
Abstract
Brain metabolism is a fragile balance between nutrient/oxygen supply provided by the blood and neuronal/glial demand. Small perturbations in these parameters are necessary for proper homeostatic functioning and information processing, but can also cause significant damage and cell death if dysregulated. During embryonic and early post-natal development, massive neurogenesis occurs, a process that continues at a limited rate in adulthood in two neurogenic niches, one in the lateral ventricle and the other in the hippocampal dentate gyrus. When metabolic demand does not correspond with supply, which can occur dramatically in the case of hypoxia or ischemia, or more subtly in the case of neuropsychiatric or neurodegenerative disorders, both of these neurogenic niches can respond—either in a beneficial manner, to regenerate damaged or lost tissue, or in a detrimental fashion—creating aberrant synaptic connections. In this review, we focus on the complex relationship that exists between the cerebral vasculature and neurogenesis across development and in disease states including hypoxic-ischemic injury, hypertension, diabetes mellitus, and Alzheimer’s disease. Although there is still much to be elucidated, we are beginning to appreciate how neurogenesis may help or harm the metabolically-injured brain, in the hopes that these insights can be used to tailor novel therapeutics to regenerate damaged tissue after injury.
INTRODUCTION
The metabolic processes enabling the brain’s many complex, resource-intensive computations and functions, remain poorly understood, despite extensive study. From early on, it was recognized that cerebral blood flow changes locally in response to neuronal activity. In a series of classical experiments conducted in the late 1800s, Charles S. Roy and Charles S. Sherrington, working out of Cambridge in the United Kingdom, examined the vasomotion of cerebral vessels in response to a barrage of neural stimuli, concluding that the brain must be capable of regulating its vascular supply in order to meet local changes in neuronal activity [1]. Half a century later, Fred and Erna Gibbs, and their associate Harry Maxwell at the University of Illinois College of Medicine, injected a dye into the internal carotid artery supplying the brain, then extracted the dye back from the venous system as it returned to the heart, and examined changes in dye concentration to approximate the blood flow and oxygen consumption rates of the human brain [2]. Following up on these findings, experimental pharmacologists and neurosurgeons, interested in how the brain would cope with changes in systemic blood pressure, determined—somewhat unexpectedly—that cerebral blood flow remains remarkably constant across a wide range of cerebral perfusion pressures [3–5]. Consistent with Roy and Sherrington’s earlier findings, this effect is mediated by changes in cerebral vascular tone [6, 7].
The critical importance of cerebral perfusion regulation is clearly demonstrated when these processes go awry. Failure of cerebral autoregulation, as seen in patients with fulminant liver failure or severe head trauma for example, may result in catastrophic changes in intracranial pressure (ICP) [8, 9]. Meanwhile, more subtle abnormalities in regional blood flow may compromise cognitive function, and have long been known to associate with a variety of neuropsychiatric diseases [10, 11].
The neocortex and hippocampus are susceptible to transient decreases in oxygen or glucose, leading to memory loss and executive function deficits following global cerebral hypoperfusion [12, 13]. The hippocampus is of particular interest, as it remains one of the only mammalian brain regions that continues to give rise to new neurons throughout life, including in humans [14, 15]. For this reason, the hippocampus’s potential regenerative response following HI-injury differs from that of other brain regions [16]. The high degree of neuroplasticity afforded by ongoing neurogenesis may protect the hippocampus from injury exacerbation following hypoxia or ischemia, and may also provide us with insight for potential therapies for hypoxia/ischemic to explore in other brain regions. The subventricular zone (SVZ) of the lateral wall of the lateral ventricle also continues to produce new neurons throughout life, normally destined for the olfactory bulb [17]. However, under conditions of cortical ischemia, these neurons can be diverted to the site of injury, where they may play a role in regeneration and recovery [18].
In this review, we will examine recent research into how newborn cells in these areas may influence the progression and outcome of the injury. Our review begins with a discussion of the normal development of the neuro-glio-vascular system in the mammalian central nervous system, after which we consider the neurogenic response to pathological changes in nutrient/oxygen supply. We then move to the adult condition, in which we track neurogenesis over the normal lifespan and changes that occur in association with common vascular diseases related to aging. We focus on recent strides made over the past couple of decades, with classic and historical studies serving as the foundation for our discussion.
THE ANATOMICAL FORMATION OF HIPPOCAMPAL VASCULATURE, NEURONS AND GLIAL CELLS
The hippocampus arises from two adjacent clusters of cells located on the dorsomedial and dorsoposterior walls of the primordial telencephalon, and its individual components can be grossly appreciated as early as embryonic day E16-E18 in the developing rodent [19, 20]. By E21, the laminae of Ammon’s horn have developed and the dentate gyrus (DG) begins to take form out of medially-derived neuroepithelial cells near the dentate notch [21]. Out of these cells, the upper and lower blades of the DG take shape from E22 and reach their fully mature, adult form by P20-P30. Within the inner core of both blades sits a pool of neural progenitor cells (NPCs) responsible for generating the bulk of dentate granule cells (DGCs) during early postnatal life, and that will continue to produce new functionally integrated adult-born DGCs (abDGCs) throughout life [22–24].
