
Abstract
Depression and anxiety are the most common mood disorders affecting 300 million sufferers worldwide. Maladaptive changes in the neuroendocrine stress response is cited as the most common underlying cause, though how the circuits underlying this response are controlled at the molecular level, remains largely unknown. Approximately 40% of patients do not respond to current treatments, indicating that untapped mechanisms exist. Here we review recent evidence implicating JNK in the control of anxiety and depressive-like behavior with a particular focus on its action in immature granule cells of the hippocampal neurogenic niche and the potential for therapeutic targeting for affective disorders.
Anxiety and depression are among the largest causes of disability worldwide [1]. They have complex and varied etiologies with genetic, epigenetic and environmental factors contributing to disease susceptibility. Maladaptative changes in normal stress responses leading to long lasting physical changes at the level of synapses and circuits are believed to be among the underlying causes. Antidepressant drugs have targeted the same core mechanisms for several decades, yet treatment-resistant depression is still a major problem, indicating the need for a paradigm shift [2]. Many theories of depression have been proposed, including dysregulation of monoaminergic neurotransmission, neurotrophic factors and hippocampal neurogenesis [3, 4]. However, the signalling molecules that govern mood and its underlying circuitry are largely unknown and identifying these will be essential for a comprehensive understanding of mood disorders and development of new treatments.
ADULT HIPPOCAMPAL NEUROGENESIS AND DEPRESSION
Prominent studies have shown that adult hippocampal neurogenesis is required for the action of antidepressant drugs [5–8], as well as for electroconvulsive shock therapy [9]. This discovery prompted extensive studies of adult born hippocampal granule cells in the context of mood, leading to the hypothesis that depressive and anxiety symptoms can be alleviated by boosting neurogenesis in the hippocampus while impaired neurogenesis may be causal in triggering symptoms initially. Yet, this theory remains controversial as other studies have shown beneficial effects of antidepressants that were independent of neurogenesis changes [10, 11]. Our lab identified that inhibition of c-Jun N-terminal kinase (JNK) in the hippocampal neurogenic niche increases the number of adult born granule cells, while alleviating anxiety behaviour [12]. More critically, we demonstrated that inhibiting JNK solely in adult born granule cells was sufficient to alleviate anxiety and depressive behavior [12]. These findings implicate JNK as a central player that controls anxiety and depression from a subpopulation of adult born granule cells in the hippocampus.
First described in the hippocampus [13], the generation of new neurons in adult brain, or “neurogenesis” occurs at a rate of about 700 new granule cells added daily per hippocampus in humans, representing 0.004% of dentate gyrus neurons [14]. Notably, adult hippocampal neurogenesis is confined to the dentate gyrus sub-region of the hippocampus in all species [15]. In rodents, the extent of neurogenesis in the dentate gyrus is higher [16], accounting for up to 0.06% of dentate gyrus granule cells in two month old mice [17, 18]. Increased neurogenesis in the dorsal hippocampus has been shown to improve spatial memory [19, 20] whereas in the ventral hippocampus, it is associated with suppression of anxiety-like behaviour [7, 21–23]. Neurogenesis in mice can be boosted by exercise or by exposure to an enriched environment [17, 22, 24], leading to improved memory [25, 26]. However, running induces neurogenesis in the dorsal and not the ventral hippocampus [27], and running-induced suppression of anxiety has been dissociated from hippocampal neurogenesis [7, 10, 11, 28].
