].
In a study in the nematode Caenorhabditis elegans, it was found that muscle-specific lipolysis, induced via muscle-specific protein kinase A (PKA) activation, prolongs lifespan by promoting the production of lipid species with signaling functions (lipokines) and activation of lipid-sensing transcription factors in the CNS [109]. Although it remains undetermined whether such signaling is associated with changes in brain function and neurodegeneration, this study highlights the role of bioactive lipid species originating from skeletal muscle and hence the diversity of muscle-released factors that mediate muscle-to-brain communication [109]. Such lipid-mediated signaling may also occur in response to exercise because utilization of energy stores and lipolysis occurs during muscle contraction [12, 67].
Another example of muscle-to-brain signaling possibly mediated by lipid species comes from the observation that thermal stress induces migration (thermotaxis) of C. elegans to more favorable temperatures, a process governed by thermosensory neurons and interneurons. Worms that are null for heat shock factor 1 (hsf-1) are defective in thermotaxis but this behavior is restored by hsf-1 expression solely in skeletal muscle, and this may entail estrogen-mediated signaling of muscle to thermosensory neurons [110].
Beyond muscle-released lipid signals, many well-known metabolites are secreted by skeletal muscle [111], including L-lactate [112]. At resting levels as well as during exercise, skeletal muscle secretes lactate into the circulation [113–115]. Lactate can be transported intracellularly via monocarboxylic acid transporters (MCTs) but can also signal by binding to the lactate receptor called hydroxycarboxylic acid receptor (HCAR1), a G-protein-coupled receptor [114]. Morland et al. [116] found that mouse HCAR1 is enriched in cells of the BBB as well as in neurons and astrocytes whereas it is not detected in skeletal muscles. HCAR1 activation led to increased cerebral vascular endothelial growth factor A (VEGFA) expression, a mediator of angiogenesis that is known to improve neurogenesis and cognition [90, 117]. The authors found that, similar to high-intensity exercise, subcutaneous injection of L-lactate (to mimic exercise-induced raise in lactate serum levels) leads to higher brain VEGFA levels [116], capillary density [116] and adult neurogenesis [118, 119] in wild-type but not in HCAR1 knockout mice. Furthermore, exercise-induced lactate as well as its intraperitoneal injection increases hippocampal BDNF expression and improves spatial learning and memory [120], suggesting yet another mechanism for lactate’s systemic actions.
Alternatively, lactate can be uptaken by MCT intracellular transporters and converted into pyruvate by lactate dehydrogenase, concomitantly to the production of NADH from NAD+. Such an increase in NADH alters the cellular redox status and influences the activity of NAD-dependent histone/protein deacetylases. The authors found that MCT-mediated intracellular transport of lactate increases BDNF expression via SIRT1-dependent induction of PGC1α, which in turn induces FNDC5 [120], which is known to induce BDNF expression [58]. Altogether, this study implies that the SIRT1/PGC1α/FNDC5/BDNF signaling axis contributes to exercise-induced brain plasticity in response to lactate in mice [120]. Overall, lactate can function as a neuroprotective agent by increasing the expression of BDNF and VEGF in the brain and by acting as a signaling molecule that binds to the HCAR1 receptor [121]. Moreover, lactate-induced signaling appears to be deranged in a mouse AD model (APP/PS1 mice) [122]. Similarly, both the lactate content and MCT2 transporter expression decrease in the cerebral cortex and hippocampus of these AD mice [123].
In addition to contribute to lactate-induced signaling in the brain, as described above, analysis of mice with skeletal muscle-specific conditional knockout indicates that VEGF produced by myofibers contributes to maintain normal blood flow in the hippocampus and is necessary for the hippocampal response to acute exercise [124, 125]. Specifically, VEGF promotes proliferation of neural progenitors in the dentate gyrus in response to exercise [125, 126]. Rather than resulting from direct signaling of VEGF to neural stem cells, exercise-induced proliferation of neural progenitors likely results from the impact of VEGF on the vasculature, which is in close proximity to newborn neurons and is a constituent of the stem cell niche [127]. In addition to direct muscle-brain signaling, VEGF may impact the brain indirectly, via VEGF-induced adaptations in skeletal muscle [126].
Another exercise-induced metabolite that regulates histone deacetylase activity is β-hydroxybutyrate (BHB), a ketone body that is released from the liver and oxidized for energy production in the brain under low-glucose conditions. Recently, Kwak et al. found that the BHB levels are low in skeletal muscles of sedentary old mice compared to young counterparts but are increased by endurance exercise in old mice [128]. On this basis, the authors proposed that BHB can be secreted from skeletal muscles following endurance exercise. To test this hypothesis, skeletal muscle cells were treated with the exercise mimetic forskolin. Both forskolin-treated cells and the cell culture media displayed a high BHB concentration compared to control untreated cells [128], suggesting that BHB is secreted from exercising skeletal muscles. Administration of BHB in vivo improved the viability of both myofibers and glial cells under normal conditions and when treated with doxorubicin, a chemotherapy drug that causes toxicity via reactive oxygen species [128].