As these processes take place, the coordinated sprouting, maturation, infiltration and branching of a network of blood vessels is also occurring. Mesodermally-derived angioblasts give rise to endothelial cells, which form blood islands around E6-E6.5, and in turn give rise to the major blood vessels of the body by E9-E12 [25, 26]. Angiogenesis in the brain can be traced back to branching and elongation of capillary sprouts from a vascular plexus in the ventricular zone at E11 [27, 28]. Growth factors such as vascular endothelial growth factor (VEGF) and angiopoietin control the co-maturation of not only vascular endothelial cells, but also the neurons and glia these prospective vessels will supply [29, 30]. From approximately E18.5, cerebral blood vessels mature from short capillaries with irregular lumens and containing few or no erythrocytes to smooth-walled capillaries containing abundantly flowing erythrocytes [31]. Hippocampal endothelial cell proliferation occurs extensively around E17, and relies critically on choline-dependent production of angiopoietin and VEGF [26]. Of course, endothelial cells are just one component of this vascular system, which will soon be ensheathed in a mesh of pericytes and vascular smooth muscle cells (VMSCs), derived from mesenchymal cells that proliferate extensively in the vicinity of the vessels from E11.5-E18.5 [31]. Likewise, astrocytes, which contact vessels extensively via endfeet, develop from ventral neural tube glial precursors from E11 that give rise to bipotential progenitors also capable of differentiating into oligodendrocytes [32, 33].
In the hippocampal dentate gyrus of the adult brain, at least two distinct populations of GFAP+ cells exist, astrocytes that regulate metabolic homeostasis, and radial glial-like (RGL) cells that serve as the pool of self-renewing, multipotent progenitors giving rise to newborn neurons and some astrocytes [34]. These RGL cells of the dentate gyrus, which persist as actively-dividing progenitors throughout life, are distinct from the radial glial cells derived from the embryonic cortical ventricular zone neuroepithelium, which undergo terminal division to give rise to cortical neurons and astrocytes principally between E15 and P0 [35, 36]. Hippocampal RGL cells likewise differ from astrocytes in several aspects: expression of stem cell markers including Nestin, Hes5, and Sox2, but not mature astrocyte markers such as S100β, the fact that RGL cells exhibit a polarized morphology, and different susceptibility to viral targeting [37–39]. Despite these differences, RGL cells share similar electrophysiological and some anatomical characteristics with astrocytes [40]. Likewise, both astrocytes and RGL cells of the dentate gyrus contact the vasculature via their fine processes [41, 42]. The functional significance of these vascular contacts remains uncertain, but those of RGL cells seem to guide fate specification and proliferation of progenitor cells via hematogenously-derived cues, while those of astrocytes may link hippocampal glucose metabolism to glutamatergic synaptic transmission at the doublecortin (DCX) stage [39, 43, 44].
In considering the development of the hippocampus and its vascular supply, we must also mention the establishment of the BBB, the tightly-regulated structural gate between the parenchyma and the vasculature of the central nervous system that differs considerably from the leaky vascular structure within other organs of the body. Adjacent vascular endothelial cells of the brain are connected together via tight junctions composed of claudins, occludins, junctional adhesion molecules (JAMs) and cadherins, allowing for high selectivity for the compounds and ions allowed to cross between the peripheral blood and the brain parenchyma [45, 46]. This unique seal is likely not fully present from the outset of embryonic angiogenesis, developing gradually over the course of embryogenesis, although this is still controversial [47]. Intravascularly-delivered horseradish peroxidase (44 kDa) can indeed be detected at high levels in the brain parenchyma from E15, with permeability peaking at E18 and decaying over the course of development thereafter [48]. On the other hand, the BBB remains impermeable to the larger (66.5 kDa) serum protein albumin throughout E15-E21 [49]. Pericytes are essential for the establishment and maintenance of the BBB, as transgenic mice whose pericytes harbor mutated or deleted PDGFRβ—critical to pericyte function and survival—exhibit heightened barrier permeability at E18 and P5. Pericytes are thought to ensure structural stability and physiological transcriptional regulation of endothelial cells, as well as facilitating the recruitment of polarized astrocytic endfeet to the barrier interface [50, 51].
Neuro- and glio-genesis under conditions of neonatal/perinatal hypoxia-ischemia
Given this foundational understanding of normal development, we turn to the most common vascular-related pathology encountered in the fetal/neonatal period: HI injury. In humans, neonatal hypoxic-ischemic encephalopathy (HIE) occurs in approximately 1.5 per 1,000 live births and is associated with considerable morbidity and mortality. Thirty to 40% of neonates surviving moderate HI injury, and more than 90% surviving severe HI injury have long-term neurological impairments [52, 53]. The pathogenesis is multifactorial—initiated by oxygen/glucose deprivation from initial ischemia, and resulting in excitotoxic neuronal injury and apoptosis and necrosis, reactive oxygen species (ROS) generation and mitochondrial dysfunction, and neuroinflammation with BBB breakdown [54, 55]. Potentially exacerbating the injury, pericytes have been shown to potently constrict capillaries in the presence of ischemia, limiting the capacity for microvascular reperfusion [56]. Numerous endogenous responses and exogenous therapies have been shown to mitigate the damage caused by these mechanisms in animal models, including antioxidants, anti-inflammatory mediators, and hypothermia [57–59]. Here, we focus on the neurogenic response.
Our present understanding of how the neonatal hippocampus response to HI injury is complicated by variations in experimental hypoxia and/or ischemia induction protocols, ages of animals used, and post-injury time points examined. In general, following an acute period of cell death, there is an increase in the number of proliferative cells found not only in the neurogenic zones of the SGZ and SVZ, but throughout the other subfields of the hippocampus and cortex. In the hippocampus, this is especially true if HI injury happens early in development (before P9) or later in the juvenile period (P21). Widespread hippocampal neuronal loss peaks by 1-week post HI injury and is followed by a ∼3-fold increase in proliferation and neurogenesis, as well as generation of new microglia throughout the subfields of the hippocampus, with the dentate gyrus exhibiting the highest cell turnover rate [60, 61].