ANXIETY AND DEPRESSION – THE HIPPOCAMPAL CONNECTION
Anxiety is a normal stress response that allows the individual to react to a threatening environment. Anxiety disorders arise when this stress response occurs in the absence of real threat. It shows a high level of comorbidity with depression and rates of secondary depression following a prior psychiatric illness, in particular anxiety, have grown significantly in modern times [29]. In depressed individuals, disease-associated structural changes take place in the hippocampus, indicating a central role for this region in the underlying pathology. Neuroimaging studies have suggested that hippocampal volume may be reduced in patients suffering from depression and anxiety [30, 31]. These findings from small patient cohorts have been validated by meta-analysis (involving 1728 major depressive disorder (MDD) patients and 7199 controls) and conclude that hippocampal volume decreases on average by 1.42% in MDD. Moreover, this decrease in hippocampal volume correlates with recurrence frequency [31]. Post-mortem studies in MDD subjects revealed loss of neuropil, i.e. neuritic matter consisting of unmyelinated axons, dendrites or glial cell processes, suggesting that neurite atrophy and synapse loss accounted for diminished volume in this region [32, 33]. Specifically in the hippocampal Cornu Ammonis-3 (CA3) region, reduced dendrite and spine density was associated with higher levels of trait anxiety and longitudinal depression scores [34, 35], while in the dentate gyrus sub-region, the number of neural progenitor cells and granule cells was reduced in MDD subjects [36, 37]. While these anatomical hallmarks of MDD suggest that levels of adult hippocampal neurogenesis may be reduced in these individuals, definitive proof that neurogenesis is impaired in humans suffering from affective disorders is still lacking. This is because no studies have used birth dating approaches to study neurogenesis in post mortem brains from humans that suffered from these disorders. Also, there are currently no methods to directly measure neurogenesis in situ in humans.
Stereological measurement of neurogenesis in post mortem brain has provided some evidence for a link between depression and adult hippocampal neurogenesis. For example, depressed subjects that received selective serotonin reuptake inhibitor (SSRI) or tricyclic anti-depressant (TCA) drugs, showed increased neural progenitor number in the anterior (ventral) dentate gyrus compared to untreated subjects [36]. Another study showed that hippocampal volume was restored in patients that received antidepressant drugs [37]. Interestingly, the meta-analysis found no significant recovery of hippocampal volume in patients undergoing antidepressant treatment [31], possibly due to confounding interactions such as severity and duration of disease. Nonetheless, these clinical studies have provided clear evidence that structural changes occur at many levels in the hippocampus of individuals suffering from depression and anxiety and have given insight into the underlying pathology.
Similarly in animal models, the hippocampus emerges as a central regulator of depression and anxiety [38–42]. The ventral hippocampus extends connections to prefrontal cortex (PFC) [43] and forms reciprocal connections with amygdala [41], among other regions [44–46]. These circuits are highly conserved and are required for synchronized hippocampal-PFC activity and normal adaptive, anxiety behaviour [47]. Maladaptive changes in circuits emanating from the hippocampus are likely to contribute to the pathological state where anxiety is present in the absence of threat [48]. The signalling molecules driving these circuits and maladaptive changes therein are largely unknown.
c-JUN N-TERMINAL KINASES (JNKs)
JNK was originally identified as stress-activated protein kinase (SAPK) that responded to a range of cellular stressors including DNA damage, oxidative stress, cytoskeletal toxins, infection and inflammation [49]. SAPK was renamed “JNK” when it was identified as the kinase responsible for phosphorylation and transcriptional activation of c-Jun [50, 51]. In brain, JNK expression is complex, with three JNK genes; JNK1 (MAPK8), JNK2 (MAPK9) and JNK3 (MAPK10) and 10 splice variants (JNK1α1, JNK1α2, JNK1β1, JNK1β2, JNK2α1, JNK2α2, JNK2β1, JNK2β2, JNK3α1, JNK3α2) being expressed [52]. JNKs are associated with inflammation, neurodegeneration and insulin resistance [53–55]. JNKs are particularly important kinases in brain where they display elevated activity in the absence of stress, indicating that brain JNKs also serve non-stress related functions. This distinguishes brain JNK from JNK in other tissues such as heart, lung, kidney, spleen and liver, where its activity is low in the absence of stress [56]. JNK1 constitutes a major component of physiologically active JNK in brain, whereas JNK2 and JNK3 isoforms exhibit lower basal activity and increased stress responsivity [57]. Yet, knockout or knockdown of all three JNK isoforms has been necessary to reveal functions for JNK in trophic deprivation-induced neuronal death [58], autophagy [59] and pituitary function [54] indicating functional redundancy between isoforms. JNK has also been shown to regulate neuropathic pain [60] and inhibition of JNK protects from neuronal death following cerebral ischemia [61, 62]. Interestingly however, with the exception of JNK regulation of pituitary function and insulin resistance [63, 64], until recently, there was relatively little known about the physiological role of JNK in adult brain based on in vivo study and behavioural analysis. Here we outline recent evidence that associates JNK with the control of mood.
