
Abstract
Attempts to develop a disease modifying intervention for Alzheimer’s disease (AD) through targeting amyloid β (Aβ) have so far been unsuccessful. There is, therefore, a need for novel therapeutics against alternative targets coupled with approaches which may be suitable for early and sustained use likely required for AD prevention. Numerous in vitro and in vivo studies have shown that flavonoids can act within processes and pathways relevant to AD, such as Aβ and tau pathology, increases in BDNF, inflammation, oxidative stress and neurogenesis. However, the therapeutic development of flavonoids has been hindered by an ongoing lack of clear mechanistic data that fully takes into consideration metabolism and bioavailability of flavonoids in vivo. With a focus on studies that incorporate these considerations into their experimental design, this review will evaluate the evidence for developing specific flavonoids as therapeutics for AD. Given the current lack of success of anti-Aβ targeting therapeutics, particular attention will be given to flavonoid-mediated regulation of tau phosphorylation and aggregation, where there is a comparable lack of study. Reflecting on this evidence, the obstacles that prevent therapeutic development of flavonoids will be examined. Finally, the significance of recent advances in flavonoid metabolomics, modifications and influence of the microbiome on the therapeutic capacity of flavonoids in AD are explored. By highlighting the potential of flavonoids to target multiple aspects of AD pathology, as well as considering the hurdles, this review aims to promote the efficient and effective identification of flavonoid-based approaches that have potential as therapeutic interventions for AD.
Keywords
ALZHEIMER’S DISEASE: THE NEED FOR NOVEL THERAPEUTICS
At the time of writing, there are at least 50 million people living with dementia worldwide [1]. Due to increasing life expectancies, this is predicted to triple by 2050, and yet there are still no therapeutics capable of preventing or slowing the onset of dementia. The most common cause of dementia, Alzheimer’s disease (AD), is a neurodegenerative disease that is traditionally characterized by deposition of amyloid plaques and neurofibrillary tangles (NFTs), caused by amyloid β peptide (Aβ) and tau aggregation respectively. More recently, chronic neuroinflammation and gliosis have joined plaques and tangles as hallmarks of AD. One of the major obstacles to preventing AD progression is that by the time a clinical diagnosis has been given, irreversible brain atrophy has already occurred (Fig. 1), and pathological cascades are well developed.
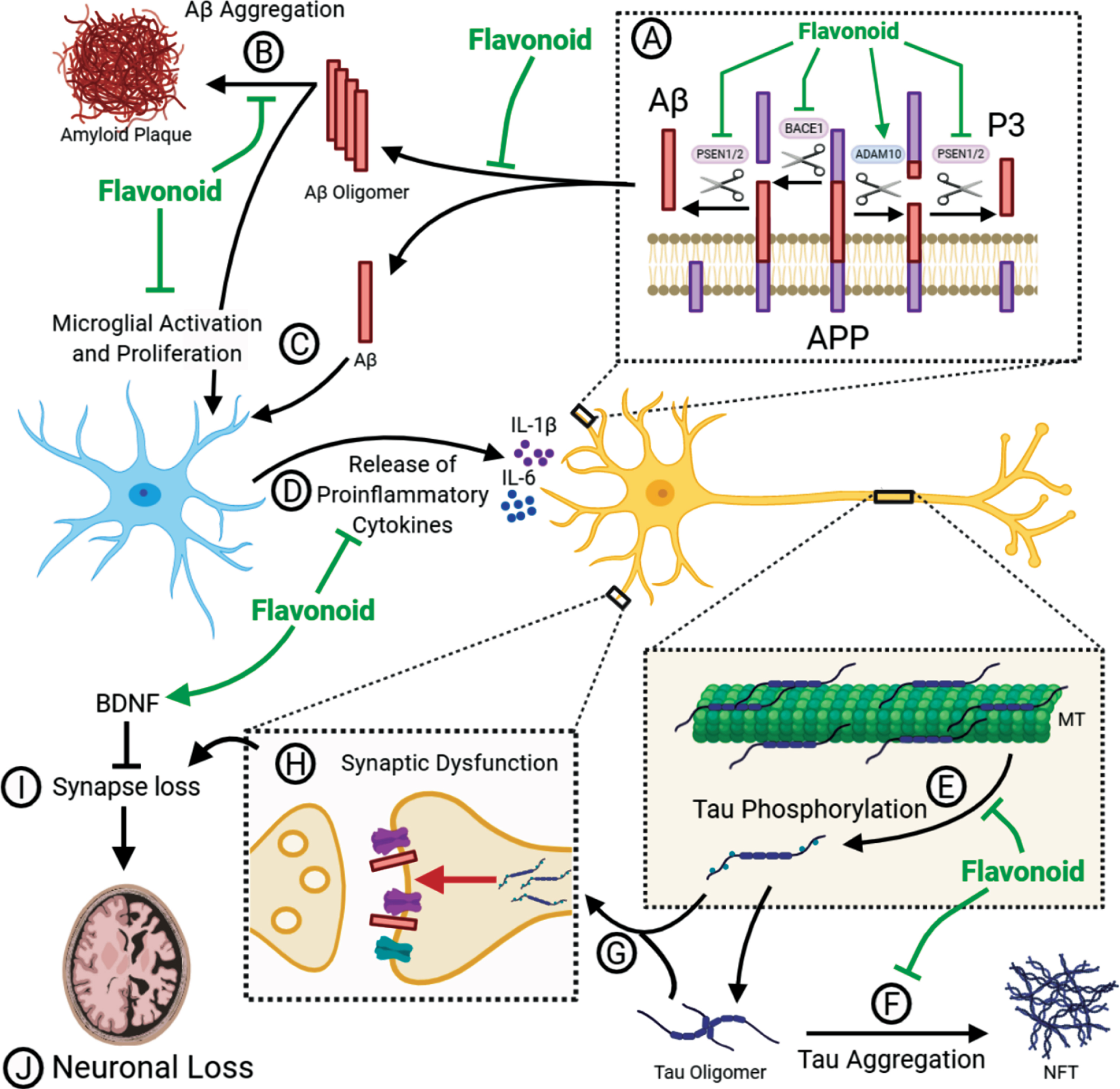
Flavonoids can act as multi-modal inhibitors of AD pathology. (A) APP is processed in two pathways. In the non-amyloidogenic pathway, APP is cleaved by α-secretase (ADAM10) to produce α-C-terminal fragment (α-CTF) and secretory APP (sAPPα) whereas the pro-amyloidogenic pathway involves cleavage of APP β-secretase (BACE1) to produce β-CTF and sAPPβ. α-CTF and β-CTF is then cleaved by γ-secretase to release P3 and Aβ, respectively, as well as the APP intracellular domain (AICD). Flavonoids have been shown to inhibit βγ-secretase processing as well as promoting α-secretase processing. This causes a shift towards the non-amyloidogenic pathway and reduces the levels of Aβ produced. (B) Aβ can self-aggregate to form oligomers and eventually amyloid plaques. Flavonoids may be able to inhibit the formation of amyloid plaques by binding to Aβ and inhibiting aggregation or promoting the formation of non-toxic off-target oligomers. (C) Toxic Aβ monomers and oligomers have been shown to induce microglial activation and proliferation. Animal models have shown that flavonoid intervention can reduce the levels of gliosis in the brain. (D) Activated microglia secrete pro-inflammatory cytokines such as IL-1β and IL-6. Several flavonoids have been shown to reduce the levels of these cytokines in vivo. (E) The microtubule (MT) associated protein tau, which is predominantly located in the axon, is hyperphosphorylated in AD, perhaps as a result of pro-inflammatory cytokine release. This causes the dissociation of tau from the microtubule and mislocalisation to the somatodendritic region. Flavonoids can inhibit several kinases associated with tau phosphorylation, as well as attenuating the proinflammatory response. Therefore, flavonoids have the potential to reduce tau phosphorylation. (F) Hyperphosphorylated tau can self-aggregate to form toxic oligomers and eventually neurofibrillary tangles (NFT). There is evidence that flavonoids can bind to tau and inhibit its aggregation or promote the formation of non-toxic oligomers. (G) Hyperphosphorylated tau can mislocalise to post-synaptic terminals. Synaptic tau and Aβ can cause synaptic dysfunction (H) and eventual synapse loss (I). Flavonoids have been shown to upregulate BDNF to promote adult hippocampal neurogenesis and synaptogenesis. Upregulation of BDNF may, therefore, prevent the loss of synapses and the consequent loss of neurons (J).