However, not all studies of neonatal HI have observed such increased cell proliferation and neurogenesis following HI injury. Qiu et al. (2007) induced HI injury at P9 or P21 via unilateral carotid artery transection and ligation followed by brief of hypoxia, and examined neurogenesis at 5 weeks post-injury via pulse-chase labeling of dividing cells [62]. While juvenile (P21 at injury) mice exhibited elevated numbers of newly-divided neurons throughout the hippocampus, there were more limited changes in proliferation in the neonatal (P9 at injury) mice, and also limited neurogenesis (with gliogenesis favored), consistent with other work [63]. All together, these results support the notion that early life HI injury is generally associated with heightened neurogenesis and gliogenesis. Yet, differences may not be observed around the physiological peak of hippocampal neurogenesis, which occurs between P9-P12 [22], possibly because the theoretical maximal neurogenic rate has been reached during this window.
In the SVZ, similar to the SGZ, progenitor cell proliferation and neurogenesis are increased following neonatal HI injury [64]. However, the extent to which NPCs and newborn neuroblasts are able to migrate and survive in the ischemic penumbra of the injured brain has been questioned, especially in the post-natal brain [65]. Indeed, despite the increase in NPC proliferation observed in the SVZ and their migration to the distal site of injury after cortical ischemia, new neurons have been sparse or undetected in the cortex of mature rodents in these ischemia models, while the neonatal cortex has a higher capacity to accept new cortical neurons [18, 66, 67]. Though we lack studies of human neonatal neurogenesis in response to HI injury, post-mortem findings from human adults who suffered from HIE have paralleled findings in experimental animals, with an increased number of immature DGCs, as well as increased apoptotic granule cell bodies in HIE sufferers as compared to age-matched controls who died from other causes [68]. Still, one recent study accomplished time-lapse slice culture and confocal microscopy of migrating neuroblasts in the cortex of recently deceased human neonates, and demonstrated widespread migration and differentiation of immature neurons throughout the cortex [69]. This suggests that the neonatal brain is likely more capable of regeneration following neuronal loss, given the high degree of ongoing structural plasticity and neurogenesis.
The heightened neurogenesis following HI in the neonatal period raises the immediate questions of what their roles may be during and following injury. Given the well-documented production of new neurons, it is natural to wonder how they might participate in the injury and repair/recovery processes. In particular, are they overall beneficial or harmful to the organism? HI injury can lead to a variety of long-term neurocognitive and behavioral impairments [70], but how might this differ without a neurogenic response? Here, the literature is mixed, but recent evidence from adult animals suggests that new hippocampal neurons generated under these conditions may in some cases aid in functional recovery, while others—especially those that form aberrant synaptic connections—do not help and may contribute to residual neurocognitive deficits [71–74].
Hippocampal neurogenesis and hypoxic-ischemic injury in adults
Whereas studies of reactive neurogenesis during development allow researchers to address questions of mechanism, those focusing on mature organisms can address the question of function. In this regard, several lines of work have emerged aiming to determine the functional outcomes of HI injury-induced neurogenesis in adult animals.
A common model of ischemic stroke employed in adult rodents is the middle cerebral artery occlusion (MCAO), for which protocols vary with regard to where exactly the lesion is induced, and whether it is temporary or permanent. These methodological variations are important in studies of hippocampal neurogenesis because the hippocampus is largely supplied by the posterior cerebral artery, although it does receive a minor contribution from the anterior choroidal artery off of the internal carotid artery [75]. Thus, depending on the location of the lesion, the hippocampus may be directly subjected to ischemia (in the case where the entire internal carotid artery is occluded), or may instead be adjacent to ischemic tissue but not directly affected (in the case where only the MCA is occluded). Using the more severe model in which the entire common carotid artery is occluded and an arteriotomy is made, Woitke et al. (2017) subjected adult mice to this injury and labeled new neurons either using the thymidine analog 5-ethynyl-2’deoxyuridine (EdU) or a retrovirus harboring a fluorescent reporter [71]. Consistent with earlier reports, the investigators observed heightened hippocampal neurogenesis following MCAO [18, 72]. However, this increased neurogenesis failed to mitigate spatial learning deficits, instead leading to increased use of hippocampus-independent search strategies on a spatial task compared to non-lesioned controls. Furthermore, the new neurons produced after MCAO exhibited irregular morphologies, with multiple persistent basal dendrites in the hilus.
On the other hand, some evidence points to a beneficial role of new hippocampal neurons following ischemic injury. Pharmacological and non-pharmacological interventions that promote neurogenesis have been shown to foster long-term sensorimotor and spatial memory recovery following ischemic injury [73, 74, 76]. With regard to the contribution of gliogenesis to recovery, reactive astrocytes are known to proliferate from pre-existing astrocytes following HI injury, and to participate in the so-called “glial scar,” a meshwork of elongated astrocytes, immune cells, and fibrotic tissue that is produced in response to cytokines and other inflammatory mediators [77]. These reactive astrocytes are believed to be important in walling off necrotic tissue and protecting intact tissue from toxic damage by proxy, although they may in some ways impede restoration of homeostasis by obstructing neuronal regeneration and persisting in a pro-inflammatory state [77].
Hippocampal neurogenesis and stem cell therapy: potential treatments for hypoxic-ischemic brain injury
Recently, investigators have begun to explore a role for exogenous neural stem/progenitor cells that can be transplanted into the HI-injured brain. One of the first such studies to successfully employ this technique involved the implantation of human fetus-derived neural precursors isolated from fetal brains, then grown in growth-factor rich culture conditions before grafting, or directly grafted into the telencephalic vesicle of E17-E18 rats [78]. These transplanted human cells survived and migrated throughout telencephalic, diencephalic, and mesencephalic regions of the brain, differentiating into oligodendrocytes, astrocytes, and neurons. This proof-of-principle demonstration of successful integration of foreign-derived neural precursors and their progeny cells led to a surge of research aimed at harnessing stem cells for regenerative medicine.