In exercising mice and in the elderly, high BHB levels correlate with improved muscle and cognitive functions [128], presumably via its capacity to increase BDNF expression in glial cells [128] and to improve mitochondrial respiration [129, 130]. Just like lactate, muscle-secreted BHB can be transported intracellularly in neurons through MCT transporters [129]. Exercise-induced BHB inhibits the binding of class I histone deacetylases HDAC2 and HDAC3 to the BDNF promoter, hence inducing BDNF expression and glutamatergic transmission in the hippocampus [131]. Apart from functioning as an epigenetic regulator of HDACs, BHB can induce lysine-hydroxybutyrylation of histones in human and mouse cells, coupling metabolism to gene expression [132]. It was also found that BHB protects against neurodegeneration associated with Parkinson’s and Alzheimer’s disease [130, 133, 134]. In C. elegans, BHB supplementation delays amyloid-β-induced paralysis and decreases α-synuclein aggregation [133]. In vitro studies showed that supplementation with BHB protected mesencephalic neurons from methyl-4-phenyl pyridinium (MPP) toxicity (a model of Parkinson’s disease) and hippocampal neuron from Aβ1 - 42 toxicity in mice [134], primarily by reversing defects in mitochondrial respiration responsible for neurodegeneration [130, 134]. Additionally, due to its anionic nature, BHB regulates intracellular potassium levels and hence modulates neuronal excitability [135]. Consistently, BHB prevented the decline of intracellular K + in response to incubation with activators of the NLRP3 inflammasome, suggesting an important role of BHB in resisting NLRP-3-mediated neuroinflammation in mice [136].
Altogether, several myokines and myometabolites have been found to be induced by exercise and mediate muscle-to-brain signaling. As explained above, muscle-secreted factors can regulate brain function by impacting different cell types and a variety of cellular processes. Although individual studies have shown interconnection of different myokines and associated downstream signaling pathways, much remains to be learnt on how distinct myokines differently contribute to muscle-brain signaling. Moreover, apart from general actions of myokines on the CNS, it is only in part understood how distinct myokines affect different brain areas and cell types as well as the BBB, and how the downstream responses induced by myokines are reshaped depending on cell type-specific/local variables.
EFFECT OF MUSCLE-INITIATED STRESS SIGNALING ON THE BRAIN AND RETINA
Because of the high mechanical stress associated with contraction [137, 138], exercise is known to cause protein unfolding [139] and to induce adaptive stress responses [140–143], including the cytosolic and mitochondrial unfolded protein responses [144–148]. In addition to local adaptations [11, 67], activation of such stress responses in skeletal muscle has been found to induce systemic changes [111, 149–152]. We will review here below key studies that have reported effects on the CNS in response to muscle-initiated stress signaling [3, 153].
In C. elegans, it was found that a systemic stress response can be induced by expression of a temperature-sensitive myosin heavy chain B (unc-54) protein. At the non-permissive temperature, this muscle-specific protein undergoes misfolding and induces chaperone expression in the brain and intestine via an inter-tissue signaling pathway that requires the transcription factor FOXA [154]. However, the muscle-secreted signaling factor responsible for this systemic regulation of proteostasis in the brain and other tissues has not been identified.
In Drosophila, activation of the stress-sensing transcription factor FoxO specifically in skeletal muscle leads to improved muscle function due to preservation of proteostasis, as indicated by the transcriptional induction of autophagy and chaperones, and the consequent lower age-related accumulation of poly-ubiquitin protein aggregates in skeletal muscle [155]. In addition to these local effects, muscle-specific activation of FoxO also extends lifespan [155] and similar results are obtained with activation of 4E-BP, a FoxO target gene that regulates translation [155], and with drug-induced expression of FoxO in indirect flight muscles [156], indicating that skeletal muscle regulates organismal survival and resilience during aging [9, 152]. Interestingly, FoxO activation in skeletal muscle also reduces the accumulation of poly-ubiquitinated proteins in the brain and retina during aging [155]. These effects stem from systemic activation of autophagy, which derives from decreased secretion of insulin-like peptides from producing cells. Such systemic effects can arise from FoxO/4E-BP-regulated myokines and myometabolites [3]. Indeed, it was found that FoxO reduces the expression of the Drosophila activin ligand dawdle in skeletal muscle, and that reduced activin signaling improves proteostasis by sustaining the function of the autophagy/lysosome system [157]. Altogether, these studies in model organisms provide evidence for muscle-to-brain signaling and highlight how such a signaling axis can regulate proteostasis during aging.
