
Abstract
Alzheimer’s disease (AD) and Parkinson’s disease (PD) share many commonalities ranging from signaling deficits such as altered cholinergic activity, neurotrophin and insulin signaling to cell stress cascades that result in proteinopathy, mitochondrial dysfunction and neuronal cell death. These pathological processes are not unidirectional, but are intertwined, resulting in a series of feed-forward loops that worsen symptoms and advance disease progression. At the center of these loops is glycogen synthase kinase-3 (GSK-3), a keystone protein involved in many of the multidirectional biological processes that contribute to AD and PD neuropathology. Here, a unified overview of the involvement of GSK-3 in the major processes involved in these diseases will be presented. The mechanisms by which these processes are linked will be discussed and the feed-forward pathways identified. In this regard, this review will put forth the notion that combination therapy, targeting these multiple facets of AD or PD neuropathology is a necessary next step in the search for effective therapies.
INTRODUCTION
Alzheimer’s disease (AD) and Parkinson disease (AD) are the two most common neurodegenerative diseases. The prevalence of each disease increases with age and is associated with significant and progressive physical and cognitive deficits. At present, an estimated 47 million people live with Alzheimer’s disease and other dementias globally [1] whereas PD affects an estimated 10 million people at any given time [2].
Abnormal protein aggregation is a major neuropathological hallmark of both AD and PD with the abnormal processing of amyloid precursor protein (APP) into β-amyloid (Aβ) and the hyperphosphorylation of tau and consequent formation of neurofibrillary tangles (NFTs) characterizing AD, whereas the accumulation α-synuclein within protein aggregates known as Lewy bodies characterizes PD [3]. Together, oligomeric forms of Aβ, tau andα-synuclein have been hypothesized to contribute to cognitive decline in AD [4–6], as well as in PD patients that exhibit dementia [7]. In addition to these protein deposits, AD and PD share a number of pathological features that lead to neuroinflammation and neurodegeneration. These range from disrupted neurotrophin and insulin signaling to the initiation of mitochondrial and oxidative stress. Interestingly, while neuronal cell death in AD localizes predominantly to cholinergic neurons of the cortical and limbic systems [8], in PD, apoptosis is centered within dopaminergic neurons of the substantia nigra pars compacta (SNpc) and the subsequent loss of striatal innervation. This indicates that while there is synergy between the pathogenic signals that initiate cell loss in these diseases this cannot be attributed to common regions of susceptibility or neuronal subsets [9]. Given the complex etiologies of AD and PD, it is unsurprising that the dysregulation of a number of cell signaling pathways has been reported. In this regard, much of the existing research has focused on identifying novel individual therapeutic targets within these pathways. This one-target approach, however, has to date only been moderately successful and the identification of novel and efficient disease-modifying therapies has remined elusive. Levodopa for example, which has been used for treatment of PD for almost 50 years, is still the mainstay of treatment for the disease, and although recent improvements in the drug have reduced the dyskinetic side effects, 40% of patients still develop motor fluctuations within 4–6 years.
Perhaps one of the greatest difficulties with the one-target approach is that the neuropathological processes involved in AD or PD are intricately linked, with bidirectional interactions that promote further insult to the system and the consequent progressive worsening of each disease. At the center of this intricate network is the protein glycogen synthase kinase- 3 (GSK-3), a serine/threonine protein kinase with an array of physiological functions ranging from regulation of gene expression to neuronal plasticity and survival. Although GSK-3 exists as two isoforms, GSK-3α and GSK-3β, the majority of research has focused on GSK-3β as a result of its involvement in both neuropsychiatric and neurodegenerative disease. The literature evaluating a role for GSK-3 in AD and PD is extensive, and it is widely accepted that this kinase plays a critical role in the neuropathology of both diseases. A comprehensive examination of how GSK-3 links together all the major neuropathological processes, however, in these diseases has never been performed. Therefore, the purpose of the review is two-fold, first to identify how GSK-3 is involved in the positive feedback of various processes in these diseases, and second to link these processes together into one cohesive model. Given the complex bidirectional interactions that occur in AD and PD that work in concert to advance disease progression, the overarching goal of this review is to emphasize that a multi-target approach may be required to not only provide symptomatic relief to patients but to halt disease progression.
The Aβ/α-synuclein/p-tau-oxidative stress/inflammation-GSK-3 loop
GSK-3 is a multitarget kinase with over 40 biological substrates [10]. Many of these targets impact on processes known to be involved in both AD and PD (Fig. 1A). As a result, the dysregulation of GSK-3 has been widely shown to play a critical role in both diseases. At the gene level, gene association studies have attributed GSK-3 gene variants with increased risk of either AD [11–13] or PD [14]. Indeed, the GSK-3β (GSK-3 β) and tau (MAPT) gene variants have been shown to interact to increase risk of late onset AD or idiopathic PD [13, 14]. Specifically, Kwok and colleagues [13, 14] showed risk for developing each disease was dependent on the individual’s GSK-3β and MAPT haplotype, with the homozygous GSK-3β T/T haplotype increasing risk in those with at least one copy of the MAPT H2 haplotype, and decreasing risk in those with the homozygous for the MAPT H1 haplotype. Importantly, this may explain why GSK-3 has not been linked to these diseases in larger-scale genome wide association studies, as the interaction between GSK-3β polymorphisms and other genes, such as MAPT, may be a critical facet to conferring disease risk. Kwok and colleagues [13] further demonstrated that the biochemical consequence of the GSK-3β T/T haplotype is to induce maximal transcription of a GSK-3β splice isoform that has increased ability to phosphorylate tau whereas the MAPT H1 haplotype increases MAPT transcript levels [14]. The authors postulated that excess tau may be able to sequester GSK-3β and shunt it toward cellular pools that will have functional effects on select substrates of GSK-3β [13].