In the domain of stroke and HI injury, a number of preclinical stem cell transplantation studies in animals have shown promise, including the demonstration that mesenchymal stem cells or human umbilical cord blood cells can promote angiogenesis following HI, likely through coordination with activated astrocytes and secretion of angiogenic growth factors [79, 80]. However, potential side effects include immune-mediated rejection and tumorigenesis (from expression of oncogenes used to generate and expand the stem cells) [81, 82]. These risks have so far precluded the use of these strategies in humans. Another potential complication for the application of stem cell-based therapy for use in HI injury is the nutrient and oxygen-poor microenvironment into which the stem cells would be implanted. Liu et al. (2014) recently addressed some of these concerns, and demonstrated that mouse embryonic fibroblasts (MEFs) could be de-differentiated into induced pluripotent stem cells (iPSCs) without the use of oncogenic factors and in the presence of hypoxic conditions [83]. Transplantation of these cells into adult mice that underwent transient MCAO resulted in improved performance on locomotion, beam walking, and rotorod testing as compared to untreated MCAO-subjected mice, indicating better functional recovery with stem cell therapy. Another option to avoid the aforementioned risks is to employ stem cell-conditioned media rather than direct injection of stem cells themselves. Evidence for stem cell-derived factors mitigating the damage response to HI has indeed been shown in culture conditions. P7 organotypic hippocampal slice cultures pre-conditioned with human stem cell-conditioned media fared better in terms of gross cellular damage than those conditioned with control Gahwiler’s media upon hypoxia induction [84]. This finding suggests that the damage may be mitigated by addition of growth factors or other factors produced by stem/progenitor cells. Still, whether treatment with stem cells or stem cell-conditioned media would result in functional improvement in humans remains to be tested, and strategies to deliver stem cells or media into the hypoxic/ischemic brain without inflicting additional damage remain challenges.
A promising non-invasive approach to promote cerebral regeneration following neonatal HI injury is the intranasal administration of mesenchymal stem cells (MSCs). Donega et al. (2014) showed that MSCs administered intranasally at 10 days after permanent common carotid artery occlusion in P9 mice led to rapid migration of these cells to the lesion site as well as dramatic reductions in lesion size and increased neurogenesis, likely via MSC-derived growth factors [85]. Further, the authors found that the obliterated hippocampus induced by the occlusion could be architecturally regenerated upon the addition of these MSCs, suggesting that the brain’s anatomical “blueprints” remain intact even after severe injury. The mechanisms underlying this complex regenerative potential, and whether such therapies could be applied successfully in humans remain to be determined. It will be important to find out whether such rebuilt brain structures retain functional capabilities as the organism develops and recovers from the injury.
In summary, hypoxia and ischemia lead to an acute phase of injury and cell death that is followed by a neurogenic and gliogenic response from both stem cell niches of the perinatal and mature animals. Regeneration through either endogenous or exogenous neuro/gliogenesis appears to be more attainable in the perinatal animal than in the adult, but there remains potential for functional recovery across the lifespan. This neurogenic response to hypoxia/ischemia is illustrated in Fig. 1. In the following section, we continue this theme by discussing the effects of aging and age-related cerebrovascular diseases on neurovascular integrity and neurogenesis.
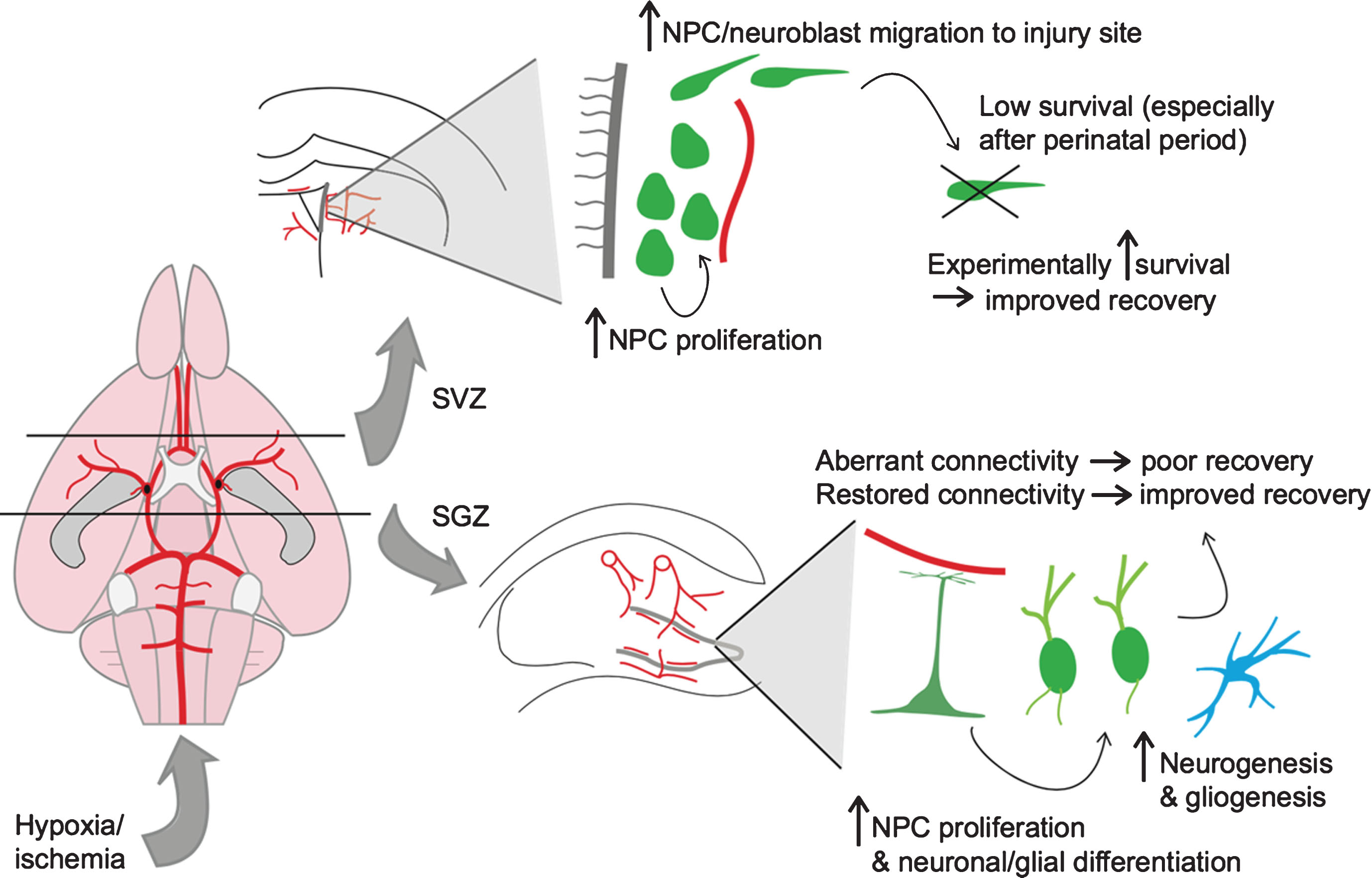