As pointed out above, induction of the cytosolic and mitochondrial unfolded protein responses occurs in skeletal muscle during contraction in mice and humans [144–148] and may contribute to the protective muscle-to-brain signaling elicited by exercise. On this basis, we have recently used an experimental strategy based on the moderate, partial inhibition of the proteasome, the major system for protein degradation in skeletal muscle, to promote the accumulation of unfolded proteins in skeletal muscle and study the consequent local and systemic adaptations [158]. As expected, RNAi-mediated knockdown of proteasome subunits in Drosophila skeletal muscles reduced proteasomal activity, which in turn elicited a compensatory transcriptional program, largely mediated by C/EBP transcription factors and transcriptional upregulation of proteases [158].
Systemically, proteasome stress in skeletal muscle reduced age-related accumulation of protein aggregates in the central nervous system (brain and retina), suggesting that muscle-secreted factors may be involved in this inter-tissue signaling. To dissect the signaling mechanisms involved, we looked for myokines that are differentially modulated by proteasome stress. Amyrel was one of the most-highly upregulated myokines, and it was responsible for the long-range protective effects of muscle proteasome stress on the CNS. Specifically, muscle-specific overexpression of Amyrel improved proteostasis in the aging brain and retina whereas its knockdown partially impeded the protective stress response induced by RNAi for proteasome components in skeletal muscle [158]. Amyrel is an unusual myokine because it is not a cytokine or a growth factor but it is an amylase enzyme [159]. Although its function had been described in the intestine, there was no prior knowledge for the function of an amylase in skeletal muscle. Indeed, expression of Amyrel is minimal in non-stressed skeletal muscle but it is highly induced upon stress [158]. Likewise, AMY1/AMY2 amylases, which are mouse and human homologs of Amyrel, are profoundly induced by proteasome inhibitors (MG132) and aging in cortical brain organoids and skeletal muscle, respectively [158].
By virtue of its enzymatic activity [159], Amyrel breaks polysaccharides and oligosaccharides into the disaccharide maltose and, consistently, maltose levels increased in response to skeletal muscle-specific overexpression of Amyrel [158]. Maltose levels were previously found to increase in response to various insults to protein homeostasis such as cold [160], heat-shock [161], toxic Aβ expression [162], and inbreeding [163]. Possibly, maltose preserves proteostasis by virtue of its chemical chaperone activities. Interestingly, we found that maltose also induces a transcriptional response that consists of several proteases and chaperones with protective functions [158]. In particular, Amyrel-induced expression of the small heat shock proteins Hsp23 helps preserve proteostasis in the CNS during aging (Fig. 2 and ref. [158]). Moreover, Drosophila brains and retinas with reduced levels of SLC45 intracellular maltose transporters did not show an induction of sHsps expression and subsequently failed to preserve brain proteostasis with aging, confirming that maltose is indeed responsible for the transcriptional upregulation of chaperones in brain cells [158]. Interestingly, the disaccharide maltose is present in the human serum [164, 165] but little is known about its signaling functions in humans. The finding that a SNP with significant association to Parkinson’s disease is in proximity of a SLC45 family member, SLC45A3 [166], suggests that maltose transport may impact proteostasis and neurodegeneration also in humans.