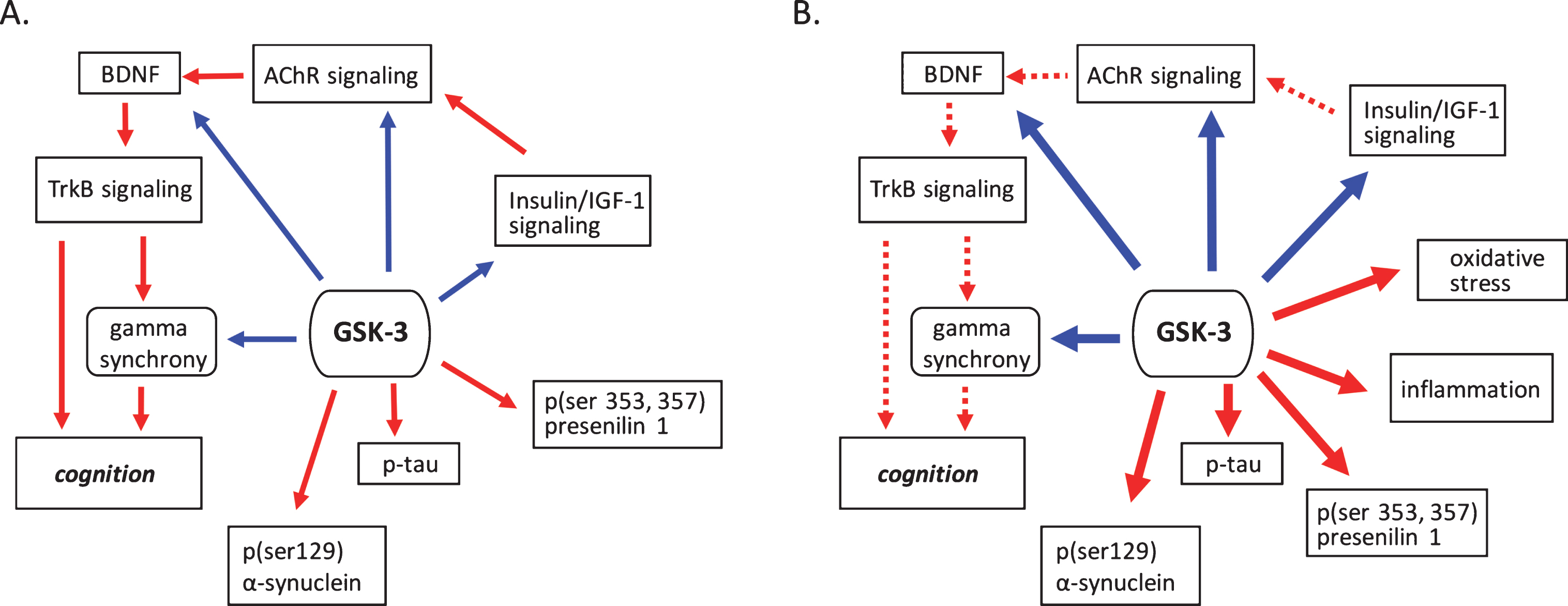
GSK-3 regulates processes implicated in neurodegenerative disease. A) Under normal conditions GSK-3 is known to suppress processes that positivetly regulate cognition such as cholinergic, BDNF, and insulin signaling. GSK-3 also is involved in the suppression of gamma frequency oscillations. The phosphorylation of tau, α-synuclein and presenilin are also mediated by GSK-3. B) Under pathological conditions GSK-3 activity is upregulated, increasing its negative influence on cholinergic, BDNF, and insulin signaling, and at the same time promoting phosphorylation events critical to the development of proteinopathies. Increased GSK-3 activity also induces oxidative stress and the onset of inflammatory processes. Positive regulation (red arrows) and negative regulation (blue arrows) of each pathway and/or process are shown. Changes in the overall activity of these pathways compared to normal conditions are represented by the solid arrows (increased activation) and dotted arrows (decreased activation). AChR, acetylcholine receptor; BDNF, brain-derived neurotrophic factor; GSK-3β, glycogen synthase kinase-3β; IGF-1, insulin growth factor 1; TrkB, tropomyosin receptor kinase B; TrkB.T1, truncated tropomyosin receptor kinase.
With regard to enzymatic function, the activity levels of GSK-3 are elevated in post-mortem cortical tissue from both AD and PD patients [15–18], as well as in animal model systems [17–19]. In AD specifically, this elevation of GSK-3 activity induces a number of downstream consequences, including Aβ and tau oligomerization [20], as well as impacts on other downstream substrates of GSK-3 (Fig. 1B). In AD, the formation of extracellular Aβ and intracellular tau oligomeric species in both cortical and hippocampal regions are thought to be a leading cause of cognitive dysfunction [21, 22] with the synergistic interaction of Aβ and phosphorylated tau recently hypothesized to drive metabolic decline and the progression to dementia [23]. GSK-3 inhibitors block the production of Aβ oligomers by interfering with cleavage of the membrane-bound amyloid precursor protein (APP) by γ-secretase [20]. Tau, by contrast, is a direct substrate of GSK-3 for phosphorylation, with this process having been shown to be exacerbated by the accumulation of Aβ [24]. In addition to the development of NFTs, hyperphosphorylation of tau induces the production of soluble oligomeric as well as insoluble protein species (paired helical filaments) [25], which promotes inflammasome activation and tau interaction with microtubules. The consequence of this is cytoskeletal destabilization and the promotion of insulin resistance [26] (Fig. 2).