Neurogenic response to hypoxic-ischemic injury. Shown on the left is a schematic of the rodent cerebral vasculature, seen from a ventral perspective. The black lines represent coronal sections through the brain at the level of the lateral ventricles (top) and hippocampus (bottom). On the top right is a depiction of the neurogenic response to hypoxic-ischemic (HI) injury in the subventricular zone (SVZ) of the lateral ventricle. On the bottom right is a depiction of the neurogenic/gliogenic response to HI injury in the subgranular zone (SGZ) of the hippocampus.
HIPPOCAMPAL NEUROGENESIS AND VASCULAR DISEASES IN OLD AGE
In the previous section, we focused on acute disruptions in brain oxygenation and perfusion that perturb metabolic homeostasis and trigger a neurogenic response. In this section, we shift our attention to longstanding remodeling of the cerebrovascular system that occurs with aging and age-related vascular diseases, with consequent changes in neurogenesis that may lead to age-related cognitive decline and more severe pathologies such as dementia.
Hippocampal neurogenesis declines with age
The aging brain produces fewer new neurons, despite a relatively well-maintained pool of NPCs [86, 87]. The ever-decreasing rate of neurogenesis across the lifespan in both neurogenic zones is thought to correspond to a decreased proliferative potential of NPCs with age, and impairments in dendritic development and morphogenesis among newly born hippocampal neurons of aged animals [88–90]. Possibly as a result of this decline in neurogenesis, the prevalence of hippocampal atrophy in the human brain increases with age [91] and may be associated with age-related cognitive decline [92]. The decline in neurogenesis with age has motivated a search for strategies to restore juvenile levels of neurogenesis in older animals. In animal models, interventions to rescue age-related decline in neurogenesis, including: increasing NPC proliferation [93], inhibiting stem cell apoptosis [94], increasing the dendritic connections of new neurons [95], and increasing environmental complexity [96], were sufficient to improve performance on hippocampal-dependent tasks in aged animals. It remains to be determined whether similar interventions might increase hippocampal neurogenesis and in turn improve age-related cognitive decline in humans.
Neurogenesis is a process with high metabolic demands. To mechanistically understand the decline in neurogenesis among aged animals, another line of work has focused on whether this decline may be due to age-related changes in the cerebral vasculature. Arteries become stiffer, and less compliant with age (particularly in the presence of atherosclerotic disease) leading to increased vascular resistance [97]. As detailed in a recent review [98], an overall increase in cerebrovascular resistance, reduces the brain’s ability to recruit an increased vascular supply (vasodilatory reserve). Moreover, the number of cerebral microvessels also decline with age. One would expect decreased neurogenesis to result from such a diminished vascular supply. Furthermore, reduced vasodilatory reserve is thought to impair neurovascular coupling [99–101], leading to decreased cognitive ability that is likely neurogenesis-related [102, 103]. Aging is also associated with increased permeability of the BBB [104, 105]. Heightened BBB permeability directly leads to decreased adult neurogenesis, likely via increased local concentrations of blood borne chemokines such as CCL11 [105]. Increased neuro-inflammation also drives changes in microglial activation [106, 107], which may in turn regulate circuit-integration of adult-born neurons [108]. Finally, age-related changes in interneuron signaling, known to play a critical role in the circuit-integration of adult-born neurons, is also important for vasodilation in response to neuronal activity [108, 109]. In Fig. 2, we outline how the various cellular components of the neurovascular unit may be affected by aging and age-related vascular pathology.