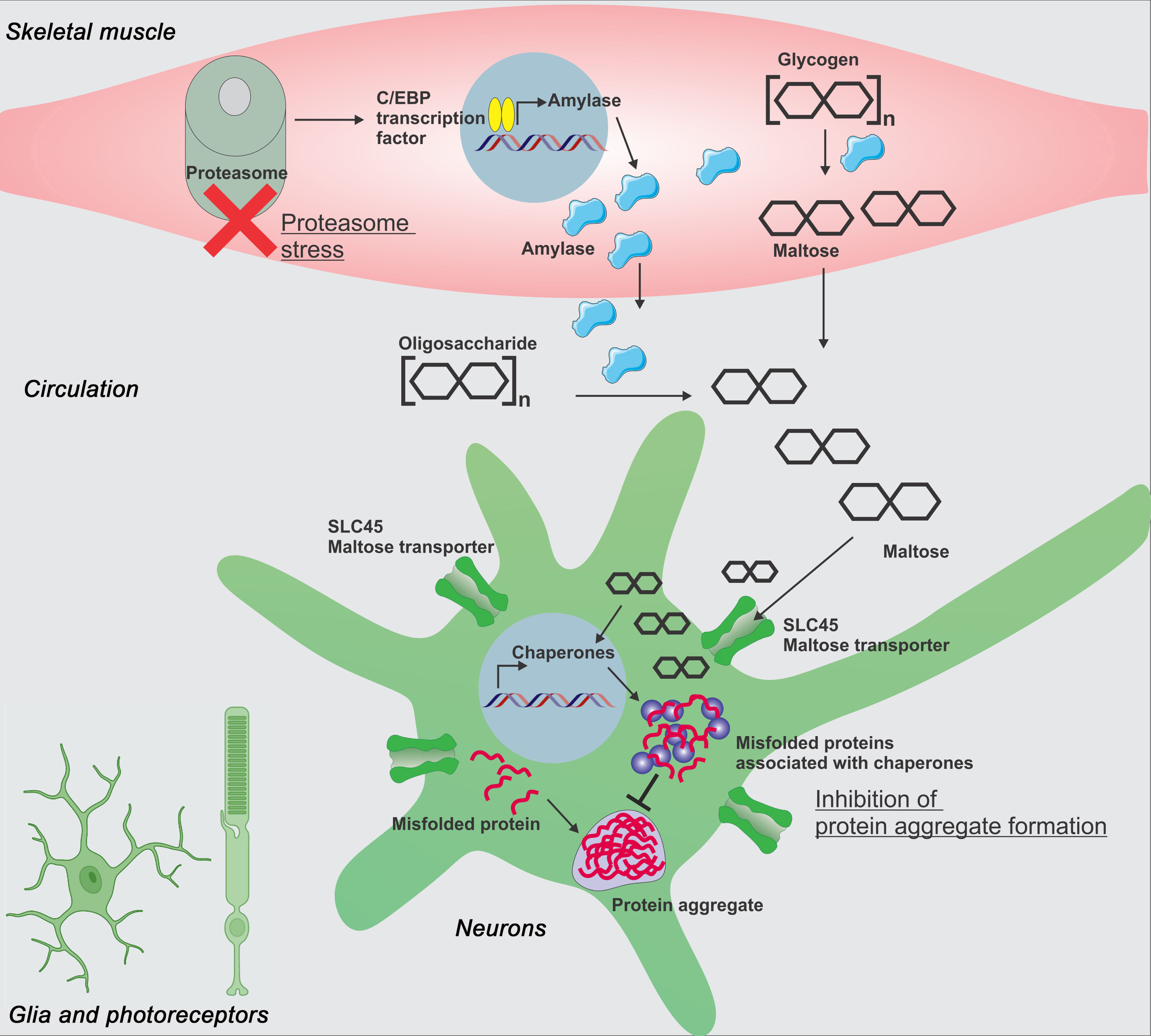
Proteasome stress in skeletal muscle induces a long-range protective response that preserves proteostasis in the aging brain and retina. Moderate perturbation of the proteasome (proteasome stress) in skeletal muscle increases Amyrel expression in muscle in a C/EBP-dependent manner. Amyrel is a stress-induced, muscle-secreted amylase that increases body levels of maltose via its enzymatic activity. In turn, maltose preserves proteostasis in the brain and retina through SLC45 intracellular maltose transporters. In addition to its chemical chaperone activity, maltose elicits a transcriptional response in neurons which leads to higher small heat shock protein levels. Chaperones in turn preserve proteostasis and prevent neurodegeneration by shielding unfolded and aggregation-prone proteins, thus avoiding their detrimental interaction with native functional proteins. Although these studies have primarily investigated the impact of Amyrel/maltose signaling in neurons and photoreceptors, glial cells are also likely to be protected by such adaptive stress signaling.
To gain insight into the function of maltose in humans, we treated human HEK293 cells and human cortical organoids with maltose and found that maltose preserves proteostasis when cells and organoids are challenged by thermal stress [158]. Moreover, maltose promoted the recovery of neuronal activity, which declines upon heat shock [158]. Interestingly, maltose also preserved the transcriptional profile of heat-shocked cortical organoids, including expression of genes involved in protein quality control such as CRYA (α-crystallin, homologous to Drosophila Hsp23) [158]. Altogether, this study has identified a muscle-to-brain signaling axis based on a stress-induced amylase enzyme, the disaccharide maltose, and the SLC45 intracellular disaccharide transporters [158].
Previously, the disaccharide trehalose was found to protect from neurodegeneration in Parkinson’s and Huntington’s disease models [167] and to extend lifespan in C. elegans [168]. Although autophagy induction and anti-oxidant functions have been proposed as responsible for such protective effects, the mechanisms underlying the action of trehalose remain elusive [167]. Because trehalose is not detected in vertebrates [167], it remains largely unexplored whether any endogenous disaccharide has neuroprotective and signaling roles in vertebrates. Our findings suggest that maltose may be important for preserving proteostasis in the CNS during aging. However, it is also possible that Amyrel regulates proteostasis via additional mechanisms. In particular, a recent study has reported that Amyrel has an additional enzymatic activity, 4-alpha-glucanotransferase [169], suggesting that other neuroprotective metabolites may also be produced by Amyrel and related AMY amylases.
Altogether, the studies here reviewed provide evidence on how initiation of stress signaling in skeletal muscle can be transmitted to the brain and induce adaptive stress responses that have presumably evolved for protecting the CNS from incoming homeostatic challenges perceived by peripheral tissues. Therapeutic activation of such resilience pathways during normal aging may help preserve brain function and protect from age-related neurodegeneration.