The amyloid cascade hypothesis (ACH) postulates that it is the accumulation of Aβ oligomers that initiates a downstream cascade causing neuroinflammation, tau-induced toxicity, synaptic dysfunction and neuronal loss [2]. Support for this hypothesis comes from the identification that mutations in the amyloid precursor protein (APP), presenilin-1 (PS1) [3, 4] and presenilin-2 (PS2) [5, 6], which either enhance the accumulation of Aβ or promote an increased ratio of the longer, more aggregation prone Aβ1–42 over Aβ1–40 [7–9], are sufficient to cause early onset familial AD (FAD) [10]. APP can be processed in either a non-amyloidogenic pathway, driven by the α-secretase enzyme ADAM10, or in a pro-amyloidogenic pathway involving the β-secretase enzyme BACE1. Following cleavage by either of these enzymes, there is a second cleavage event by γ-secretase which yields either p3 (non-amyloidogenic) or Aβ (pro-amyloidogenic) (Fig. 1A). Presenilin forms the catalytic subunit of the γ-secretase enzyme complex [11]. According to the ACH, the failure to clear Aβ1–42 initiates consequent pathological pathways that lead to the onset of dementia [2]. This hypothesis has been the foundation for the development of many Aβ-targeting therapeutics, with the idea that by targeting this initial Aβ accumulation, the resulting toxic pathways can be inhibited.
Disappointingly, therapeutics targeting APP processing directly, such as BACE1 inhibitors, have failed at Phase III [12], likely due to off target effects [13]. Moreover, methods to reduce soluble and/or aggregated forms of Aβ via antibody strategies have had disappointing outcomes [14, 15], although, it should be noted that recent reports claim that the Aβ-targeting antibody Aducanumab meets its primary endpoint. It is becoming increasingly clear that reducing the levels of Aβ in the brain is unlikely to provide significant clinical benefit when delivered at the stage of mild cognitive impairment (MCI) or later. It is likely that, in order to be maximally effective, the use of Aβ-targeting therapeutics will be required from the start of, and throughout, the long prodromal phase of AD. This limitation necessitates a strategic shift, either switching attention to targets that correlate better with later stage disease progression, such as neuroinflammation or tau, or sticking with the existing targets but putting the focus on biomarker development to enable better patient stratification for earlier intervention. An additional approach is to identify and validate safe and cost-effective lifestyle interventions which could be widely implemented throughout mid-life to reduce AD risk at a population level.
Flavonoid rich diets: Impact on cognitive decline and AD
Epidemiology has long suggested that diets rich in polyphenols, such as the Mediterranean diet, might slow age-related cognitive decline and some studies indicate reduced risk of dementia [16, 17]. Moreover, consumption of flavonoids, a large family of dietary polyphenol compounds, decreases cognitive decline with aging [18, 19]. Although direct evidence of a reduction in AD is lacking, the available epidemiological data does suggest that flavonoid consumption has the potential to ameliorate AD pathology and provide symptomatic benefit.
There are more than 5000 types of flavonoid, and they are found almost ubiquitously in plants and thus are widely available in the human diet. The six main subclasses of flavonoids are: anthocyanidins, flavones, isoflavones, flavonols, flavanones, and flavan-3-ols (flavanols) (Fig. 2). While flavonoids are well known for their ability to act as antioxidants, it is clear that they are also capable of regulating intracellular responses principally through modulation of protein kinase signalling pathways [20]. Furthermore, there is now a substantial and growing body of evidence supporting the ability of flavonoids to interfere in AD-related pathways [21].
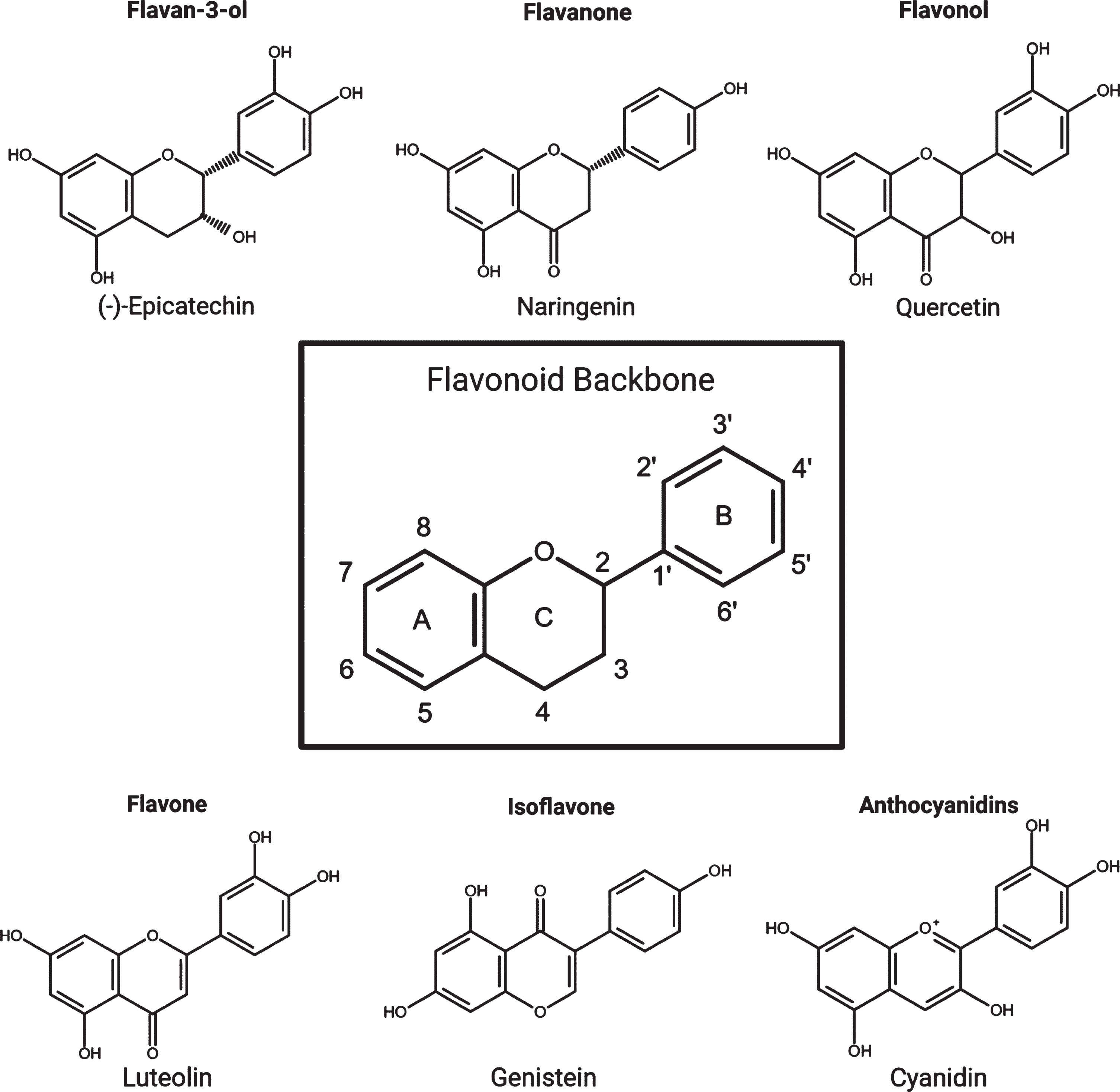
The flavonoid backbone and the six main subgroups with examples from each.