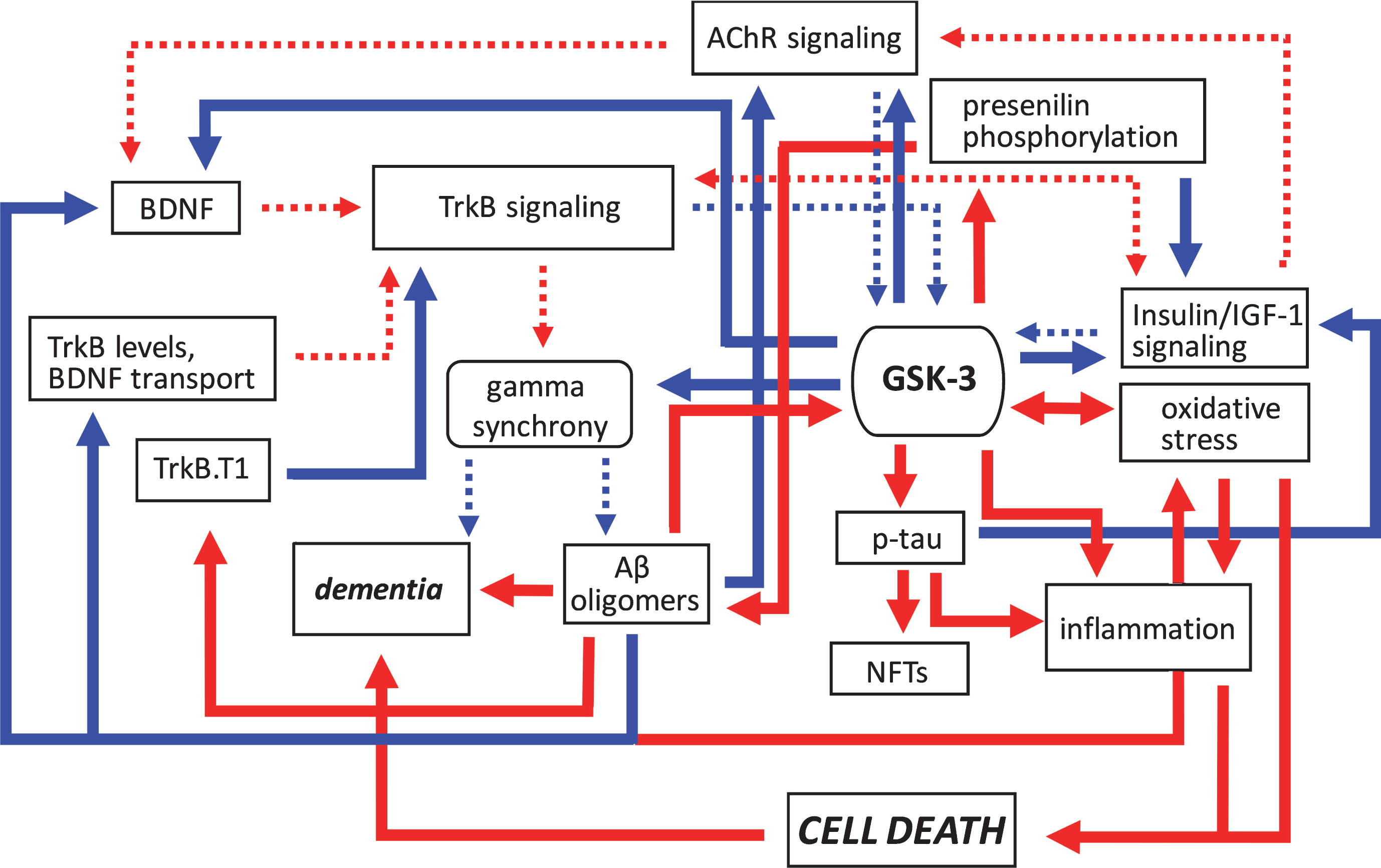
Pathogenic feedforward loops in Alzheimer’s disease converge on GSK-3. In AD, several cellular, and bidirectional, processes work in concert to increase GSK-3 activity. These include deficits in cholinergic, BDNF/TrkB, and insulin or IGF-1 signaling, mitochondrial dysfunction leading to oxidative stress, and production of Aβ oligomers. This increase in GSK-3 activity further suppresses cholinergic, BDNF/TrkB, and the insulin/IGF-1 signaling pathways, and additionally promotes tau hyperphosphorylation and inflammation, and the disruption of gamma synchrony. The production of Aβ oligomers also increases due to GSK-3-mediated presenilin phosphorylation. Together these processes contribute to the onset of dementia. Positive regulation (red arrows) and negative regulation (blue arrows) of each pathway and/or process are shown. Changes in the overall activity of these pathways compared to normal conditions are represented by the solid arrows (increased activation) and dotted arrows (decreased activation). Aβ, amyloid-β; AChR, acetylcholine receptor; BDNF, brain-derived neurotrophic factor; GSK-3β, glycogen synthase kinase-3β; IGF-1, insulin growth factor 1; NFTs, neurofibrillary tangles; TrkB, tropomyosin receptor kinase B; TrkB.T1, truncated tropomyosin receptor kinase.
In PD, GSK-3-mediated phospho-tau is also present [27], although the phosphorylation profile of tau appears to differ from that of AD [28]. In cerebral cortices from patients with Dementia with Lewy Body disease, phosphorylated tau colocalized with phosphorylated α-synuclein in the Lewy bodies, with phosphorylation of α-synuclein occurring predominantly at serine 129 [29]. Serine 129 phosphorylation has been shown to accelerate neurodegeneration in an animal model of PD [30], promote fibril formation in vitro [29], and was more recently demonstrated to be a site phosphorylated by GSK-3 [27]. The effects of synucleinopathy are further exacerbated by findings showing that α-synuclein increases activation of GSK-3 and GSK-3-mediated tauopathy in both cellular and animal models of PD [18, 31], resulting in a positive feed-forward mechanism that exacerbates GSK-3 mediated pathogenesis (Fig. 3).