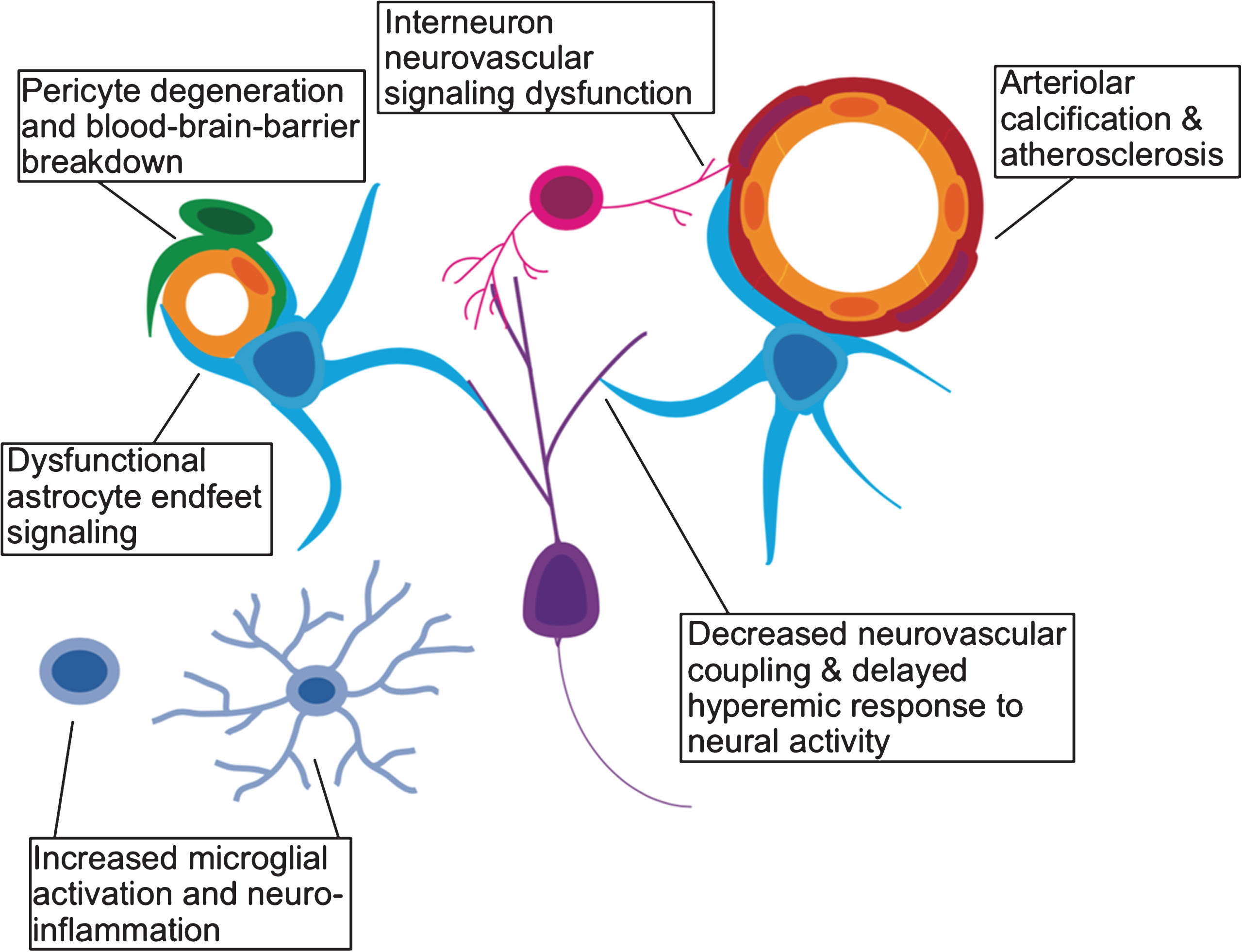
Effects of aging on the neurovascular unit. A schematic representation of the various components of the neurovascular unit including excitatory neurons (purple), interneurons (pink), microglia (light blue), astrocytes (cyan), pericytes (green), vascular endothelium (yellow), and vascular smooth muscle (red).
Neurogenesis and vascular disease: Hypertension and diabetes mellitus
Beyond aging, vascular pathology also contributes substantially to age-related cognitive decline in humans [110]. Conditions known to contribute to cerebrovascular disease, such as diabetes mellitus (DM), and hypertension [111, 112], or to involve significant vascular pathology, like Alzheimer’s disease (AD) [113, 114], are associated with cognitive decline in older adults. It is possible that impaired neurogenesis is one of the causes of this decline, and that interventions aimed at ameliorating neurogenic impairment may slow or prevent cognitive aging. To better understand whether neurogenesis is involved, we review recent updates on two typical vascular diseases.
Neurogenesis in hypertension
Chronic hypertension in humans (recently re-defined by the American College of Cardiology/American Heart Association guidelines as chronically elevated blood pressure of greater than 129 mmHg/89 mmHg) has a well-documented association with cognitive decline—especially long-term hypertension beginning in middle age—and may contribute to hippocampal atrophy in humans [91, 115]. These effects are likely mediated by a variety of mechanisms related to brain homeostasis, including impaired cerebral perfusion. Hypertension is known to induce endothelial dysfunction and cerebral artery stiffening, leading to impaired amyloid clearance, BBB breakdown, and microvascular dysfunction [116].
A variety of animal models have been used to address whether hypertension affects adult neurogenesis, with mixed results. Pedroso et al. recently employed two separate models of hypertension: chronic intermittent hypoxia (CIH, a model of hypertension that develops in patients with obstructive sleep apnea), and Dahl salt sensitive rats (DSS, a model of genetic susceptibility to high sodium diet-induced hypertension) [117]. In the CIH model, the authors observed increased proliferation of astrocytes, but not neural stem cells in the SGZ. By contrast, they noted no changes in neurogenesis or gliogenesis in the DSS hypertension model, which was recently confirmed by others [118]. In contrast, Kronenberg et al. found that hypertension increased neurogenesis [119]. Here, two genetically-susceptible rat models of hypertension were employed: 1) spontaneously hypertensive/hyperactive rats (SHR, a model of essential hypertension created by selective inbreeding of hypertensive rats), and 2) stroke-prone SHR rats (SHRSP, a model of essential hypertension with cerebrovascular disease created by selective inbreeding of SHR rats). They observed an increased number of newborn hippocampal neurons when compared to genetic control WKY strain animals, though it must be noted that the use of WKY strain animals as an appropriate control for SHR/SHRSP rats has been questioned [120]. With animal models of renovascular-induced hypertension, scientists have observed more consistent impairment in neurogenesis [121, 122].