However, the development of flavonoids as therapeutic interventions for AD has been hindered by considerable uncertainty surrounding bioavailability, metabolism and basic pharmacokinetics. In this review emerging mechanistic evidence for flavonoid-mediated modulation of AD pathways is evaluated, with a focus on studies where genuine consideration has been given to the bioavailability of flavonoids. The metabolism and bioavailability of flavonoids in both humans and animal models, including the increasingly evident role of the gut-microbiome to these outcomes will be highlighted. Considering this, approaches to improve the translatability of flavonoids into therapeutics for AD will be discussed. Finally, we will examine whether supplement or more conventional drug design approaches based on flavonoid scaffolds represents the best way forward.
INHIBITING AD PATHOLOGY WITH FLAVONOIDS: MECHANISMS OF ACTION
There are a very large number of studies describing protective effects of various polyphenols against AD-relevant insults following their administration to cell lines but, unfortunately, we are unable to include them all here. Likewise, several studies show that flavonoids can act as acetylcholinesterase (AChE) inhibitors but, as AChE inhibitors are symptomatic interventions and not preventative or disease modifying, they will not be discussed here (for review see [22]). A major advance in the last decade has been the investigation of flavonoids in mouse models of AD. Here, the analysis of these in vivo studies will be mainly limited to those that have employed an oral route of polyphenol administration in drinking water or food, as well as via oral gavage; a summary of the outcomes of those studies that used oral methods of delivery is shown in Table 1. The doses investigated range from 1–500 mg/kg/day which in humans would be equivalent to 0.07–35 g intake for a 70 kg adult. While little is known about the exact binding partners of most flavonoids, common molecular mechanisms have emerged which impact on Aβ and tau pathology, neuroinflammation, oxidative stress and neurogenesis (Fig. 1).
A summary of the results of flavonoid interventions in AD mouse models. Only interventions that used an oral method of delivery such as in water, diet, oral gavage (O.g.) or intragastric administration (I.g.) were included. The method of delivery was classified as ‘orally’ where further detail was not supplied. Treatment times of 28–30 days were rounded to 1 month. A decrease in Aβ pathology was classified as a decrease in the levels of soluble or insoluble Aβ1–40 or Aβ1–42, a reduction in visible Aβ plaques or a shift in APP processing. A decrease in gliosis involved decreased levels of microglia or astroglia. Pro-inflammatory markers included iNOS, COX2, NF-κB, TNF-α, M-CSF ICAM-1, TLR4, NLRP3, IL-16, IL-1β, IL-6, IL-17A, IL-12p70 and the JAK2/STAT3 pathway. Oxidative stress markers included Nrf2, SOD1, GPx, GSH, H2O2, MDA, CAT or HO-1. Synaptic markers included PSD95, SNAP23, SNAP25, Arc, Homer, Synaptotagmin, Synapsin, Spinophilin, Gephyrin, Synaptophysin and glutamatergic receptor subunits. Evidence for pro-survival/neurogenesis included increases in BDNF, activation of the TrkA pathway and inhibition of p75NTR pathway as well as decreased neuronal apoptosis and corresponding markers such as caspase-3. Improvement in the Morris Water Maze was classed as a reduced escape latency, an increased time spent in the target quadrant and an increase in the number of platform crossings. () A significant effect was found; (cross)no significant effect was found; (–) this variable was not investigated. * Oxidative stress was identified as a decrease in anti-oxidative enzymes
Flavonoid inhibition of APP processing and Aβ deposition
Several flavonoids have shown the capacity to ameliorate Aβ pathology, both in vitro and in vivo (Table 1). This effect may come as a result of a shift towards the non-amyloidogenic processing pathway, enhanced degradation of Aβ and inhibition of aggregation (Fig. 1A, B). The resultant effect is a net reduction in Aβ42. A well-established example of flavonoid-mediated inhibition of APP processing is seen with epigallocatechin gallate (EGCG), a much studied catechin that is abundant in tea. EGCG has been shown in vitro to dose dependently increase the proportion of mature ADAM10 (mADAM10) relative to pro-ADAM10, correlating with a shift towards non-amyloidogenic processing and a decrease in Aβ production [60, 61]. The increase in proteolytically active mADAM10 by EGCG was found to occur through at least two pathways: oestrogen receptor α (ERα)-mediated activation of the PI3K/Akt pathway, and activation of the pro-ADAM10 cleavage enzyme furin through a PI3K-independent mechanism [62]. The finding that EGCG shifts APP processing towards the non-amyloidogenic pathway has been confirmed in vivo in several different mouse models of AD [32, 35–37]. Oral administration of EGCG was found to promote α-secretase activity while down regulating β- and γ-secretase activity, and as such decreased the levels of both soluble and insoluble Aβ.
Another example is that of the flavone baicalein, which increases non-amyloidogenic cleavage of APP while inhibiting pro-amyloidogenic cleavage through a GABAA receptor-dependent pathway [63]. GABAA receptor activation has been shown previously to promote non-amyloidogenic processing of APP [64]. Furthermore, baicalein may regulate GABAA receptor activation [65], potentially through inhibition of GABA transaminase which degrades GABA [66].
Moreover, the modulation of APP processing by flavonoids is known to occur at very low concentrations that could potentially be found in the brain. For example, (–)-epicatechin ((–)-EC) inhibits βγ-secretase dependent APP processing with a low IC50 of 20.5 nM, which correlates with reduced Aβ pathology in TASTPM mice treated with (–)-EC [39]. Furthermore, both quercetin (100 nM) and quercetin-3-O--rutinoside (rutin) (50 nM, 100 nM) significantly increased degradation of BACE1 in SH-SY5Y cells over expressing APPs we [67]. These effects at low concentrations enhance the likelihood that flavonoids could modulate APP processing in vivo.
Another mechanism through which flavonoids could decrease the levels of Aβ in the brain is by enhancing degradation of Aβ. Neprilysin (NEP), a zinc-metalloprotease, is an Aβ-degrading enzyme which is decreased in affected areas of the AD brain [68]. EGCG (1–3μg/ml) increases NEP release from astrocytes in an Akt/PI3K-dependent manner, resulting in degradation of exogenous Aβ [69]. Consequently, intragastric administration of EGCG to SAMP8 mice, a senescence model of AD, increased NEP expression and reduced Aβ level same lio rating spatial learning [29]. Similarly, oral administration of a combination of green tea catechins, including EGCG, to a transgenic human APP over expression mouse caused an increase in NEP levels, as well as decreased Aβand improved spatial learning [70]. Hence, EGCG can promote the degradation of Aβ, as well as inhibiting production.