REGULATION OF MOOD AND SLEEPING PATTERNS BY MUSCLE-DERIVED SIGNALS
Exercise induces many organismal behavioral adaptations suggesting that signals released by contracting muscle can influence brain function [12, 67, 68]. Here, we review how exercise-induced signaling regulates sleeping and depression.
It is well known that individuals that regularly exercise are less likely to develop depression and mood fluctuations [170–174]. By using transgenic mice with skeletal muscle-specific PGC1α overexpression, Agudelo et al. found that these mice were resistant to developing depression [175]. Stress can activate the kynurenine pathway of tryptophan degradation in the liver, kidney, and immune cells, increasing the levels of kynurenine, which is known to modulate neurotransmission and is associated with neurodegenerative and psychiatric diseases [176]. In agreement, intraperitoneal administration of kynurenine raised its plasma levels like chronic mild stress and this induced a depressive state in wild-type but not in transgenic mice with skeletal muscle-specific PGC1α overexpression [175]. PGC1α elevates skeletal muscle expression of kynurenine aminotransferase (KAT) and this occurs also in response to exercise in mice and humans. KAT increases the conversion of kynurenine into kynurenic acid, a metabolite incapable of crossing the blood-brain barrier, and hence the brain is protected from kynurenine-induced depression [175]. Conversely, mice with skeletal muscle-specific loss of PGC1α do not resist stress-induced depression, and this further worsens upon kynurenine administration [175]. Because PGC1α expression in skeletal muscle declines with aging [177], this study also sheds light on a possible mechanism via which depression may increase in the elderly [178].
Exercise is also known to decrease sleep disorders and insomnia in middle- to older-aged adults [179], suggesting that muscle-to-brain signaling may also regulate sleep. A remarkable study supporting this model comes from the analysis of mice with the muscle-specific genetic disruption of the circadian transcription factor Bmal1 (brain and muscle ARNT-like factor). Detailed analysis of these mice revealed impaired glucose uptake and metabolism in muscle [180] and an aberrant sleep pattern that recapitulated sleep disorders found in Bmal1 whole-body knock-out animals [181]. Muscle-specific restoration of Bmal1 expression rescued non-rapid eye movement sleep amount in Bmal1 knock-out mice, and muscle-specific over-expression of Bmal1 in wild-type mice reduced the time required to recover from sleep loss [181]. These and other observations indicate that muscle is a key peripheral clock [182–185], and that muscle-to-brain signaling is an important determinant of sleep behavior. To determine the mechanism involved, Harfmann et al. utilized a targeted metabolomics approach and found changes in TCA cycle metabolites and compensatory increases in amino acids [180]. These metabolites are likely secreted from muscles and taken up by the brain to regulate normal sleep patterns and length. Future studies to validate the involvement of these and other factors in muscle-to-CNS signaling will provide important insights into the role of skeletal muscle in regulating sleep. Because derangement of sleep patterns is also associated with neurodegeneration [186], understanding how muscle regulates sleep will also shed light on mechanisms responsible for the influence of skeletal muscle on neurodegeneration.
Altogether, these studies highlight the importance of muscle-brain signaling for normal brain function and for preventing the development of psychiatric disorders.
CENTRAL CONTROL OF FEEDING BEHAVIOR by MYOKINES
The CNS regulates feeding choices by integrating external sensory cues with internal signals that inform the brain of the energy status of peripheral tissues [187]. Although the role of many peripheral tissues such as the liver, pancreas, gut, and white adipose tissue is well established [187], relatively little is known on how skeletal muscle contributes to the central control of feeding behavior by the brain.
Circumstantial evidence indicates that exercise and muscle-secreted factors profoundly impact the total amount of ingested food as well as feeding choices [188, 189]. For example, running prevents hyperphagia and obesity in rats by normalizing meal patterns, food intake, and body weight [190] and by decreasing the preference for highly palatable, energy-dense, fatty foods [191, 192]. Likewise, it was found that moderate exercise reduces the preference for high-fat diets in obese mice, whereas preference for sucrose and milk was increased. This exercise-induced change in food preference appeared to be mediated by dopaminergic neurons of the ventral tegmental area-nucleus accumbens [193]. Here below, we discuss recent studies that support a role for skeletal muscle in influencing feeding behavior via myokine signaling.
In Drosophila, it was found that skeletal muscle regulates feeding initiation via decapentaplegic (Dpp), a myokine homologous to human BMP2, BMP4, and related BMP/GDF family members. Similar to other myokines that are modulated by nutrients and nutrient-sensing pathways [3, 194], Dpp is transcriptionally induced by mTOR signaling in skeletal muscle via the transcription factor Mnt and reaches the brain through the circulation to communicate the energy status of skeletal muscle and consequently adjust feeding behavior [195]. Specifically, muscle-restricted Dpp RNAi promotes foraging and feeding initiation, whereas Dpp overexpression reduces these food-seeking behaviors. Mechanistically, muscle-derived Dpp regulates Dpp receptor signaling in dopaminergic neurons, which have known roles in promoting feeding initiation [195]. Specifically, reduction in Dpp receptor signaling promotes tyrosine hydroxylase (TH) expression and dopamine biosynthesis in the brain, and this promotes feeding initiation [195]. Conversely, muscle-specific Dpp overexpression downregulates brain TH expression via the transcriptional repressor Schnurri and reduces feeding initiation. Altogether, this study demonstrates a role for myokine signaling in the regulation of brain dopamine biosynthesis and feeding behavior [195].