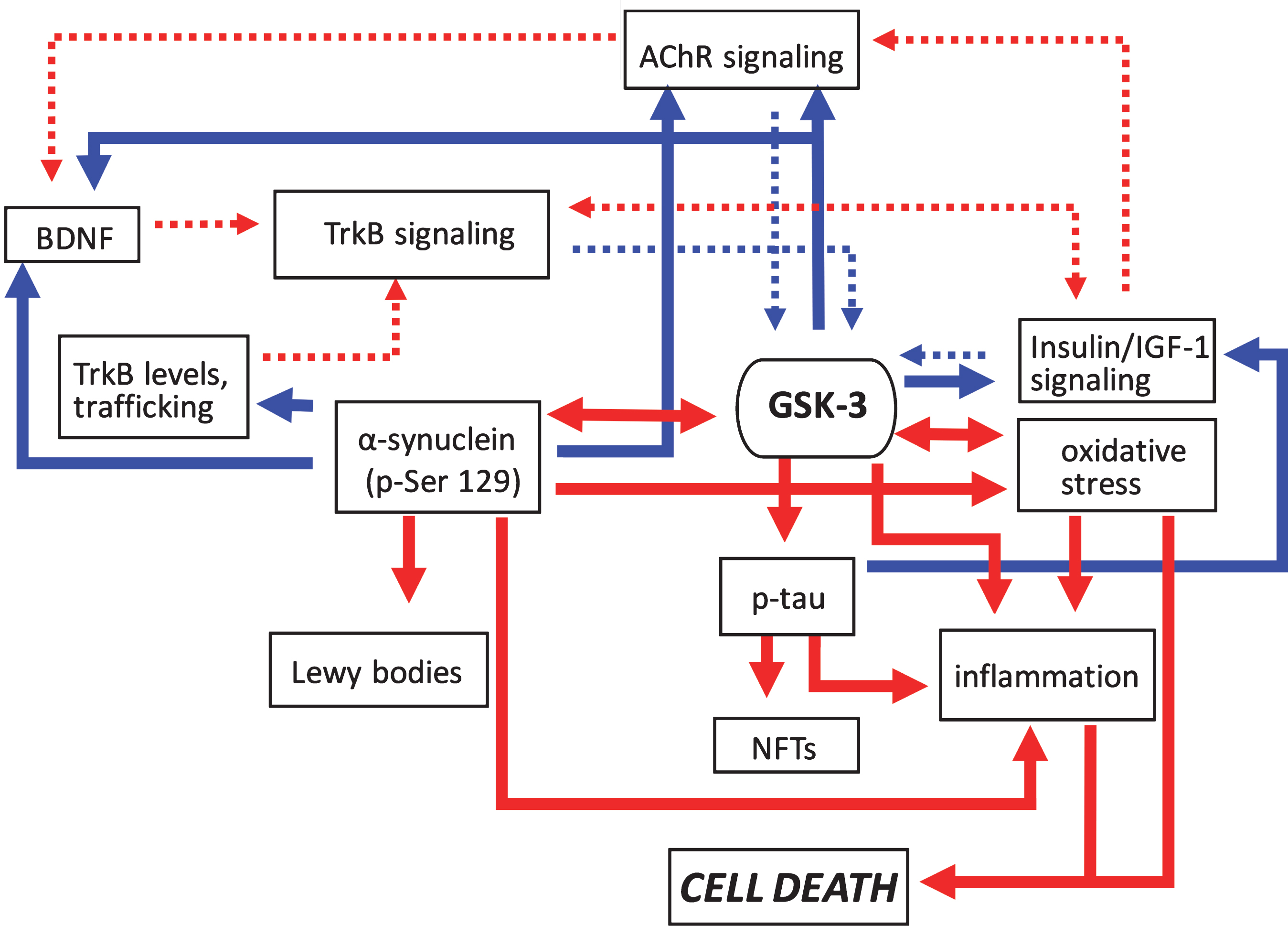
Pathogenic feedforward loops in Parkinson’s disease converge on GSK-3. In PD increased GSK-3 activity can arise as a result of deficits in cholinergic, BDNF/TrkB, and insulin or IGF-1 signaling, and mitochondrial dysfunction leading to oxidative stress. Consequently, increased GSK-3 activity results in enhanced tau and α-synuclein phosphorylation leading to proteinopathy, and induces inflammatory responses. GSK-3 activation also has additional impacts on oxidative stress. Positive regulation (red arrows) and negative regulation (blue arrows) of each pathway and/or process are shown. Changes in the overall activity of these pathways compared to normal conditions are represented by the solid arrows (increased activation) and dotted arrows (decreased activation). AChR, acetylcholine receptor; BDNF, brain-derived neurotrophic factor; GSK-3β, glycogen synthase kinase-3β; IGF-1, insulin growth factor 1; NFTs, neurofibrillary tangles; TrkB, tropomyosin receptor kinase B; TrkB.T1, truncated tropomyosin receptor kinase.
Inflammasome activation by GSK-3-induced phospho-tau has been critically linked to oxidative stress, and widely characterized in both AD [32] and PD [33]. Indeed, the induction of tau hyperphosphorylation, mediated by an upregulation in GSK-3 activity, in cortical neurons has been demonstrated to be in response to increased oxidative stress [34]. GSK-3 has also been shown to directly mediate other important inflammatory processes associated with neurodegeneration, such as the stimulation of proteins such as interleukin-6, interleukin-1β, and tumor necrosis factor [35] however this role of GSK-3 has been reviewed in detail elsewhere [36]. The role of GSK-3 in induction of mitochondrial stress in AD stems from its phospho-regulation of the mitochondrial fission protein dynamin-related protein 1. In studies from AD patients, rodent and in vitro models, Aβ oligomers were shown to accumulate in the mitochondria, leading to mitochondrial fission and the consequent production of catalytic reactive oxygen species (ROS) [37, 38]. Suppression of GSK-3-mediated dynamin-related protein 1 phosphorylation prevents mitochondrial fragmentation-induced Aβ oligomers in hippocampal neurons [39], further centralizing the role of GSK-3 in AD pathogenesis.
In PD, increased oxidative stress resulting from ROS production has also been proposed to contribute and to disease neuropathology, with reduced expression or dysfunction of mitochondrial complex I considered to be a key mediator of neuronal loss [40]. In postmortem PD brain there is accumulation of α-synuclein within mitochondria in the SNpc and striatal regions [41] where it interacts with mitochondrial complex I and can promote oxidative stress [41, 42]. Indeed, reactive production of dopamine-quinone is further associated with the significant loss of dopamine within the striatum of PD patients prior to cell death in the SNpc [43]. In vitro α-synuclein phosphorylation at ser 129, a site phosphorylated by polo-like kinase 2 (PLK-2) [44], G-protein receptor kinases [45], leucine-rich repeat kinase 2 (LRRK2) [46], and GSK-3 [27], is highly correlated with ROS production [47] and has been shown to play a significant role in mitochondrial dysfunction in a drosophila model [48]. Of relevance, upregulated mitochondrial GSK-3 activity in human neuroblastoma cells decreased mitochondrial complex 1 activity, that was accompanied by increased ROS production and altered mitochondrial morphology [49]. In addition, MPP+ treatment of neurons induced cell death that was shown to be associated with time- and concentration-dependent activation of GSK-3 and mediated by depolarization of mitochondrial membrane potential [50]. These findings critically tie GSK-3 to cellular dysfunction in PD.
The BDNF-Aβ/α-synuclein-GSK-3 loop
A second feed forward mechanism of GSK-3 action centers on the neurotropic factor, BDNF. BDNF is a widely expressed protein within the central nervous system that plays a critical role in neuronal survival, maintenance, and plasticity. These effects are mediated through interactions with the high affinity full length catalytic isoform of the tropomyosin-related kinase B (TrkB) receptor [51]. Upon binding of BDNF to TrkB, the receptor dimerizes and autophosphorylates resulting in activation of the PLCγ, PI3K, and Erk/MAPK pathways [51]. In addition to TrkB, BDNF can also bind to three truncated isoforms that lack the intracellular kinase domain (TrkB.T1, TrkB.T2 and TrkB.T-Shc) [52]. TrkB.T1, the most predominant truncated isoform, was first characterized as a dominant-negative receptor to indirectly inhibit BDNF function through dimerization with full length TrkB. Since this time TrkB.T1 has been shown to be involved in the suppression of full length TrkB cell surface expression [53] and the sequestration and translocation of BDNF [54, 55].