Alternatively, flavonoids have the potential to target Aβ through direct binding and inhibition of aggregation which has been demonstrated in vitro in aggregation assays [71–76] (Fig. 1B). A limitation of these studies is that they are typically undertaken at high stoichiometric Aβ-flavonoid ratios that would not easily be achieved in vivo. Nevertheless, such in vitro aggregation assays are useful in identifying flavonoid structures with anti-aggregative properties that could be used as molecular scaffolds for the design of therapeutics. Consideration of the physiological stoichiometry, impact of the cellular environment and posttranslational modifications of the target protein will enhance the overall translatability in vivo.
While in vivo experiments have shown that flavonoid treatments reduce the levels of insoluble, aggregated Aβ, it is not yet possible to determine whether this is due to direct flavonoid binding to the target protein or interactions with other proteins such as kinases, which modulate the tendency for amyloidogenic proteins to aggregate. Likewise, it is tempting to relate the changes in spatial learning and memory observed in through flavonoid intervention in APP-based models of AD[23–25, 30–32, 36, 41–43, 46, 49–53, 54–59, 77–85]to reductions in Aβ (Table 1). However, the behavioural recovery observed could be due to recruitment of other mechanisms, either downstream of Aβ signalling or involved in physiological cognition, particularly as not all studies with reductions in Aβ-pathology correlated with behavioural improvements [38, 51] and vice versa [77, 83, 84]. Indeed, there is evidence to suggest that flavonoids modulate other disease-associated mechanisms, therefore having the potential to act as multi-modal AD therapeutics.
Flavonoid inhibition of tau phosphorylation and aggregation
Tau is a microtubule-associated protein that, physiologically, binds to microtubules predominantly in axons [86, 87]. The primary function of tau is thought to be the stabilisation and organisation of microtubules, aiding in the transport of cargo to and from synapses. However, in AD, tau is hyperphosphorylated [88, 89], and dissociates from the microtubule (Fig. 1E). Hyperphosphorylation of tau promotes its mislocalization to the somatodendritic region [86] (Fig. 1G) where it has been shown to impair nuclear import [90], and synaptic function [91] (Fig. 1H). Tau accumulation in the somatodendritic region has also been associated with local translation of tau mRNA, which can be induced by Aβ application via a FYN/ERK/S6 pathway [92]. Hyperphosphorylated tau also aggregates into oligomers, eventually forming insoluble straight/paired helical filaments and neurofibrillary tangles (NFT) which are one of the characteristic hallmarks of AD (Fig. 1F). Several kinases are thought to contribute to the hyperphosphorylated state of tau in AD, especially the proline directed kinases; GSK-3β [93, 94], CDK5 [95, 96] and members of the MAPK family [97]. The reported activity of flavonoids within MAPK and other protein kinase signalling pathways raises the potential for flavonoids to modulate tau phosphorylation, thereby impacting on the development of tau pathology. This is highlighted in Fig. 3, which demonstrates the extent to which flavonoids have the potential to influence tau phosphorylation [98–109].
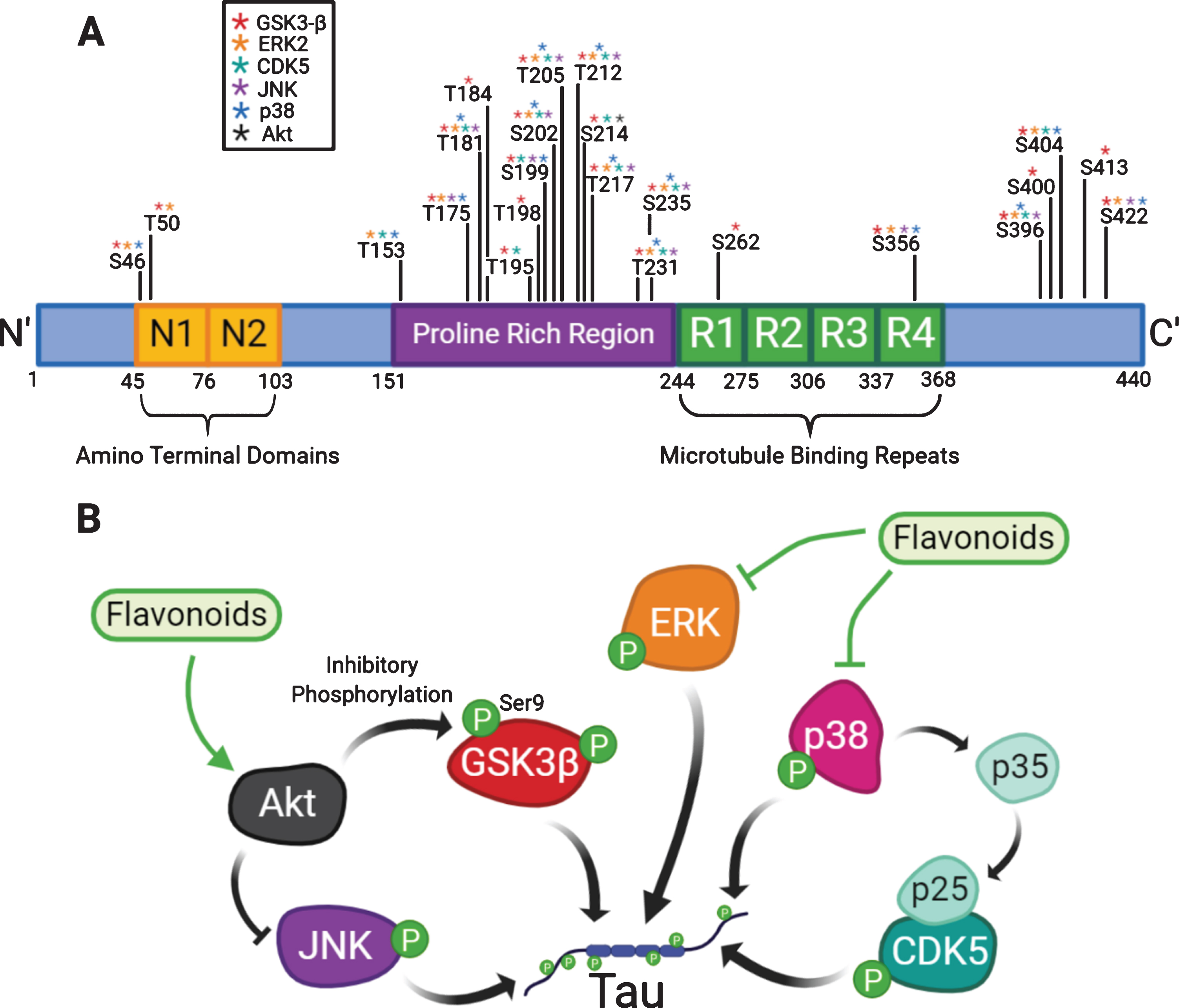
Flavonoids can modulate kinases involved in the hyperphosphorylation of tau. (A) Tau contains an amino-terminal domain, a proline rich region and microtubule binding repeats (R1–R4). The diagram highlights the identified phosphorylation sites on tau which have been shown to be phosphorylated by kinases that can be modulated by flavonoids: GSK3β, ERK2, CDK5, JNK, p38 and Akt [98–109]. (B) A diagram explaining how flavonoids may attenuate tau phosphorylation by modulation of kinases.