JNK1 REGULATES ANXIETY AND DEPRESSIVE-LIKE BEHAVIOUR IN MICE
To gain insight on JNK1 function in adult brain, Jnk1-/- mice were subjected to a battery of behavioural tests. The most prominent results emerged from tests for anxiety where Jnk1-/- mice displayed a low anxiety phenotype in the open field, elevated plus maze and light-dark tests [12] (Table 1). In the forced swim test of behavioural despair and the sucrose deprivation test to determine anhedonic behaviour, Jnk1-/- mice displayed reduced immobility and reduced anhedonia respectively, indicating a low depressive phenotype [12] (Table 1). These behavioural phenotypes were replicated when mice underwent intracerebroventricular infusion for six weeks with a peptide inhibitor of JNK (DJNKI-1, chemically identical to XG-102) (Fig. 1A; Table 1) [12].
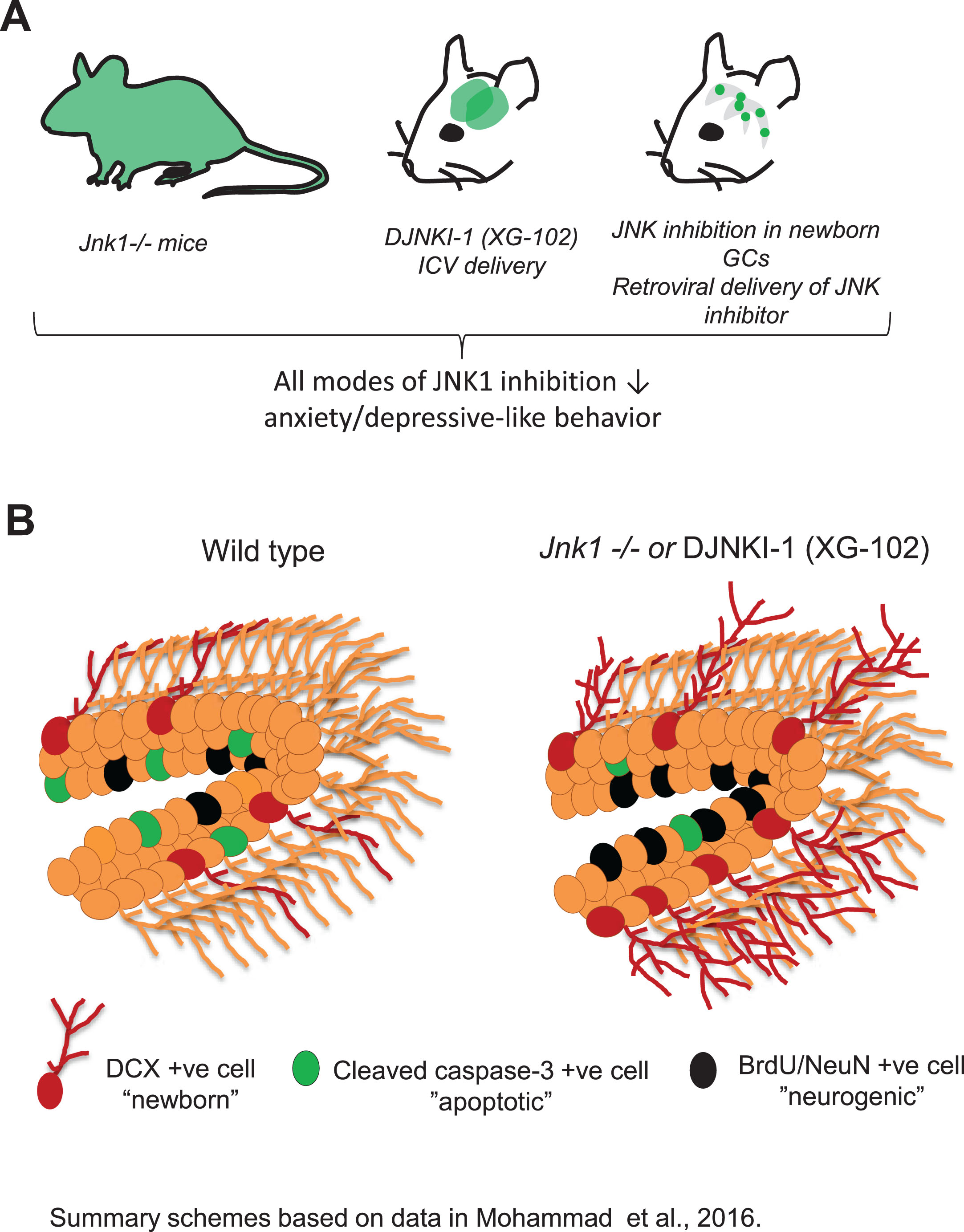
Inhibition of JNK solely in newborn granule cells of the hippocampus alleviates anxiety and depressive behaviour whereas global inhibition of JNK1 in brain boosts neurogenesis. A. Genetic ablation of Jnk1, intracerebroventricular (ICV) osmotic mini-pump infusion of DJNKI-1 (XG-102) over six weeks, or retroviral delivery of JNK inhibitor (MLV-NLS-JBD) to granule cells of the dentate gyrus for six weeks, evoke an anxiolytic and anti-depressant effect. The key finding was that specific targeting of adult born granule cell JNK was sufficient to switch behaviour to a low anxiety and low depressive state independent of neurogenesis changes [12]. B. Levels of neurogenesis were assessed in Jnk1-/- mice or DJNKI-1-treated mice. Neurogenesis was increased at several levels. There was increased proliferation (BrdU cell number, Ki67-positive cells), survival (decreased numbers of cells positive for cleaved caspase-3) and increased numbers of doublecortin (DCX)-positive and BrdU/NeuN-positive cells, representing newly born neurons. Thus global JNK1 inhibition increases hippocampal neurogenesis. However inhibition of JNK solely in immature granule cells using retroviral delivery alleviates mood without increasing neurogenesis, telling us that JNK inhibition evokes separable effects in the hippocampal neurogenic niche, both of which act to lower anxiety and depression.