In mice, it was found that skeletal muscle contributes to the central control of feeding behavior in response to partial inhibition of the electron transport chain, which activates the mitochondrial unfolded protein response (UPRmt). Specifically, Chung et al. [196] used transgenic mice deficient specifically in skeletal muscle for Crif, a gene encoding a component of the large subunit of the mitoribosome, which resulted in reduced oxidative phosphorylation in skeletal muscle, activation of the C/EBP transcription factor CHOP, and UPRmt induction. Transcriptomic data identified GDF15 (growth differentiation factor 15) as a TGF-β ligand that is induced by CHOP in skeletal muscles [196]. Mice lacking Crif in skeletal muscle display increased fatty acid oxidation and heightened phosphorylation of hormone-sensitive lipase and ERK1/2, indicative of increased lipolysis [196]. Consequently, administration of GDF15 decreased body weight and improved glucose tolerance in obese mice by increasing lipolysis, suggesting an important role for GDF15 in systemic metabolism [196, 197]. Because GDF15 is secreted at high levels from skeletal muscle, discovering receptors for GDF15 in non-muscle tissues was important to gain insights into its systemic effects. Hsu et al. [197] identified glial cell-derived neurotrophic factor (GDNF) receptor alpha-like (GFRAL) as a brainstem-restricted GDF15 receptor that is not expressed in any other part of the brain or any peripheral tissue. GDF15-mediated reduction in body weight and food intake is observed in wild-type mice but is completely impeded in Gfral knock-out mice, strengthening the idea that GDF15 influences systemic metabolism through signaling mediated by brain-stem GFRAL receptors [197].
Consistent with the fact that the parabrachial nucleus and the central amygdala regulate meal termination in normal conditions [198] and also control food intake and body weight in disease states [199–201], GDF15 increased the activity of neurons in the parabrachial nucleus and in the central amygdala, as indicated by immunostaining for Fos, a marker of neuronal activity [197]. Another interesting observation was that the Gfral knock-out mice showed normal food intake and body weight if provided a normal diet but gained weight when on a high-fat diet. Circulating GDF15 increases during exercise [202] but also in various disease states such as cancer, cardiovascular disease, kidney dysfunction, inflammation, and obesity [200], suggesting that GDF15-GFRAL may help relay information from stressed tissues, including contracting skeletal muscle, to the brain. In agreement with this model, exogenous and endogenous GDF15 activates the hypothalamic-pituitary-adrenal (HPA) axis and the circulating levels of glucocorticoid stress hormones [203].
Beyond these examples, many other myokines may contribute to the central regulation of feeding behavior by the brain in response to muscle-derived cues. For example, FGF21 is a myokine induced by exercise and cellular stress [204, 205]. It was found that mitochondrial uncoupling in skeletal muscle due to UCP1 expression induces the integrated stress response and FGF21 expression [206]. Likewise, inhibition of muscle autophagy induces a protective stress response centered on FGF21 upregulation [207]. Specifically, these mice display increased fatty acid oxidation and browning of white adipose tissue and were protected from diet-induced obesity and insulin resistance [207]. The systemic metabolic adaptations induced by muscle-derived FGF21 may also include regulation of feeding behavior. In support of this hypothesis, it was found that FGF21 receptor signaling in the brain is needed for mounting feeding choices to cope with protein restriction in mice [208]. Specifically, brain knock-out mice for the FGF21 co-receptor β-Klotho do not select protein-containing foods when presented with this choice even after being on a protein-restricted diet [208]. Other studies have found that FGF21 administration reduces sweet and alcohol preference in mice and humans [63, 209, 210]: this effect requires the FGF21 co-receptor β-Klotho in the brain and is associated with lower dopamine concentrations in the nucleus accumbens [209]. Importantly, also the actions of FGF21 on other organs such as the liver appear to be mediated by FGF21 signaling to the hypothalamus and activation of the HPA axis, which promotes hepatic gluconeogenesis via glucocorticoids [211, 212].
Although these studies did not examine the relative contribution of muscle-derived FGF21 to these behaviors compared to FGF21 derived from other peripheral tissues, FGF21 secreted by skeletal muscle may acquire prominent roles in particular in conditions known to upregulate muscle FGF21 levels, such as exercise and mitochondrial dysfunction [205]. Apart from these functions in feeding behavior, muscle-secreted FGF21 may also protect from neurodegeneration via its capacity to promote brain mitochondrial function, synaptic plasticity, and cognition [62, 213–215].