Deficits in BDNF/TrkB signaling is involved in the neuropathology of both AD and PD [56] with many reports demonstrating aberrant BDNF expression levels or signaling in both diseases in humans and animal models. For example, in post-mortem studies of individuals who had AD, significant reductions in BDNF levels in hippocampus and cortical areas have been documented [57] with robust diminishment in the full length active isoform of TrkB in NFT-bearing neurons [58]. These findings are supported by animal studies with diminished BDNF mRNA or protein and/or TrkB signaling in the cortex or hippocampus of transgenic AD mice [59, 60]. These BDNF deficits have been suggested to be a biomarker of impaired memory and cognitive function in both AD and healthy individuals [61, 62]. One mechanism by which BDNF may mediate cognitive function is through the regulation neural oscillatory activity, with the gamma frequency rhythms highly associated with higher brain function [63]. In AD, patients display delayed sensory-evoked or event-related gamma activity as well as increased fronto-parietal gamma synchrony [64]. Similarly, in AD models, cortical gamma activity and hippocampal gamma amplitude modulation by theta phase is impaired [65].
Several studies have evaluated the impact of BDNF signaling on GSK-3 activity with two major signaling pathways having been identified that involve Akt or protein kinase C (PKC). The binding to TrkB by BDNF induces the downstream activation of PI3K, and the subsequent phosphorylation and activation of Akt, which in turn phosphorylates GSK-3α and GSK-3β at serine 21 and serine 9 respectively to inactivate the proteins [66]. Similarly, GSK-3 phosphorylation following BDNF treatment of cerebellar neurons was attenuated with a PKC inhibitor [66]. In AD and PD, one consequence of increased GSK-3 activation is the induction of a positive feedforward mechanism that progressively worsens the existing BDNF signaling deficit, an effect potentially mediated through direct mechanisms or indirect processes involving Aβ or α-synuclein as described previously (Figs. 2 and 3). Added to this, α-synuclein has been reported to interact with PKC, to inhibit its kinase activity [67] and thus its function as a negative regulator of GSK-3βl.
Evidence suggests that GSK-3 activity may have a direct impact on neuronal BDNF levels, with inhibitors of GSK-3 increasing both BDNF mRNA and protein in cortical neurons in culture [68]. Although the mechanisms underlying this regulation remain unknown, GSK-3 has been demonstrated to phosphorylate and inhibit the transcriptional activity of myocyte enhancer factor 2D (MEF2D) [69] that is critically involved in activity-dependent bdnf promoter I transcription in hippocampal neurons [70]. GSK-3 inhibition has been demonstrated to enhance prefrontal cortical-hippocampal coherence in rats [71] and in an AD model GSK-3 activation suppressed gamma power in the entorhinal cortex [72], a region particularly vulnerable to Aβ deposition in AD [73]. Aβ in turn not only increases activation of GSK-3 [72, 74], thus promoting another positive feedback loop of progressive oscillatory disruption and increased deposition of Aβ, but additionally was shown to attenuate BDNF mRNA expression and signaling in cortical cultures [75, 76] and impair BDNF retrograde transport [77] (Fig. 2). In addition, exposure of cortical cultures to Aβ increased truncated TrkB-T1 receptor levels with a corresponding decrease in full length TrkB [59]. Furthermore, Aβ peptide-induced cleavage of TrkB, shown to be mediated by calcium-dependent calpain proteases, produced a new truncated receptor and an intracellular fragment [78]. Thus, Aβ may play a direct role in the dysregulation of circuit dysfunction and connectivity in AD through inhibition BDNF/TrkB signaling, with GSK-3 acting as a central mediator.
In regard to PD, as stated earlier GSK-3 activation increases α-synuclein expression and phosphorylation of serine 129 [27, 31] promoting aggregate formation [79] and is also implicated in unfolded protein response-mediated cell death [80]. Importantly, α-synuclein was also demonstrated to suppress BDNF expression [31], and to interact directly with TrkB receptors [81]. In addition, α-synuclein reduced TrkB axonal trafficking and suppressed TrkB protein levels via up-regulation of TrkB ubiquitination [81]. These interactions between GSK-3, Aβ and α-synuclein, and the resulting suppression of BDNF signaling, establish a complex interconnection of pathways that promote AD and PD neuropathology and hasten the neurodegenerative process (Fig. 3).
The insulin/IGF-1-GSK-3 loop
Insulin and insulin-like growth factor-1 (IGF-1) are closely related hormones that control different aspects of growth, glucose and lipid metabolism. This is accomplished via activation of two tyrosine kinase receptors, the insulin receptor (IR) and the IGF-1 receptor. Insulin and IGF-1 fully activate their own receptors, as well as bind and activate each other’s receptors with reduced affinity. Evidence suggests that, for the most part, insulin predominantly mediates metabolic responses whereas the major role of IGF-1 is the promotion of growth. However, under certain conditions, such as insulin resistance, insulin and IGF-1 can elicit very similar responses in both humans and animals [82, 83]. Like the BDNF receptor TrkB, the IR and IGF-1R are tyrosine receptor kinases that suppress the activity of GSK-3 through phosphorylation and activation of Akt and PKC. Thus impairments in brain insulin/IGF-1 signaling contribute to neurodegeneration via impacts on tau and α-synuclein phosphorylation; increased expression of APP in AD; the induction of oxidative stress; and the onset of inflammatory processes (Figs. 2 and 3).
There is emerging evidence in favor of AD as a metabolic disease, whereby the brain develops resistance to the effects of insulin and insulin growth factor-1 (IGF-1) and loses the ability to efficiently utilize glucose [84, 85]. In addition, individuals with type 2 diabetes mellitus show approximately double the risk of developing AD [86]. The role of insulin or IGF-1 signaling deficits in the neuropathology of PD has been less well studied. PD patients exhibit increased insulin resistance, as well as significantly reduced IR mRNA and increased levels of insulin receptor substrate (IRS) serine phosphorylation in the SNpc [87–89]. Reductions in insulin and IGF-1 signaling PD brains have also been documented, with an apparent relationship between serum or frontal cortical IGF-1 deficits with α-synuclein accumulation, Lewy bodies and dementia [90, 91].