A number of studies have demonstrated a role for JNK in tau phosphorylation [110, 111], and inhibiting JNK in primary cortical neurons, a transgenic Aβ mouse model and human fibroblasts derived from AD patients all showed a decrease in phosphorylation of tau at Ser202, Ser205 and Ser422 [108]. Several flavonoids have been shown to inhibit JNK activity in vitro [112–115], although not typically at concentrations found in vivo. Despite this, there is evidence from animal models of AD that oral or intragastric flavonoid administration can inhibit the activity of JNK [30, 40, 116–118].
The MAPK member p38 kinase has also been proposed to contribute to hyperphosphorylation of tau both directly [119] and indirectly, for example through activation of the CDK5 activator p35 [120] (Fig. 3). As such, increased levels of p38 kinase and CDK5 were found to correlate with tau hyperphosphorylation in a htau transgenic mouse model [121]. Furthermore, both p38 kinase and JNK were found associated with NFTs in AD brains [122] and in a patient with P301L-associated FTDP-17 [123]. Moreover, as will be discussed later, several lines of evidence suggest that neuroinflammation induces p38 kinase-dependent phosphorylation of tau [120, 124–126]. However, despite evidence supporting p38 kinase-mediated phosphorylation of tau as well as flavonoid interactions with p38 kinase [127] and flavonoid modulation of p38 kinase activity in a variety of systems [40, 128, 129], there have been few attempts to directly link the two mechanisms. For example, administration of proanthocyanidins by oral gavage reduced lead-induced tau phosphorylation which correlated with levels of activated p38 kinase and JNK, but no direct association was made [130] (Table 1).
An alternative mechanism for the inhibition of tau phosphorylation by flavonoids is through activation of the PI3K/Akt pathway which inhibits the key tau kinase GSK-3β (Fig. 3) by phosphorylation at Ser9. Delivery of naringin [54], hesperidin [43, 80], EGCG [30], (–)-EC [131], and baicalin [25] through oral routes to mouse models of AD have all been found to inhibit GSK-3β phosphorylation at Ser9. Additionally, several in vitro and in silico models have pointed towards direct inhibition of GSK-3β by flavonoids. For example, crystal structure studies of GSK-3β in complex with the flavonol morin revealed direct inhibition of GSK-3β through interactions with the ATP binding pocket [132], which correlates with studies demonstrating reduction in tau phosphorylation in 3×Tg mice carrying mutations in APPswe, PS1 M146V and taup301L[133]. Similarly in silico docking studies with quercetin and other citrus flavonoids suggested direct interaction with the GSK-3β active site and direct inhibition was confirmed in vitro [124, 131]. Furthermore, unlike other serine/threonine protein kinases, GSK-3β phosphorylation prefers pre-phosphorylation 4 amino acids C-terminally to the target phosphorylation site [135–138]. This adds an additional layer of regulation, as inhibition of other tau-kinases can consequently inhibit GSK-3β phosphorylation of tau.
The few studies that have looked to directly link flavonoid treatment with tau phosphorylation have shown promising results. Oral delivery of grape seed polyphenolic extract (GSPE), which contains high concentrations of proanthocyanidins, particularly catechin and (–)-EC oligomers, has been shown to inhibit tau phosphorylation at several key sites, such as: Ser202/Ser205 (AT8), Ser396/Ser404 (PHF-1), Thr212/Ser214 (AT100) [139] and Thr181 [140]. Alongside this, significant reductions in the presence of Sarcosyl-insoluble tau follow GSPE treatment, suggestive of a reduction in aggregation [139, 140]. It has been suggested that the mechanism through which GSPE elicits these effects is primarily through inhibition of ERK1/2, as GSPE treatment did not correlate with inhibition of GSK3β. A role for ERK1/2 in phosphorylation of tau in AD is supported by in vitro studies and immunohistological analysis of post-mortem brains [97, 141, 142].
Therefore, several flavonoids have the capacity to reduce tau phosphorylation, either through inhibition of GSK-3β via activation of the PI3K/Akt pathway or modulation of MAPKs that are capable of phosphorylating tau directly (Fig. 3). However, there is a clear lack of studies exploring the direct link between flavonoid-mediated modulation of tau-phosphorylating kinases and the phosphorylation state of tau itself. As such, more studies are required to determine whether flavonoid-mediated inhibition of tau phosphorylation is achievable at the nanomolar concentrations that might be detected in brain. It will be essential to investigate this in order to determine the full therapeutic potential of flavonoidsin AD prevention.
Similar to Aβ, flavonoids have also been suggested to inhibit tau aggregation (Fig. 1F). Flavanols are the predominant group associated with inhibition of tau aggregation, although the flavone baicalein has been shown to inhibit tau oligomerization, albeit with a relatively high IC50 (∼35μM) [143]. GSPE inhibits tau aggregation of the 306VQIVYK311 hexapeptide [140, 144], which is thought to be essential in aggregation of full length tau [145] and destabilises tauopathy patient-derived fibrils [146]. Interestingly, the oxidation of (–)-EC by H2O2 has been shown to facilitate inhibition of tau aggregation [147]. Studies giving more consideration to stoichiometric ratios have demonstrated that (–)-Epicatechin-3-gallate (ECG) inhibits tau aggregation with an IC50-of 1.8μM [148] and EGCG inhibits the aggregation of a tau peptide at substoichiometric levels (0.1X) [149]. While more studies are needed to determine the impact of this inhibition of tau aggregation in a cellular context, these studies suggest that flavonoid-mediated inhibition of tau aggregation is a possible approach for reducing AD pathology.
Flavonoids, therefore, have the potential to interfere in the pathological phosphorylation of tau, as well as in the formation of tau oligomers and NFTs. Considering that tau phosphorylation and aggregation occurs consequently to Aβ pathology, and therefore, is a more tractable target for impairing AD progression, a reduction in tau pathology could suggest intervention is possible at MCI and later stages of disease.
Flavonoid modulation of neuroinflammatory response in AD
The role of neuroinflammation in the development and progression of AD has long been debated. However, astrogliosis, microgliosis and chronic inflammation are now well accepted pathological hallmarks of AD, alongside plaques and tangles [150]. Genome wide association studies (GWAS) [151] and transcriptomics [152, 153] have highlighted the involvement of the innate immune system in AD. In particular microglia have been linked to Aβ and tau spreading and propagation [154], synaptic phagocytosis [155–157], and the release of pro-inflammatory cytokines that promote AD pathology [150, 158].
An increasing number of studies have implicated microglia as the missing link between Aβ accumulation and tau pathology (Fig. 1C). In transgenic mice expressing the P301S mutation associated with frontotemporal dementia (FTD), microglial activation and synapse loss occurred prior to development of tau tangles [159]. Furthermore, suppression of the immune response with FK506 reduced tau pathology and increased lifespan. Histological analysis of post-mortem brain correlated increased microglial activation with AD pathology, suggesting that microglial activation occurred following Aβ plaque deposition but prior to NFT formation [160]. Analysis of the transcriptional profile of APPswe/PS1dE9 and Tau22 transgenic mouse models, which exhibit amyloid and tau pathology respectively found that while Aβ pathology activated the expression of several AD risk genes associated with microglia, this did not occur in the tau transgenic mouse [161]. Thus, providing further evidence that microglial activation lies upstream of tau phosphorylation and downstream of Aβ. Furthermore, silencing genes associated with the microglial inflammasome (Nlrp3–/–and Asc–/–) in the Tau22 transgenic mouse model of tauopathy led to reduced tau phosphorylation and aggregation [162]. Significantly, this pathway could be stimulated by Aβ-fibrils and was essential to Aβ- associated induction of tau phosphorylation in Tau22 mice following hippocampal injection of APP/PS1 brain homogenate [162]. This work presents a possible timeline of AD pathology, with Aβ accumulation initiating activation of NLRP3 inflammasomes (Fig. 1C) in microglia, which consequently cause release of IL-1β (Fig. 1D) and thus triggers tau phosphorylation (Fig. 1E). As IL-1β release has been shown to cause tau phosphorylation in a p38-MAPK-dependent manner [125, 126], this presents an additional target for flavonoid intervention.