It was previously found that muscle-specific activation of FoxO/4E-BP signaling improves skeletal muscle function and systemic proteostasis but also reduces feeding behavior in adult Drosophila [155]. A decrease in feeding behavior was also found by modulation of this pathway in Drosophila larval muscles [216]. Although the FoxO/4E-BP-induced myokines responsible for the modulation of feeding behavior have not been identified in Drosophila, it is interesting to note that transgenic activation of 4EBP1 in mouse skeletal muscle (which was accompanied by higher FoxO1 levels) protects from age- and diet-induced insulin resistance and metabolic decline via enhanced secretion of FGF21 by skeletal muscle [217]. Therefore, based on the known roles of FGF21 in this process [63, 209, 210], FoxO/4E-BP signaling in skeletal muscle may regulate feeding behavior via muscle-to-brain signaling mediated by FGF21 and related myokines.
Altogether, these studies indicate that skeletal muscle contributes to the central regulation of feeding behavior by the brain via muscle-secreted signaling factors. Because myokines influence the control of feeding behavior exerted by the CNS together with other circulating factors, future studies should determine how signals originating from muscles are integrated with those originating from other peripheral tissues.
IMPACT OF INACTIVITY AND ATROPHY on MUSCLE-BRAIN SIGNALING
Contrary to adaptive protective muscle-to-brain signaling induced by exercise [12, 67, 68], lack of physical activity is extremely deleterious for the entire organism, including the CNS and the skeletal muscle itself [218, 219]. Cast immobilization of the hindlimbs mimics muscle disuse and results in muscle mass loss in 5XFAD mice that are predisposed to AD [220]. Conditioned media from cultures of atrophied muscles from cast-immobilized mice identified several secreted factors that are differentially regulated. In particular, hemopexin was found to be the most significantly induced protein compared to non-cast 5XFAD and wild-type control mice [220]. Concomitantly, hemopexin transcript levels increased in the hind limb muscles but not in the hippocampus. Higher hemopexin protein levels were detected in the plasma and hippocampus of cast immobilized 5XFAD mice compared to non-cast 5XFAD mice, suggesting that secreted hemopexin transits through the circulation [220]. Consistent with the known role of hemopexin in inflammation, cast-immobilized 5XFAD mice showed marked memory decline already at a young age. Similar memory deficits were also observed upon intracerebroventricular infusion of hemopexin in both 5XFAD and wild-type young mice [220]. The authors propose that the mechanism of action of hemopexin may at least in part depend on lipocalin-2, a glycoprotein that is markedly upregulated in the hippocampus of 5XFAD mice infused with recombinant hemopexin, compared to vehicle-infused counterparts, and that is known to promote neuroinflammation [220].
These results pinpoint the importance of maintaining skeletal mass and function to preserve cognitive abilities. The association between skeletal muscle function and the risk of neurodegeneration is indeed consistent with demographic studies showing that old subjects with low handgrip strength and low gait speed have a significantly higher chance of displaying cognitive impairment compared to age-matched subjects that better maintained muscle mass and function [221–226]. In addition to hemopexin, this and other studies [51, 227] have found myokines that are differentially expressed in atrophied versus control muscles, highlighting that the production of myokines is affected by signaling pathways that impact muscle wasting, as also recently found with an integrated transcriptomic/proteomic analysis of different modes of muscle atrophy in mice [228].
There are also alternative mechanisms via which diseased muscles could impact the CNS. Loss of proteostasis is increasingly appreciated not only as a component of age-related neurodegeneration but also as a driver of skeletal muscle aging (sarcopenia) and age-related myopathies in Drosophila, mice, and humans [152, 155, 229, 230]. In this framework, it has been proposed that aggregation-prone pathogenic proteins could be released by skeletal muscle and other peripheral tissues and subsequently be uptaken by cells in the CNS, where they could contribute to derange protein quality control and to promote neurodegeneration [8, 231, 232]. A study in C. elegans has found some evidence for inter-tissue transmission of pathogenic huntingtin between skeletal muscle and neurons and such myofiber-to-neuron spreading of huntingtin led to neurodegeneration and reduced lifespan [232]. For these studies, Kim et al. [232] utilized transgenic expression of huntingtin exon 1 with poly-glutamine tract fused with either the N-terminal or C-terminal part of the fluorescent protein Venus. These split versions of Venus-huntingtin were then expressed specifically in pharyngeal skeletal muscles and neurons, respectively. In this experimental setup, Venus fluorescence is detected only when the N-terminal and C-terminal fragments are located within the same cell, which indicates transcellular transmission of Venus-huntingtin fragments from one tissue to the other. Bimolecular fluorescent complementation indeed revealed transmission of pathogenic huntingtin between pharyngeal skeletal muscles and neurons, and vice versa [232]. However, it remains unclear whether this is a regulated process or rather occurs because of cellular damage, and whether this is a relevant mechanism of neurodegeneration also in higher organisms. Nonetheless, this study suggests that unexpected mechanisms such as this might contribute to muscle-to-brain signaling.