In healthy humans, a significant correlation between insulin resistance, tau phosphorylation and poorer performance in cognitive tasks has been recently shown [92] with a similar relationship demonstrated in rats [93, 94]. The suppression of GSK-3 activity in response to insulin has been established [95] and the induction of a diabetic-like state and insulin resistance in non-human primates or rodents by streptozotocin administration, or exposure to a high fat diet, is associated with increased tau and amyloid pathology, neuroinflammation and cognitive dysfunction that is coincident with increased activation of GSK-3 [96–98]. Importantly, the effects of IR signaling inhibition on GSK-3 activation are potentially exacerbated via tau effects on insulin signaling. The dysregulation of normal tau protein interactions resulting from hyperphosphorylation likely play a role in the induction of insulin resistance [99, 100]. Insulin was not only demonstrated to accumulate in hyperphosphorylated tau-bearing neurons in AD, but tau hyperphosphorylation in neurons directly resulted in the accumulation of insulin oligomers and deficits in insulin signaling, suggesting a causal role for tauopathy in AD-associated insulin resistance [100]. With respect to APP processing GSK-3 phosphorylates presenilin-1 at serine 353 and 357 residues [101] and increases the Aβ42/40 ratio [20, 102]. There is an inverse correlation between GSK-3 activity and presenilin-1 phosphorylation with IR mRNA and protein expression in rodent and AD brain [103], thereby promoting the suppression of insulin signaling and the further upregulation of GSK-3 activity. Reports also indicate that insulin signaling may also be reduced by Aβ directly. Specifically, intra-hippocampal administration of Aβ40/42 attenuates insulin signaling and induces spatial working memory deficits in mice [104]. Furthermore, a recent in vitro study showed that Aβ exacerbated α-synuclein-induced neurotoxicity that was mediated via impairments in insulin signaling [105]. A direct role of α-synuclein in the regulation of insulin signaling is less clear however. In human SK-N-SH cells, and in transgenic mice, overexpression of α-synuclein increased serine phosphorylation of IRS-1 and suppressed Akt signaling [106], and α-synuclein was shown to increase activation of insulin-degrading enzyme [107], a protein involved in the degradation of small peptides such as insulin. Importantly, these inhibitory effects of α-synuclein on insulin signaling would also have additional consequent actions on GSK-3 via loss of inhibitory regulation.
Studies from diabetic models also suggest that, in addition to its role in the regulation of presenilin, GSK-3 may have a direct effect on insulin signaling. Specifically, inhibitors of GSK-3 can augment insulin action on glucose uptake [108, 109] and improve insulin resistance in obese mice [110]. Similarly, genetic knockdown improved insulin sensitivity in a model of insulin resistance [111]. Furthermore, in vitro GSK-3 has been shown to phosphorylate and suppress the activation of IRS-1 [112, 113].
It is also noteworthy that interactions between the TrkB and IR/IGF-1R pathways have been reported. Specifically, BDNF enhanced peripheral insulin signaling in diabetic mice via increased IR expression and PI3K activity [114], an upstream regulator of Akt. This suggests that a similar interaction may exist in brain, an idea supported by studies in cultured neurons that showed increased IRS-1 and IRS-2 phosphorylation following exposure to BDNF [115]. Furthermore, a reciprocal relationship may exist as IGF-1 application to cultured neurons increased TrkB mRNA and protein expression and enhanced BDNF/TrkB signaling [116, 117]. These findings collectively highlight the GSK-3 as a central component in the cross talk between bof the BDNF and IGF-1 signaling pathways in AD and PD (Figs. 2 and 3).
The cholinergic-GSK-3 loop
The involvement of dysregulated cholinergic signaling in neurodegenerative diseases has been the most well characterized in AD with the cholinergic hypothesis [118], the first theory of AD, based on initial findings showing a loss of cholinergic activity in the brains of AD patients [119, 120]. GSK-3 has recently been highlighted as a potential signaling intermediate in this pathway. Temporally, cholinergic deficits progress in AD as the disease advances becoming more prominent in later stages [121, 122]. At present, acetylcholinesterase inhibitors (AChEIs) to increase acetylcholine levels are the mainstay for the treatment of mild to moderate AD. Unfortunately, not all treatments based upon the cholinergic hypothesis are efficacious in treating the symptoms or slowing the progression of AD [123]. Thus, there is yet debate as to whether disrupted cholinergic activity is actually causal in AD, or a consequence of another pathological process [118]. Indeed, Aβ oligomers are believed to contribute to cholinergic degeneration in AD [124] suggestive of a consequential involvement. Although less well characterized, cholinergic deficits have also been reported in PD [125]. For example, in the basal forebrain cholinergic loss has been documented in the nucleus basalis and thought to contribute to cognitive deficits the disease [126]. In addition, whereas cholinergic neurons within the pedunculopontine nucleus (PPN) are unaffected in AD, there is significant degeneration of the large cholinergic neurons of the lateral part of the PPN, pars compacta [127], which is posited to be involved in the postural imbalance that may manifest in PD patients [125]. Further, the overexpression of α-synuclein in transgenic PD mouse models blocked the induction of long-term potentiation in striatal cholinergic neurons producing early motor and memory deficits [128].
There are two categories of cholinergic receptors that bind acetylcholine, muscarinic and nicotinic. The muscarinic receptor is a G-protein coupled receptor while the nicotinic receptor, which also binds nicotine, is a ligand-gated ion channel. A link between GSK-3 and cholinergic signaling has not been examined in depth, however evidence does suggest a negative bidirectional influence. For example, administration of the muscarinic agonist pilocarpine or the AChEI physostigmine induced a rapid suppression of GSK-3 activity in murine hippocampus, cerebral cortex, and striatum [129] supportive of negative role for cholinergic signaling in the regulation of GSK-3. In line with this, nicotinic receptor activation suppressed neuronal GSK-3 activity in mice [130], whereas the muscarinic antagonist scopolamine induced learning and memory deficits that was coincident with increased activation of GSK-3 [131]. In cells, GSK-3-induced tau phosphorylation was suppressed by treatment with the muscarinic agonist carbachol [132] and in an Aβ42-induced neuronal toxicity AD model, the AChEI donepezil suppressed GSK-3 activity and improved neuronal viability [133]. Effects of GSK-3 on cholinergic signaling have also been documented. In rats, activation of GSK-3 decreased striatal ACh levels, that was mediated through impaired choline acetyltransferase activity [134]. In addition, a recent study demonstrated that activation of GSK-3 disrupted cholinergic homoeostasis in both the nucleus basalis and frontal cortex of rats, with cholinergic dysfunction rescued by inhibitors of the kinase [135]. Importantly insulin has been shown to activate IR-expressing cholinergic neurons in vivo [136], with cholinergic signaling enhancing BDNF expression and neurogenesis [118]. Thus, the suppression of insulin signaling inherent in AD and PD would have direct impacts on the cholinergic system, in addition to direct and indirect impacts on BDNF (Figs. 2 and 3).