Inhibition of JNK1 lowers anxiety and depressive behaviour in mice
Summary of behavioural test results from Mohammad and colleagues [12]. DJNKI-1 refers to a peptide inhibitor of JNK, also known as XG-102. MLV-NLS-JBD encodes amino acids 1 to 277 of JIP1 in a MLV retroviral vector with upstream flanking nuclear localisation sequences (NLS). The JBD sequence effectively inhibits nuclear JNK in neurons [71, 107].
JNK1 ACTIVITY IN NEWBORN GRANULE CELLS OF THE HIPPOCAMPUS INDUCES ANXIOGENIC AND DEPRESSIVE BEHAVIOR
Changes in the rate of adult hippocampal neurogenesis have been implicated in the pathology of anxiety and depression [3, 65]. We therefore examined neurogenesis in Jnk1 knockout mice using birthdate labelling. These results showed that progenitor cell proliferation was increased in the dentate gyrus of mice lacking Jnk1 and that the number of adult born (BrdU/NeuN-positive) neurons was increased (Fig. 1B) [12]. Moreover, cell survival and dendrite maturation, as measured by arborisation, was elevated in Jnk1 knockout mice (Fig. 1B) [12]. Importantly, these effects were replicated upon mini-pump infusion to the ventricles with a peptide inhibitor of JNK1 for six weeks (Fig. 1B) [12]. Finally, to determine whether adult born granule cells themselves were involved in the anxiolytic response, we expressed the JNK inhibitor in a Murine Leukemia Virus (MLV) plasmid and injected it to the hilus. This enabled selective targeting of the JNK inhibitor to newly born granule cells in the hippocampus of adult mice [12]. To our surprise, JNK inhibition solely in these cells was sufficient to reduce anxiety and depressive behaviour, indicating that JNK activity in newborn granule cells of the hippocampus plays an important role in the regulation of mood. Notably, there was no increase in granule cell number in these mice. Thus, like others [10, 11], we showed that increased neurogenesis was not necessary for this behavioural switch, as inhibition of JNK in new born granule cells was sufficient to improve mood in mice without having any effect on number of immature granule cells (Fig. 1B) [12].