A reduction in gliosis [33, 34, 37, 38, 40, 41, 44, 48, 55, 56, 59, 78, 83, 117, 118, 163] as well as neuroinflammatory markers [25, 30, 32, 34, 37, 38, 40, 42, 46, 48, 55, 56, 58, 78, 83, 117, 118, 163] has been shown in many animal models of AD orally administered with flavonoids (Table 1). Additionally, several flavonoids have been identified that inhibit microglial activation and IL-1β release. The isoflavone genistein reduced mRNA levels of IL-1β and TNF-α through a G-protein coupled oestrogen receptor (GPER)-dependent mechanism in primary microglia and BV2 cells [164]. Pre-treatment with the flavanone pinocembrin inhibited LPS-induced activation of BV2 cells and consequent release of pro-inflammatory mediators including IL-1β [165]. In vivo, intra-gastric administration of procyanidins reduced morphine-induced activation of NLRP3 and IL-1β, likely through inhibition of p38 kinase [166]. Furthermore, a number of in vivo studies of flavonoid intake in AD mouse models have shown a reduction in IL-1β following oral or gastric administration of EGCG [32, 34], formononetin [42] and eriodictyol [40]. Alongside this, many studies have reported decreases in levels of activated microglia following flavonoid treatment [167]
In addition to IL-1β, other pro-inflammatory cytokines have been suggested to play a role in AD. Tumour necrosis factor-α (TNF-α), which can be reduced by flavonoids [167], has also been implicated in AD [168, 169]. TNF-α, and to a lesser extent IL-1β was found to upregulate γ-secretase cleavage of APP through a JNK-dependent pathway, albeit in stably transfected HEK293 cells [170]. Furthermore, IL-6, which has been shown to be down regulated following administration of baicalin [78], formononetin [42] and eriodictyol [40], induces AD-like phosphorylation of tau in primary rat hippocampal neurons [120] (Fig. 1D). This phosphorylation was found to be due to activation of p38 kinase which caused upregulation of the CDK5 activator p35. Therefore, it is possible that flavonoid-induced down regulation of IL-6 in AD mouse models could reduce tau phosphorylation.
Given the accumulating evidence linking microglial activation, pro-inflammatory cytokine release and tau phosphorylation, it would be of interest to determine whether flavonoid-mediated attenuation of the microglial responses correlates with decreased tau pathology.
Flavonoid modulation of antioxidant responses in AD
Flavonoids are generally known for their antioxidant capacity [171], which may have benefit in AD as oxidative stress has been implicated in neurodegeneration [172]. Evidence from post-mortem studies on AD-brain shows that, relative to control brain, there is a net reduction in the ability to decrease H2O2 levels and stimulate anti-oxidative stress pathways [173, 174]. H2O2 can act as a messenger molecule activating signalling pathways, such as MAPK [175, 176], or react with metallic cations, such as Fe2+, to produce HO· [177] which can damage cellular components such as lipids and proteins. Imbalances in brain-iron levels have been reported in AD [178–181] and likely contribute to cognitive decline [182], supporting the suggestion that oxidative stress promotes AD pathology. Indeed, studies in transgenic APP/PS1 mice showed that increasing oxidative stress in the brain through delivery of a pro-oxidant iron rich diet increased the deposition of Aβ [183].
While flavonoids have the capacity to act directly as reactive oxygen scavengers by donating hydrogen, it is unlikely that this is the mechanism by which theyelicit their antioxidant potential due to the low circulating concentration of flavonoids in the brain relative to better characterised ROS scavengers such as ascorbic acid [184, 185]. Instead, it is most likely that the mechanism through which flavonoids inhibit oxidative stress is through modulation of intracellular pathways such as the MAPK pathway. The MAPK pathway responds to changes in the cellular environment, such as oxidative stress, and transduces these stimuli into cellular responses through phosphorylation or activation of transcription factors. Activation of anti-apoptotic ERK1/2 and PI3K/AKT pathways, and downregulation of pro-apoptotic JNK pathways by flavonoids is neuroprotective against multiple stressors including oxidative stress at both high [112, 186] and physiological concentrations [114, 128].
Another mechanism through which flavonoids could act as antioxidants is via PI3K dependent activation of the transcription factor Nuclear factor erythroid 2-related factor 2 (Nrf2). Activation of anti-oxidant response element (ARE) by Nrf2 enhances the expression of antioxidant-related genes, such as those involved in synthesis of several anti-oxidative enzymes including glutathione peroxidase (GPx), catalase (CAT), heme-oxygenase 1 (HO-1) and superoxide dismutase (SOD) [187]. Flavonoids have consistently been shown to activate Nrf2 in non-neuronal cell lines [188–190]. Furthermore, physiological levels of (–)-EC have been shown to upregulate ARE activity in astrocytes in a PI3-K dependent manner [191], which has been shown to protect neurons against oxidative stress [192]. Quercetin [193] and EGCG [194] have also been found to promote Nrf2 signalling in primary neurons, however, the effects at physiologically relevant concentrations were not investigated and there is uncertainty as to the extent to which Nrf2 is expressed in mature neurons.
Oral ingestion, or intragastric administration, of fisetin [41], hesperidin [43, 80], quercetin [58], rutin [55, 85], apigenin [24], puerarin [84] or (–)-EC [131] has been shown to reduce markers of oxidative stress in the brains of mouse models of AD (Table 1). This was demonstrated through increased activity of anti-oxidative enzymes such as GPx, CAT and SOD and a reduction in measurable H2O2, ROS and malondialdehyde (MDA). Collectively this supports a PI3K/Akt/Nrf2/ARE mechanism for conferring neuroprotection against oxidative stress by flavonoids which may be beneficial in ameliorating AD pathology.
Flavonoids promote BDNF induced neurogenesis in AD
Brain-derived neurotrophic factor (BDNF) is known to play an important role in neural development, differentiation and synaptic plasticity through binding to its receptor TrkB. BDNF levels are also known to be decreased in the AD brain relative to healthy controls [195–197]. Furthermore, decreased levels of BDNF have been linked to delta-secretase mediated cleavage of both Aβ and tau and subsequent neurodegeneration [198, 199]. BDNF also plays an important role in neurogenesis. The existence of adult hippocampal neurogenesis (AHN) in humans has been highly contested through the years, yet new advances in tissue processing techniques have now confirmed AHN [200, 201]. Moreover, it has been shown that, while neurogenesis decreases with aging, this decline is greatly exaggerated in AD [200]. Targeting BDNF/TrkB signalling to promote synaptic plasticity and neurogenesis may in turn slow the deterioration of the learning and memory abilities of AD patients.
There is evidence that flavonoids can promote BDNF/TrkB signalling and related downstream effects. Physiological concentrations of (–)-EC (100 nM) activate the cAMP response element binding protein (CREB) and consequently upregulate levels of BDNF in primary cortical neurons [202]. Both quercetin and (–)-EC have been shown to upregulate the expression levels of BDNF following oral administration to several rodent models of AD [131, 203–205]. Furthermore, oral delivery of 7,8-dihydroxyflavone (7,8-DHF), a TrkB agonist, has been shown to enhance long term potentiation (LTP), reduce synaptic loss, and improve performance in the Morris water maze (MWM) in vivo [23, 77]. Alongside the increased levels of BDNF seen with quercetin treatment, increases in neural progenitor cells (NPCs) and neural stem cells (NSC) associated with neurogenesishave also been shown [205].