The need for a multi-target strategy
In the search for novel therapies for both AD and PD, most studies have focused on targeting one of the pathological loops outlined above, and even though preclinical findings appeared promising, the subsequent clinical translation has been limited. In AD, AChEIs are the primary mode of treatment for patients with mild to moderate AD, however the effectiveness of these drugs on improving cognitive performance remains uncertain. Only a subset of AD patients responds to AChEIs [137], and of those that do respond, improvements in cognition and functionality appear to be short-lived [138, 139]. In clinical trials evaluating the therapeutic effects of AChEIs in PD, there were improvements in postural instability [140], as well as modest efficacy in improving cognitive impairment [141]. Thus, AChEIs may prove beneficial as adjunct therapy in PD to alleviate disease-associated symptoms.
Immunotherapeutic strategies to combat proteinopathies in AD and PD have also been employed. In AD, strategies aiming to attenuate the pro-inflammatory immune response or to facilitate Aβ clearance have been repeatedly attempted, with some studies even demonstrating increased progression of disease pathology [142]. For example, the administration of nonsteroidal anti-inflammatory drugs (NSAIDs) was shown to have adverse effects in AD patients with more advanced pathogenesis [143], and active or passive immunization against Aβ were either ineffective or very poorly tolerated [142]. Attempts to reduce Aβ load through the use of beta-secretase 1 (BACE) inhibitors have also been unsuccessful with the most recent failure that of verubecestat, which did not improve cognition in AD patients, nor suppress cognitive decline [144]. Importantly, even with the successful elimination of Aβ in AD there is still question as to whether this will result in a cure as one study showed that the clearance of Aβ plaques via Aβ42 immunization did not prevent progressive neurodegeneration in AD patients [145]. Efforts to limit tau aggregation with inhibitors have also been performed in clinical trials with no effect [146], however trials evaluating the effects of immunotherapy on tau are currently ongoing. Similarly, in PD, clinical trials using immunotherapy to reduce α-synuclein are also underway, and other mechanisms to inhibit α-synuclein aggregation including chaperone-related manipulations, mechanisms to inhibit serine 129 phosphorylation, and small molecule inhibitors are also being evaluated [147]. Unfortunately, if the AD clinical trials are any indication, a one-target approach must be viewed with cautious optimism, and the additional challenges associated with these methodologies in PD considered, such as targeting the appropriate molecular species of α-synuclein and addressing individual variability in pathogenic α-synuclein conformers [148].
In addition to immunotherapy other approaches have looked into targeting other pathways involved in AD or PD pathology. Liraglutide, a glucagon-like peptide-1 (GLP-1) analog used to treat type 2 diabetes, shows promise in alleviating some of the features of AD, such as cognitive impairment, insulin signaling pathology, tau hyperphosphorylation, and inflammation in rodent and non-human primate models [149, 150]. Similarly, liraglutide was recently shown to be neuroprotective in a PD mouse model [151]. There have been conflicting reports regarding the beneficial versus harmful effects of another diabetes drug, metformin, in AD. Whereas some evidence suggests a therapeutic effect of metformin in AD-like pathology in diabetic as well as AD models [152, 153], other animal and clinical studies highlight the potential of the drug to advance the disease [154-156]. Similarly in PD, despite some preclinical and clinical evidence of therapeutic effects of metformin [157–159] the hastening of disease progression is also a concern [155, 160].
Less research has focused on enhancing BDNF-TrkB signaling as a therapeutic approach in AD or PD, in part likely due to BDNF exhibiting poor pharmacokinetic properties. As a result, the development of small molecule TrkB agonists have become a worthwhile goal [161]. To date several of these molecules have been identified and shown to have positive effects in preclinical models. For example, in an AD rodent model, following treatment with the “TrkB agonist” 7,8-dihydroxyflavone neuroprotective effects and improved cognition were observed [162, 163], however with no effects on APP processing [164]. Similarly, deoxygedunin was neuroprotective in both the 6-OHDA and MPTP PD models [165]. However, the specificity of these compounds has been recently challenged, with concerns raised as to whether TrkB is the actual pharmacological target [166]. Specifically, employing a series of quantitative assays to measure TrkB receptor activation, TrkB-dependent downstream signaling, and gene expression, the authors were unable to reproduce the effects induced by BDNF using the small molecule TrkB agonists. Nonetheless, given the pivotal role of neurotrophin signaling in neurodegenerative diseases, a continued focus on developing selective TrkB agonists is wholly warranted. However, other more indirect mechanisms to induce BDNF and/or TrkB expression may also be worth considering. Serotonin receptors, for example, have been shown to play a positive role in learning and memory and are associated with increased BDNF signaling [167]. Similarly, wnt signaling, known to be impaired in AD and PD [168, 169] increases BDNF expression and regulates BDNF signaling in neurons and microglia [170, 171]. Indeed, in vitro wnt1 or wnt1-like agonists have been shown to be neuroprotective [172, 173] and the drug simvastatin, shown to induce neurogenesis via enhanced wnt signaling [174], was associated with reduced risk and incidence of dementia in PD [175, 176]. Increased wnt signaling and the consequent increase in β-catenin levels are not without its own potential issues, however, and are addressed in further detail below.