THE DJNKI-1 INHIBITOR OF JNK
To inhibit JNK, the peptide inhibitor of JNK, DJNKI-1 (also known as XG-102) was used. This is the minimal sequence from the JNK scaffold, JNK interacting protein-1 (JIP1) that binds to the common docking domain of JNK and competes for substrate binding [53, 66]. DJNKI-1 also encodes the transactivator of transcription (TAT) sequence from the HIV vector which facilitates cell penetration [67, 68]. Further, to improve resilience against enzymatic digestion in vivo, DJNKI-1 utilizes D-amino acids, providing stability in cells for at least two weeks [69]. DJNKI-1 is considered to be a specific inhibitor of JNK and does not inhibit the closely related ERK or p38 kinases [70]. In our hands, DJNKI-1 more potently inhibits JNK1 isoforms over JNK2 and JNK3 in vitro (Coffey lab unpublished data), however its bioactivity against JNK isoforms in vivo has not been characterised. For this reason, it is significant that we cross-validated the findings with DJNKI-1 in Jnk1-/- mice.
Our lab has earlier shown that a longer version of the JNK inhibitor peptide, amino acids 1–277 of the JNK scaffold JIP1 (JBD for JNK binding domain), increased dendrite arborisation in cultured neurons, recapitulating findings in Jnk1-/- mice where dendrite arborisation was increased in the cerebellum and motor cortex [71, 72]. Thus, it was not altogether surprising that in adult hippocampus, JNK1 constrained the arborisation of doublecortin (DCX)-positive, immature granule cells (Fig. 1B) [12]. Indeed inhibition of JNK1 proved to be a powerful stimulus for adult hippocampal neurogenesis, promoting all four stages: proliferation, survival, differentiation and maturation (Fig. 1B).
HOW COULD JNK INHIBITION IN GRANULE CELLS ALTER BEHAVIOUR?
Dentate gyrus granule cells form a major cell group of the hippocampal trisynaptic circuit. They receive excitatory input from the entorhinal cortex via perforant path axons and transmit this to CA3 pyramidal neurons via their mossy fibers. The circuit is completed by projections from CA3 to CA1 which projects back to the entorhinal cortex. Indeed, the immature granule cells of the hippocampus are particularly sensitive to perforant path input [73]. They display a lower activation threshold than resident granule cells due to their particular configuration that equips them with increased excitatory/inhibitory balance [73]. Significantly, immature granule cells negatively regulate the overall activity of resident dentate granule cells by recruiting local GABAergic inhibitory neurons [42, 73, 74]. Consistent with this, increasing neurogenesis in mice leads to reduced activity of the dentate gyrus hippocampal subregion [42, 74, 75]. Conversely, blocking neurogenesis using x-ray irradiation or chemical genetic ablation, increases dentate gyrus gamma burst amplitude and synchronized firing [76]. The ventral hippocampus forms direct monosynaptic connections to structures that regulate mood such as medial prefrontal cortex which promotes anxiety behaviour [47, 77], reciprocal connections to and from amygdala, which governs fear processing [78], and to the medial shell of the nucleus accumbens [79, 80] which controls reward seeking behaviour and susceptibility to stress [81]. Thus boosting the number of adult born granule cells, or increasing their activity, is expected to reduce the neural activity of glutamatergic projections from the ventral hippocampus to downstream structures that regulate anxiety and reward responses [42]. Indeed, it has recently been shown that excitatory output from the ventral hippocampus to the medial prefrontal cortex is required for anxious behavior [47], while reduced activity in this region is associated with stress resilience in the chronic social defeat model of depression [81]. One possibility is that JNK evokes anxiogenic behaviour by controlling properties of immature granule cells leading to increased ventral hippocampal output, though this theory requires testing.