Although most studies have focused on changes in the expression of myokines in muscle, the modulation of myokine secretion is a less-explored mechanism that may also contribute to modulate muscle-to-brain signaling in response to exercise as well as inactivity. An in-silico survey of Drosophila myokines revealed that ∼80% have signal peptides that guide their secretion via the ER-Golgi but that ∼20% lack such motifs [230]. These findings suggest that some myokines may be secreted via unconventional routes, such as vesicles (e.g. exosomes) released by skeletal muscle: these vesicles may transport signaling factors that are not predicted to be secreted and potentially also increase the signaling range of canonical myokines [3, 233–235].
An additional unconventional route of myokine secretion may consist in secretory autophagy, i.e. a version of macroautophagy whereby the content of autolysosomes is released into the extracellular space via direct fusion with the plasma membrane [236, 237]. This may occur in response to external and internal stimuli that stimulate autophagy, such as stress responses and exercise [238, 239]. In this model, secretory autophagy may regulate the release of specific pools of myokines and of metabolites produced by lysosomal degradation. In agreement with this model, lysosomal enzymes are known to be secreted by myofibers [3, 240] as exemplified by the identification of lysosomal cathepsin B as a neuroprotective factor secreted by skeletal muscle following exercise [57]. However, inactivity may grossly impact such autophagy-mediated mode of myokine/myometabolite secretion and hence result in blatant derangement of muscle-to-brain signaling. The use of new mapping technologies to identify circulating factors derived from a specific tissue source [241] may help define how myokine secretion is reshaped by exercise and aging. Altogether, much remains to be learnt about the mechanisms that regulate myokine secretion, and on the impact of inactivity on muscle-to-brain signaling.
CONCLUSIONS
The understanding of skeletal muscle-to-CNS signaling and its impact on the regulation of neurodegeneration and brain-dependent behaviors represents a promising avenue for healthy aging interventions. Throughout the review, we have highlighted how myokines can have a general impact or rather differentially regulate distinct cell types in the central nervous system. Whereas most of the reviewed literature is based on model organisms (e.g. mice, rats, and Drosophila) and experimental disease models, these studies have identified evolutionary-conserved myokine signaling pathways that may well play similar roles in muscle-brain signaling in humans (Table 1). Transferability of this knowledge into humans may lead to the development of therapies based on the delivery of recombinant versions of protective myokines, myometabolites, or boosters of their endogenous expression and/or release that may find application in the treatment of dementia and related conditions.
List of major myokines and myometabolites known to act on the central nervous system
Most of these myokines have been characterized in mice and other mammalian models (human, monkeys) ad in the fruit fly Drosophila (when indicated). All are evolutionary well-conserved and likely to play similar roles in humans.
Because of the protective effects of exercise on a number of organ systems including the CNS, there is growing interest in the identification of exercise mimetics, i.e. therapeutics that mimic or enhance the effects of exercise [242–247]. In addition to mimicking local adaptive responses induced by exercise in skeletal muscle and other tissues, these interventions may rely on the induction of exerkines, i.e. exercise-induced circulating factors secreted by skeletal muscle and non-muscle tissues, and/or potentiation of their signaling in the CNS [242–247]. Because several myokines and myometabolites are secreted by skeletal muscle in response to exercise, there could be overlap and synergy of efforts in the identification of exerkine- and myokine-based therapeutics. However, exerkines are produced also by non-muscle tissues: therefore, efforts to identify exercise mimetics may more globally reproduce the benefits of exercise. On the other hand, as reviewed here, exercise is only one of the stimuli that regulate expression and release of protective myokines, in addition to nutrient sensing, stress signaling, and others. Therefore, studies centered on myokines and myometabolites may highlight muscle-brain signaling axes that preserve CNS function and protect from age-related neurodegeneration not only in response to exercise but also in response to other protective interventions, such as dietary restriction/intermittent fasting and adaptive stress signaling. Moreover, analysis of detrimental myokines produced in response to inactivity and muscle disease states may also provide complementary means for therapeutic intervention.
Altogether, concerted efforts in understanding the role of peripheral signals originating from muscle and non-muscle tissues in response to exercise and other stimuli may help reap the full therapeutic potential of myokine and exerkine-based interventions.
Footnotes
ACKNOWLEDGMENTS
The illustrations were in part made with BioRender.
FUNDING
This work was supported by research grant to F.D. from the National Institute on Aging (R01AG055532 and R56AG63806). Research at St. Jude Children’s Research Hospital is supported by the ALSAC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST
The authors have no conflict of interests to report.