GSK-3 as a therapeutic target
In this review the central role of GSK-3 in mediating and promoting feed-forward processes to advance disease progression of AD and PD highlights the potential of this protein as a therapeutic target. Indeed, numerous preclinical studies using inhibitors of GSK-3 have shown success towards normalizing some of the deficits inherent in these neurodegenerative diseases. In this regard the search for novel inhibitors is ongoing. Yet, is the sole targeting of GSK-3 with highly potent inhibitors necessarily the best approach? To date, monotherapies have shown limited efficacy in stalling disease progression, and the few clinical studies that have evaluated GSK-3 inhibitors do not appear to be any more successful in this regard. Perhaps one of the more well studied GSK-3 inhibitors is lithium. Some human studies suggest that lithium, a non-selective GSK-3 inhibitor, may have some benefits in improving the cognitive symptoms of AD [177, 178] whereas other evidence suggests no effect of lithium treatment on tau phosphorylation [179]. Similarly, treatment with tideglusib (NP12), a small molecule non-ATP-competitive and irreversible GSK-3 inhibitor, was neuroprotective and alleviated AD pathology in an AD model [19] but did not show any clinical benefits following 26 weeks of administration [180].
The screening of potential candidate molecules in the search for potent and selective GSK-3 inhibitors for treatment in AD and PD is ongoing. The long-term effects of GSK-3 inhibition as a therapeutic approach in AD and PD has yet to be determined, yet a vital physiological function of GSK-3 as a master regulator of cell fate raises concerns regarding undesired pharmacological effects in patients. Specifically, the role of GSK-3 in cellular apoptosis depends on the cellular or signaling context [181]. Most studies have evaluated the pro-apoptotic effects of GSK-3, yet the idea that GSK-3 can also inhibit apoptosis came from the demonstration that GSK-3β knockout mice are not viable due to massive hepatocyte apoptosis and TNF hypertoxicity [182]. Whereas mitochondria-mediated intrinsic apoptotic signaling is initiated by processes that cause cell damage, such as oxidative stress, and are facilitated by GSK-3; the extrinsic apoptotic pathway, mediated through activation of death receptors, is inhibited by GSK-3 [181]. Indeed, lithium and other GSK-3 inhibitors can facilitate apoptotic signaling [183] highlighting the need to keep this dual role of GSK-3 in cell fate at the forefront of our minds when developing novel therapies. In line with this, GSK-3 is a critical effector of the wnt signaling pathway that is critically involved in the regulation of cell survival [184]. One of the predominant and most widely studied substrates of GSK-3 in this pathway is the protein β-catenin, that mediates cell–cell adhesion and gene transcription. In response to wnt signaling GSK-3 activity is suppressed and β-catenin is stabilized, entering the nucleus to initiate gene transcription [185]. Of importance, increased levels of β-catenin in various cancers have been widely documented [186]. Local cellular GSK-3 is also involved in the migratory processes of various cell types, with opposing effects of local versus global cellular GSK-3 inhibition on cell motility [36]. This suggests that the long-term impacts of GSK-3 inhibition may be unpredictable, due to the differing functional effects of the protein in distinct cellular pools.
Despite a variety of pathogenic processes being individually targeted in AD and PD, the identification of novel and effective therapies has thus far been disappointing. While some aspects of disease pathology appear to improve in some instances, generally in preclinical studies, similar effects in the clinic are not apparent. When one examines the inter-relationship between the multiple feed-forward loops in AD or PD perhaps this should not be entirely surprising as correcting one imbalance may have lesser impact given the concerted contribution of others. This highlights not only the need for preclinical studies to use a combination of readout measures in study designs to evaluate novel therapeutics, but also draw attention to the need for a different therapeutic approach. Ideally, given that GSK-3 lies at the hub of many of the aberrant pathways in AD and PD, one would think that their normalization could be achieved through targeted and potent inhibition of this kinase, yet, thus far clinical studies evaluating GSK-3 inhibitors in AD have also failed, once again drawing attention to the idea that monotherapies may simply not be sufficient. We propose that a multi-target strategy targeting the major feed-forward loops in AD and PD, and which could incorporate less potent inhibitors of GSK-3, may hold the most promise for advancing therapies. The idea of multi-target therapy in AD was first proposed a decade ago [187], following identification of the dual cholinesterase and BACE-1 inhibitor, memoquin, that possessed both anti-Aβ and anti-oxidant properties [188]. More recently, this same group has advocated for the use of a dual GSK-3 and BACE-1 inhibitor ligands for AD treatment [189] and the development of multi-target inhibitors holds much promise for the future treatment of neurodegenerative diseases. Clinical studies evaluating the effects of combination therapy with the NMDA antagonist memantine and cholinesterase inhibitors in patients with AD have demonstrated good tolerability and, in two instances, improved outcomes [190–192]. On the other hand, idalopirdine, a 5-HT6 receptor antagonist, used in a recent clinical trial as adjunct treatment with cholinesterase inhibitors to improve cognition in AD patients was unsuccessful [193]. Similarly, the effects of antioxidant supplements, such as vitamin E, resveratrol, curcumin, among others as adjunct therapy in AD have also been evaluated with mixed results [194].
CONCLUSIONS
Clearly there is still much work to do in the search for the appropriate combination of therapeutic drugs in AD and PD that are not only efficacious but are safe and well tolerated. However, after the successive string of disappointing clinical trial outcomes it is time to step back and critically reassess the monotherapeutic strategies. While not without its own challenges, simultaneously targeting the multiple processes that are dysregulated in these diseases, such as GSK-3 inhibition, neurotrophin signaling, insulin signaling, and oxidative stress to halt the feed forward processes that contribute disease pathology may have significant potential to slow disease progression. For example, a recent small clinical trial showed that administration of the polyphenol resveratrol, a sirtuin 1 activator, for 52 weeks reduced neuroinflammation in AD patients and this was associated with improved cognition and reduced CSF levels of Aβ [195]. No effects were observed on tau levels in this study however, suggesting that perhaps a co-intervention to target this facet of the disease may have merit. It is also possible that less potent drugs may be sufficient in combination therapy, which may be advisable given current evidence of advanced disease progression with diabetic drugs. Similarly, while the incorporation of GSK-3 inhibitors into this multi-target approach has great merit, we caution the use of highly potent and/or irreversible inhibitors due to the potential for significant negative effects with long-term use. One possible approach is to focus more research on targeting select important downstream substrates of GSK-3, instead of the protein itself where possible, to limit impacts on pathways not pertinent to AD or PD pathology.
DISCLOSURE
No conflicts of interest.
Footnotes
ACKNOWLEDGMENTS
This work was supported by a grant from the W. Garfield Weston Foundation (to MLP).