From the molecular standpoint, there are several mechanisms whereby JNK could influence the behaviour of new born granule cells leading to changes in the activity of circuits that control mood. New born granule cells exhibit specialized features during a “critical period” from two to six weeks after birth [73]. Within this timeframe, new granule cells show a lower threshold for activation by perforant-path excitatory hippocampal input. This distinguishes them from the resident mature cells that account for ∼90% of dentate neurons and are less easily excited [73, 82]. Increased excitability of immature granule cells is dependent on high expression during the critical period, of the NKCC1 cotransporter which carries Cl- inwards. High expression of NKCC1 (SLC12A2) results in reversal of the Cl– potential, meaning that upon GABA-A receptor activation, Cl– flows outwards, down its concentration gradient and the neurons become depolarized, rather than hyperpolarized [82]. As these neurons mature, NKCC2 (SCL12A1) expression increases and internal Cl– concentrations are restored to lower levels. At this point, granule cells display typical GABA-A responses, i.e. Cl– ions are driven outwards leading to hypolarization [83, 84]. JNK has been shown to phosphorylate NKCC1 in vitro in [85], however whether this alters the activity or expression levels of the cotransporter is not known.
Other key changes that occur to immature granule cells during the critical period include dendritic arborisation and spine formation, together representing increased capacity for synaptic integration. In this regard, we have shown that dendritic field size is increased in immature granule cells in Jnk1-/- or JNK inhibitor-treated mice [12]. NMDA receptor NR2B subunit expression is also increased during this period and facilitates synaptic plasticity [86]. Whether JNK controls NR2B expression is not known. All in all one can envisage several possible mechanisms whereby pathological activation of JNK (as outlined in Table 1) could alter properties of immature granule cells, either indirectly by altering gene expression, or directly by switching on or off protein function. More precise understanding of what JNK does in immature granule cells to alter mood will be greatly facilitated by phosphoproteomic study and analysis of circuits and gene expression.
WHAT IS KNOWN ABOUT JNK REGULATION IN ANIMAL MODELS OF DEPRESSION?
Studies in animal models of depression have shown that JNK activity is elevated in the hippocampus and prefrontal cortex (Table 2) [87–93]. For example, chronic treatment with corticosterone [94], strongly activates JNK in the hippocampus [93]. Elevated levels of cortisol in depression signifies impaired feedback regulation of the hypothalamic pituitary adrenal axis [95] and contributes to long lasting synapse loss [35]. Similarly, in chronic variable stress models of depression and in acute stress models (cold followed by rewarming) [87], forced swim and tail suspension tests [91] and immobilization stress [88], JNK is hyper-activated in the hippocampus and prefrontal cortex (Table 2). Significantly, in the social defeat stress model of depression, JNK activity is increased in animals that are susceptible to stress and reduced in animals that are resilient [90], suggesting that JNK activity predisposes to depression and inhibition of JNK is protective. In contrast to these classic models of depression, exposure of adult rats to 21 days of social isolation (a model for depression and negative symptoms of schizophrenia [96]), does not activate JNK but slightly decreases its activity in the hippocampus and PFC [88, 92] (Table 2). While the stress response was validated in these two studies by showing increased corticosterone levels, the behavioural outcome was not monitored. The reported decrease in JNK activity may therefore represent a protective, adaptive response to social isolation in this relatively short duration isolation model. Finally, cognitive impairment induced by chronic corticosterone administration is reversed by the DJNKI-1 inhibitor [93], while DJNKI-1 also lowers baseline anxiety and depressive behaviour [12]. Together these data provide evidence that JNK is activated in rodent models of depression and that preventing this activation alleviates anxiety and depressive behaviour. Significantly also, reduced JNK activity is associated with resilience to stress.