All together, these results suggest that different etiologies and pathophysiologies of hypertension may differentially modulate the neurogenic response. However, further work is needed to draw a firmer conclusion. In particular, further clinical data from post-mortem human hippocampi, will be vital in understanding whether the effects on neurogenesis suggested by these animal models do indeed occur in humans.
Hippocampal neurogenesis in diabetes mellitus
Another prevalent disease, diabetes mellitus (DM), has been extensively studied for its role in cerebrovascular disease [116]. DM has been associated with hippocampal atrophy and cognitive impairment in older adult humans [123, 124]. These impairments hint at the possibility that DM might affect hippocampal neurogenesis and affect cognition. Animal models of DM show disrupted neurogenesis, with decreased proliferation of NPCs and less production of new neurons [125], although human studies have found mixed results regarding DM and hippocampus volume, likely due to variability in glycemic control, physical activity, and end organ function [126–128].
DM contributes to microvascular dysfunction through many of the same pathways as those induced by hypertension, including increasing BBB leakage by damaging pericytes, which are particularly sensitive to glucotoxicity [116, 129, 130]. However, as DM also results in defects in cerebral metabolic signaling beyond its vascular effects, more work will be needed to determine the relative contribution of aberrant gliovascular signaling to cognitive impairment and neurogenesis. Furthermore, whether the decreased neurogenesis contributes to neurocognitive impairments observed in diabetic patients and in animal models of longstanding DM remains to be determined. Nonetheless, factors that promote insulin sensitivity and good glycemic control, including caloric restriction and physical exercise, have been shown to be neuroprotective and to enhance neurogenesis, suggesting that these may counteract the negative effects of DM on neurogenesis, among their other positive roles [131, 132]. However, more clinical data is needed to determine whether neurogenesis is a promising therapeutic target for diabetic patients. Table 1 illustrates the interrelationship between aging, cerebrovascular integrity, and neurogenesis/neuroplasticity.
Neurogenesis, vascular pathology, and response to injury across the lifespan
Shown is the progression of physiological neurogenesis, cerebrovascular pathology, and response to vascular injury across developmental stages. Blood brain barrier (BBB).
Neurogenesis and vascular disease: Alzheimer’s disease
Alzheimer’s disease, known for its classical pathological hallmarks of amyloid beta (Aβ) plaques and neurofibrillary tangles, appears to generally spare the DG from these gross cellular changes, although DG physiology may indeed be disrupted in the AD setting. While the DG is not generally characterized by a heavy amyloid plaque load, there is evidence to suggest that AD results in DG dysfunction: a mitochondria-mediated cell death pathway has been shown to be upregulated in the DGs of AD patients post-mortem, and DG-dependent memory function is impaired in patients with AD [133–135]. There is also decreased dendritic length, and branching of DGCs from post-mortem samples from AD patients relative to non-demented human controls [136, 137]. Together these defects suggest potential effects on the generation and integration of new hippocampal neurons.
Does AD affect hippocampal neurogenesis?
Evidence from animal studies:
Under physiological conditions, endogenous soluble Aβ regulates neural stem cell proliferation in the SGZ [138], and has been shown to rescue age-related decline in SGZ stem cell proliferation [139]. Both soluble amyloid-α and Aβ regulate cultured rat adult SGZ NPCs, including increasing NPC proliferation, maintaining cell viability, yet promoting glial over neuronal differentiation [140]. However, Aβ isolated from human AD patients decreases NPC survival, migration and differentiation in vitro. Studies using transgenic mouse lines over-expressing or expressing mutated forms of AD-related proteins have reported decreased neurogenesis—especially at the later stages of disease progression [139, 142]. Many of the APP model mice exhibit impaired neurogenesis in the very earliest stages of the disease process—prior to amyloid plaque formation (reviewed by [143]). In contrast, a combined APP/PS1 double transgenic mouse exhibited elevated hippocampal neurogenesis during the earlier stages of the disease [144], possibly due to more ectopic newborn DGCs in the outer granule cell layer [142, 144].
Evidence from human studies:
Although the data from animal models of AD generally support adult hippocampal neurogenesis decreasing in AD pathology, clinical data from human samples presents a much more complex picture. Unlike results suggested by in vitro studies and AD animal models, AD does not appear to affect the overall stem-cell viability in the SGZ: neural stem cells isolated from patients with AD, and from aged individuals remain viable and able to differentiate into both neurons and glia [145]. In fact, increased neurogenesis has been reported in post-mortem brains from senile AD patients [146–149], while pre-senile AD brains do not appear to have any major changes in adult hippocampal neurogenesis [150]. Further analysis with DCX as a marker for new hippocampal neurons suggested that AD-induced increase in neurogenesis was correlated with pathological changes to cholinergic signaling over the course of the disease [149]. However, several groups [150–152], among others have argued that this supposed upregulation in neurogenesis is, in fact, an upregulation in gliogenesis and astrogliosis since DCX immunoreactivity has been reported in cortical astrocytes in human AD post-mortem tissue samples [153]. However, the location and morphology of DCX immunoreactivity observed by these same authors in the DG was consistent with immature neurons [153].