Given this evidence, flavonoid-induced upregulation of the BDNF/TrkB pathway to increase neurogenesis and reduce synaptic loss is an additional mechanism through which flavonoids may be able to reduce AD pathology (Fig. 1I).
EMERGING CONSIDERATIONS FOR FLAVONOID INTERVENTIONS IN AD
Given the low success rate for translating therapeutics from in vivo animal studies to effective human interventions for AD, it is becoming increasingly important to ensure that the ‘translatability gap’ is as small as possible. In this section we consider emerging evidence as to the extent of metabolism and bioavailability of flavonoids, and how this could influence the interpretation of existing data and experimental design moving forward.
Flavonoid metabolome and brain bioavailability
While the ‘key’ metabolites of several notable flavonoids have been known for years, there is only limited knowledge of the full metabolite profiles and, more importantly for AD intervention, which metabolites reach the brain.
In this respect, important advances in the field have been the use of radio labelling to determine complete flavonoid metabolomes. Such examples include identification of the anthocyanidincyanidin-3-glucoside (C3G) metabolome in humans [206] as well as the (–)-EC metabolome in both humans and rodents [207, 208]. As a result, 24 C3G and >20 (–)-EC metabolites have now been identified in humans. In comparing these metabolomes, there are similarities in the metabolites produced, such as the presence of structurally related molecules which have been glucuronidated, methylated or sulphated, as well as structurally unrelated metabolites such as hippuric acid. Both of these human studies found that the flavonoid metabolites were much more abundant than the parent flavonoid molecule, to the extent that 14C-(–)-EC itself was undetectable in the plasma or urine. Therefore, it is likely that the bioactivity in vivo resides in one or more flavonoid-derived metabolites rather than the parent flavonoid. This is important as many in vitro experiments use only parent flavonoids to determine mechanisms of action and itis unlikely that these unmetabolized flavonoids will be available in the brain at active concentrations. In relation to (–)-EC, structurally related epicatechin metabolites (SREM) are likely to be the predominant bioactive constituents in the human brain, given the absence of the parent (–)-EC molecule in human plasma [207]. This corresponds with pharmacokinetic studies in rats which showed that chronic oral administration of (–)-EC and catechin (C) resulted in a combined concentration of ∼484 nM (–)-EC/C-glucuronides and methyl-(–)-EC/C-glucuronides, while unmetabolized (–)-EC could only be found in trace amounts [209]. Despite this many in vitro studies focus on concentrations >10μM. Flavonoids often exhibit biphasic responses, activating protein kinase signalling pathways at low concentrations (nM) and inhibiting at high concentrations (μM), or vice versa [39, 114, 193]. Therefore, it is possible that where flavonoids are shown to inhibit pro-AD pathways in vitro at high concentrations, in vivo at nanomolar concentrations they may elicit different effects.
Considering this, metabolic profiles for other flavonoids considered as potential therapeutic interventions for dementia should be established to aid mechanistic investigations. In order to determine bioavailability in the brain analysis of cerebral spinal fluid (CSF) could potentially be undertaken to confirm the appearance of flavonoid-derived metabolites.
Species differences in flavonoid metabolism limit translatability of in vitro and in vivo studies
The metabolism of flavonoids is an important consideration when designing and interpreting animal studies. While there are plenty of studies on flavonoid metabolism in rodents, the information about human metabolism is lacking. As more information becomes available, it is becoming clear that there are key differences in flavanol metabolism between species, which may further limit the translatability of in vivo studies.
An example of this is the absence of (–)-EC-3’-O-glucuronide and (–)-EC-3’-sulfate, the main metabolites of (–)-EC found in plasma and urine of humans, in rats [185, 208]. It is, therefore, not possible to determine the effect of these metabolites on the rodent brain through delivery of (–)-EC alone. Such investigations would require direct i.p. injection of these metabolites or administration by osmotic mini-pump.
Furthermore, as mentioned previously, (–)-EC was not identified in plasma or urine of humans [208], although it was in rats [207]. Likewise, C3G metabolites were found to be 42 fold more abundant than the parent C3G molecule in humans [206], whereas in mice the difference was only 6 fold [210]. Therefore, highlighting doubts about the contribution of unmetabolized flavonoids in the human brain relative to the rodent models used in pre-clinical trials.
Studies of key quercetin metabolites in gerbils, mice and rats showed that there are species dependent differences in the metabolism of quercetin [211]. Given what we now know about (–)-EC metabolism, it is likely that this would correlate with differences in quercetin metabolism between humans and rodents as well. Without knowing what flavonoid metabolites are found in humans, and particularly in the brain, it is hard to pre-determine if flavonoids would elicit therapeutic benefit in humans, despite positive findings in animal models (Table 1).
Harnessing metabolites for flavonoid intervention studies for AD
Utilising the information now available about the pharmacokinetics and bioavailability of (–)-EC, Ottaviani et al. have identified a collection of SREMs that have the potential to be utilised as biomarkers for (–)-EC intake [212]. The levels of (–)-EC-3’-glucuronide, (–)-EC-3’-sulfate and 3’-O-methyl-(–)-EC-5-sulfate, collectively termed SREMB, in the urine can be used to estimate the amount of (–)-EC ingested 24 hours previously. The standard practice for epidemiological studies interested in implications of dietary compounds on health and disease is through self-reporting of subjects to estimate consumption, which is inherently unreliable. With biomarkers to assess the actual amounts of (–)-EC in the diet, future epidemiological studies will be able to investigate the effect of (–)-EC on AD with greater accuracy and certainty.
The impact of the microbiome on flavonoid metabolism and AD
It is now known that the gut-microbiome is responsible for the generation of several flavonoid metabolites [185, 206, 208, 213–216]. The microbiome can partake in the deglycosylation, ring fission and reduction of flavonoids [215], with deglycosylation being of particular importance, as this is thought to be essential for flavonoid absorption [217], with the exception of C3G [218]. Despite making up a large proportion of circulating flavonoid-derived metabolites, few studies to date have explored the mechanistic function of microbiome-derived metabolites in relation to AD. An example of such biologically relevant metabolites are phenyl-γ-valerolactones and phenyl-γ-valeric acids, formed from microbiome metabolism of flavanols. In vitro assays have suggested that these metabolites can permeate the blood brain barrier (BBB) [219] and reduce Aβ-toxicity [220].There is further suggestion that 3-HBA and 3-HPP, derived from GSPE metabolism by the rat microbiome, can impair the aggregation of Aβ peptides, nonetheless at high molar ratios [213]. Other studies in SHSY-5Y cells show some ability of microbiome-derived metabolites of flavanols to act in a neuroprotective manner against H2O2 treatment and SIN-1-induced stress [221, 222]. There is recent evidence that flavonoid intake can alter the composition of the human gut microbiome [223]. However, it is yet to be determined what effect, if any, this may have on health and disease. An increasing number of studies have suggested a role for the gut microbiome in AD, most likely through modulation of microglial activation. One study in cognitively impaired elderly found a significant increase in abundance of pro-inflammatory bacteria as well as decreased abundance of anti-inflammatory bacteria in the gut of Aβ positive relative to Aβ-negative participants [224]. Similarly, it was also found that AD patients have a distinct gut-microbiome composition relative to age and sex-matched controls [225]. Likewise, 5XFAD mice exhibited a different microbiome composition relative to non-transgenic controls and this dysbiosis was suggested to promote microglial activation [226]. By treating these mice with GV-971, which promotes normal gut-microbiome composition, Aβ, tau and behavioural pathology was significantly reduced. Interestingly, the treatment of APPPS1-21 mice with antibiotics led to a reduction in Aβ and microglial pathology in male, but not female, mice, suggesting a sex-specific role for the microbiome in AD [227]. Therefore, modulation of the microbiome composition by flavonoids should be considered carefully as this has potential to promote or hinder AD progression.