Regulation of JNK activity in rodent models of depression
In these studies, JNK activity was measured by immunoblotting hippocampus (HC) or prefrontal cortex (PFC) with antibodies detecting active JNK (P-JNK). Chronic and acute stress models are categorized separately. Abbreviations are as follows: Lipopolysaccharide = LPS, Corticosterone = CORT, data not available = n.a.
Genetic components together with environmental influences also contribute to depression and predisposing gene anomalies are actively searched [97]. As of now, no significant genome-wide associations have been found to link the JNK pathway with MDD or bipolar disorder. Although SNPs in the JIP2 (MAPK8IP2) JNK scaffold [98] and in JNK3 (MAPK10) [99] were identified in patient cohorts, these have not shown significant genome-wide associations. Such associations may be obscured by the heterogenous nature of the disease etiology. The first genetic associations with depression are only starting to be identified and improved patient stratification and larger sample sizes will be needed to achieve a comprehensive picture of the genetic background of depression [100].
TARGETING JNK FOR DEPRESSION AND ANXIETY?
These findings from animal studies, highlight the possibility that targeting of the JNK pathway could provide a new avenue for tackling depression and anxiety. It may even provide benefit for treatment resistant depression. This raises the question as to whether unwanted side effects would be associated with systemic inhibition of JNK. JNKs, by virtue of being kinases, are involved in a variety of functions [55]. Inhibition of JNK is expected to be beneficial for several diseases including cancer, inflammatory diseases, diabetes, neurodegenerative disease, stroke [53], and more recently, depression and anxiety [12, 90]. Indeed, because JNKs are largely silent and activated by stress, there is a rationale to expect minimal side effects. However, cellular stress responses also serve important homeostatic functions. For example JNKs regulate autophagy, albeit oppositely in the periphery and CNS [59, 101]. Also, JNKs 1 and 2 are required for innate immune response signalling [102], and as discussed earlier, JNK isoforms play essential roles during brain development [55]. One can predict therefore that pan-JNK inhibitors would be precluded from use. However, because these important functions of JNKs are largely redundant between isoforms, it is anticipated that isoform-specific inhibitors, or inhibitors of specific functional modules, could be tolerated. Several years of drug development have generated a number of inhibitors against JNK, however a full panel of isoform-specific, or splice variant-specific inhibitors is not yet available [103]. In terms of tolerance, two clinical trials with small molecule ATP-competitive JNK inhibitors have been carried out [103, 104]. These were terminated due to a poor risk/benefit ratio, however these trials did not stratify patients, and it has been proposed that isoform-specific JNK inhibitors would have produced a better outcome [104]. Interestingly however, a small cohort safety trial using DJNKI-1 (XG-102, Brimapitide) reported no drug-related side effects following a single intravenous infusion [105]. Moreover, this inhibitor yielded positive results in the postoperative treatment of ocular inflammation [106], where a single injection showed similar benefit to four applications of dexamethasone over 21 days. This inhibitor is currently undergoing preclinical study for treatment of Alzheimer’s disease. Given the recent implications of JNK involvement in depression, it would certainly be of interest to monitor the effects of JNK inhibition on depression and anxiety symptoms in patients, if such trials go ahead.
CONCLUSIONS
This review highlights recent data that implicates the JNK pathway in the regulation of mood. These early findings are exciting for the following reasons. Firstly, according to current knowledge, there is no overt connection between JNK and the currently used anti-depressant drugs. Therefore, targeting of this mechanism may represent a new avenue for treatment development. Secondly, the precise localization of JNK’s anxiogenic action to the immature granule cells of the hippocampus provides the possibility for design of selective targeting approaches. In this regard, it will be advantageous to understand more aspects of JNK action in adult born granule cells from in vivo study. While much remains to be understood, we can say that JNK has entered the arena as a kinase that controls anxiety and depressive behaviour in mice, and may contribute to maladaptive changes underlying affective disorders.
Footnotes
ACKNOWLEDGMENTS
This work was funded by the EU-funded Marie Curie r’BIRTH ITN # 608346 and by the Academy of Finland project #310583. We are grateful to the members of the Coffey lab and the rBIRTH consortium for their views and opinions that guided our work on depression and neurogenesis during the course of this network.