Another group has argued that while proliferation of neuronal precursors is upregulated in AD, ultimately, the newly generated immature neurons do not survive, mature, and integrate into circuits based on quantifying relative numbers of mature and immature neurons in the DG [147]. Relative to non-cognitively impaired controls, AD brains had decreased expression of microtubule-associated protein 2 (MAP2) isoforms found in mature DGCs. Expression of MAP2c, the isoform expressed by immature DGCs was unaffected. Given that immature adult-born DGCs make up a relatively small proportion of the total DGCs at any given time [154], a slight increase in the number of newborn neurons is unlikely to result in a substantial increase in the overall number of neurons in the DG to allow for a detectable difference.
In any case, it appears likely that adult hippocampal neurogenesis is perturbed in AD, however, given the small sample sizes for most of these pathology studies, it is difficult to draw firm conclusions. It is thus premature, at this stage to assume that the decreased neurogenesis seen in many rodent models of AD accurately reflect the human pathophysiology.
Mechanistic understanding of AD and hippocampal neurogenesis: a possible pathway via the vascular system:
Many molecular changes have been identified in the setting of AD. In this section, we focus on reviewing the literature related to the vascular system. Recent evidence suggests that chronic ischemia may trigger a pathogenic cascade of neuron loss (especially in the hippocampus), amyloid-β (Aβ) deposition, tau protein pathology and irreversible dementia—all hallmarks of AD [113, 114]. Cerebral perfusion is affected at the very earliest stages of AD—before symptoms of cognitive decline are present [155, 156]. In addition to cerebral blood flow changes, pre-senile AD brains exhibit elevated endothelial and astrocyte Ki-67 expression compared to non-AD brains, suggesting that AD pathology may coincide with abnormal proliferation of endothelial cells and astrocytes [150]. Indeed, postmortem brain tissue from AD patients are hypervascularized with increased microvascular density relative to controls [157]. Ultimately, aberrant microvascular angiogenesis is thought to contribute to neurovascular uncoupling and neurovascular inflammation, that may contribute to neurodegeneration [158]. Following these vascular changes, the hippocampus undergoes significant atrophy as the disease progresses, due to neuron loss [159]. Yet there currently exists no available literature regarding whether defects in neurogenesis result from vascular dysfunction, a topic that requires future investigation.
AD and neurogenesis: No conclusion is the current conclusion
Given the discrepancies in neurogenesis findings between AD animal models and actual human pathology, it seems unlikely that the potential therapeutic benefits [151, 160] or direct pathogenic effects [161] of adult neurogenesis reported in animal models are an accurate reflection of the pathologic processes seen in human patients with sporadic AD. This is in keeping with more general concerns as to whether AD model mice are able to accurately model the disease process seen in humans (reviewed by [162]). Certainly more basic and clinical research is needed to determine if adult neurogenesis is in any way impacted by, or contributes to AD pathology in humans, and if so, whether vascular changes may be the culprit.
DISCUSSION
Here, we have explored the relationship between the brain’s capacity to generate new neurons, a form of plasticity, and cerebral blood supply. Pathological changes in blood flow or brain metabolism likely disrupt the neurogenic niche, leading to changes in neural stem cell dynamics and their microenvironment that in some cases leads to less progenitor proliferation and lower survival of newborn neurons, and in other cases heightened neuro- and glio-genesis in reaction to injury. We are just beginning to explore the potential for neural stem cells to regenerate the brain after acute or chronic vascular injury, with promising results in animal models, but uncertain implications for humans. Neural stem cell based therapies will likely benefit from an enriched understanding of the complex metabolism of these cells and how their metabolic profiles may change in aging, ischemic, or hyperemic microenvironments [163–166].
Throughout life, there are developmental changes to the physiology of how the vascular supply meets the metabolic demands of neuronal activity. These same changes in metabolic supply and demand may have a profound impact on how neurogenesis is regulated in the juvenile, adult, and aged brain. In the neonate, the still-developing neurovascular system provides a uniquely adaptable resource potentially allowing for increased ability to repair damage due to hypoxic birth-related brain injuries. As the vasculature ages, the metabolic supply is unable to keep up with demands of both neuronal activity and neurogenesis—which may be at least part of the reason neurogenesis declines with old age. Therapies aimed at augmenting neurogenesis in aging brains must therefore take this decline in metabolic support into account, or they are unlikely to be successful. Finally, we have highlighted the neurovascular changes caused by three highly prevalent diseases in the aging population—diabetes, hypertension, and Alzheimer’s disease. All three are associated with changes in adult hippocampal neurogenesis, possibly due to their neurovascular pathologies.
The neuro-vasculature remains a relatively under-studied aspect of adult-hippocampal neurogenesis. Much more work will need to be done, both in clinic and at the bench, to close many of the gaps and inconsistencies we have detailed in this review. Clinically, we will need larger, more robust pathology studies to examine neurovascular and neurogenesis-related changes throughout development, and in disease. We will also need to improve our understanding of how radial glia-like stem cells, neural progenitors, and young neuroblasts interact with the vasculature to ensure their metabolic needs are met over the course of development, so that we can better understand how various pathologies might disrupt this process. Finally, we will need to broaden our understanding of how adult-born neurons are integrated into pre-existing circuits to include how these new neurons integrate into pre-existing neurovascular signaling pathways. Although much more work still needs to be done, the relationship between the neurogenic niche and the neuro-vasculature is a particularly exciting new direction for the field, and we anticipate many intriguing discoveries in the years to come.
CONFLICTS OF INTEREST STATEMENT
The authors have no conflicts of interests to report.
FINANCIAL DISCLOSURES
The authors have no disclosures to report.
Footnotes
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (Grants NS089770 to S.G. and Grant 1F30MH110103 to G.W.K.).
All authors have contributed to the work, agree with the presented findings, and that the work has not been published before nor is being considered for publication in another journal.