Furthermore, there are important inter-individual differences in the composition of the gut microbiome as a result of various factors including age, sex, drug use and dietary intake [228]. This is significant as the composition of the human gut microbiome has so far been shown to alter the metabolism of flavonols [229, 230] and isoflavones [215]. Therefore, it may become beneficial to assess the microbiome of an individual before recommending flavonoid interventions in order to personalise the amount and type of flavonoids for maximum efficacy. Furthermore, combined treatment with pro-biotics might enhance the effects of flavonoid intake by ensuring the most efficient microbiota are present in the gut. Moreover, AD risk factors such as diabetes and obesity also influence polyphenol uptake and metabolism [231, 232]. Although this might not result from direct changes to the microbiome, they could impair an individual’s responsiveness to flavonoid intervention.
Moving forward therefore, the potential contribution of the microbiome should be taken into consideration when looking to assess the bioavailability and efficacy of flavonoids or flavonoid-derived drugs particularly when the method of flavonoid delivery bypasses the gut and other key sites of metabolism.
Alterations to Promote the Stability, Bioavailability, Specificity and Activity of Flavonoids
Like many small molecule and antibody therapies, several flavonoids are likely to exhibit low brain bioavailability, a potential limitation of their use as therapeutic interventions in humans[233]. One method to overcome this is to encapsulate the flavonoids in nanoparticles. This increases their stability and brain bioavailability as well as promoting sustained release [234]. Furthermore, nanoparticles can include targeting peptides in their coating, allowing for more specific activity. As a result, nano-encapsulation could reduce the quantity of flavonoid that would need to be administered.
Nano-encapsulation of EGCG improved bio availability in rats as well as its α-secretase promoting activity in SHSY-5Y cells [235]. Furthermore, nanoparticles loaded with EGCG and ascorbic acid were found to decrease Aβ deposition and increase synaptic expression relative to EGCG alone in APP/PS1 mice, which correlated with improved spatial learning [33]. However, it is unclear whether this is a result of the nanoparticles or the added ascorbic acid, which is a powerful antioxidant. It should also be noted that this study found that the nanoparticles may disrupt the BBB, which needs to be investigated further in vivo [33]. Nano-encapsulation of quercetin also showed significant improvements to cognition and memory in SAMP8 mice as well as lower levels of markers of inflammation relative to free quercetin [236]. These results were in-line with increased quercetin levels in the brain (∼200 ng/g free quercetin: 371 ng/g nano-encapsulated quercetin). Therefore, nano-encapsulating flavonoids may improve their efficacy in vivo, thus making them more appealing as therapeutic interventions for AD.
Their capacity to act at multiple targets associated with AD pathology makes flavonoids attractive molecular scaffolds for development of more potent inhibitors of AD pathology. Alteration of the flavonoid structure has been found to enhance inhibition of BACE-1, Aβ aggregation and acetylcholinesterase (AChE) activity, as reviewed by Jalili-Baleh (2018) [237]. However, it is possible that these changes will impact the ability of the flavonoid-based drug to reach the brain.
While the development of more potent and specific flavonoid-derived therapeutics has the potential to elicit stronger effects, the switch from nutraceutical to pharmaceutical brings its own hurdles. The increased expense and potency could mean that these compounds are no longer suitable for early and sustained delivery throughout midlife. In contrast, supplementation with naturally available flavonoids is likely to be more widely accessible, with potentially fewer side effects. As such, even if direct modulation of AD pathways is weaker, capacity for long term delivery could contribute to an overall benefit.
Incorporating the Metabolome to Promote Translatability in Identifying a Therapeutic Intervention for AD
The identification of the (–)-EC metabolome yielded a library of potential bioactive molecules, retrospective biomarkers of (–)-EC intake, and a greater understanding of the limitations of the animal models used. In the future, it would be of great value to elucidate the full human metabolomes of more flavonoids in order to limit the risk of failure at clinical trials. Utilising techniques such as the radiolabelling used by Czank et al. (2013) and Ottaviani et al. (2016) to identify the full human metabolome of these clinically relevant flavonoids may help to bridge the translatability gap between pre-clinical investigations and human interventions.
An important point to consider is that despite considerable uncertainty that parent flavonoids are present at sufficient concentrations in the brain, the metabolites themselves likely have poor bioavailability following oral delivery. To overcome this, one route would be to nano-encapsulate the metabolite of interest, thus improving its bioavailability. Alternatively, it may be more efficient to use the parent-flavonoid as a pro-drug, which with appropriate dosing will facilitate availability of metabolites in the brain. In this scenario, it will be necessary to investigate the mechanistic effects of combinations of metabolites that derive from the same parent flavonoid, to ensure a consistent mechanism of action.
While the incorporation of metabolism and brain bioavailability creates additional hurdles in experimental design, it also increases the likelihood of accurately identifying bioactive flavonoids or flavonoid-derived metabolites that can be used as therapeutic interventions for AD in humans.
CONCLUSIONS
As this review has highlighted, flavonoids have the potential to act as multimodal therapeutics to prevent the onset and/or progression of AD. Much of the research effort to date has focused on the reduction of Aβ by flavonoids. The advantage of flavonoids over conventional Aβ-targeting drugs such as antibodies and small molecule inhibitors is that it is possible for them to be administered as dietary supplements. Due to their relative affordability and low toxicity, supplementation with flavonoids could enable early protection at a young age and throughout life, without the need for pre-clinical diagnosis. However, determining if early and consistent flavonoid intake is capable of preventing AD onset would require an enormous amount of resource and most importantly, time. Until it is possible to accurately identify AD pre-symptomatically, before Aβ accumulation has initiated downstream toxic cascades, it may be necessary to shift the focus to flavonoid induced changes to later-onset pathologies such as neuroinflammation or tau phosphorylation. To date, tau pathology is clearly underrepresented in flavonoid-intervention studies carried out in mouse models of AD despite the strong basis for flavonoid-modulation of tau phosphorylating kinases.
Moving forward, it will be important to incorporate the bioavailability and metabolism into experimental planning throughout all stages of pre-clinical research. To reduce the current translatability gap, it will be essential to: Focus on physiologically relevant concentrations when determining the mechanistic functions of flavonoids in vitro, to ensure that said mechanisms are possible in vivo. Identify the complete human metabolomes of flavonoids of interest to determine functional metabolites. Compare the mechanisms of action of parent flavonoids with their human metabolites, both alone and in combination, to ensure that the same mechanisms will be engaged following in vivo administration.
Taking this into consideration should result in more efficient and effective identification of flavonoid-based molecules that could succeed as therapeutic interventions for AD.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Footnotes
ACKNOWLEDGMENTS
KH is supported by the MRC GW4 BioMed DTP, RW acknowledges support from Alzheimer’s Society (481 AS-PG-18b-009), Alzheimer’s Research UK (EG2018A-008), and an open grant from Mars Edge. All figures were created with BioRender.