
Abstract
BACKGROUND:
New bladder preserving strategies are needed for muscle invasive bladder cancer (MIBC). Combined therapy of immune-checkpoint inhibitors and radiation was shown to have synergistic antitumoral effects in preclinical studies.
OBJECTIVES:
We aim to evaluate whether the sequence of administration of this combined therapy impacts antitumoral response.
METHODS:
We developed an in-vivo syngeneic MIBC mouse model where murine bladder cancer cells (MB49) were injected subcutaneously in the right flank of C57BL/6 mice. Mice were then randomized to the following treatments: control, anti-programmed cell death ligand 1 (PD-L1) alone, radiation alone (XRT) consisting of 6.25 Gy x2 fractions, concurrent anti-PD-L1 with XRT, neoadjuvant anti-PD-L1 followed by XRT, or XRT followed by adjuvant anti-PD-L1 therapy. Tumor growth, survival, and rate of response were analyzed.
RESULTS:
Total of 60 mice were randomized. One-way analysis of variance showed statistically significant difference in tumor growth rate across the treatment arms (p = 0.029). Importantly, timing of immunotherapy (neoadjuvant, concurrent, or adjuvant) did not alter either tumor growth or survival (p > 0.05). The rate of response was also similar in each combination arm (p > 0.05).
CONCLUSION:
Combining anti-PD-L1 immunotherapy and radiation therapy offers optimal antitumoral responses. Timing of immunotherapy (neoadjuvant, concurrent, or adjuvant) does not appear to affect outcomes. Whether the toxicity profile differs across various sequential deliveries of combination therapy requires further evaluation.
Keywords
INTRODUCTION
Bladder cancer is the 5th most common cancer in Canada, with an estimated 12,200 new cases that will be diagnosed in 2020 [1]. About 25% of new patients are diagnosed with muscle-invasive bladder cancer (MIBC), for which the 5-year overall survival (OS) does not exceed 50%; while in cases of metastatic bladder cancer, the reported 5-year OS is only 5% [2, 3].
In MIBC, standard of care consists of neoadjuvant chemotherapy followed by radical cystectomy (RC) with lymph node dissection [4, 5]. However, recent data showed that bladder-sparing protocols and more notably trimodal therapy (TMT), in appropriately selected individuals, can provide comparable oncological outcomes while maintaining quality of life and avoiding the morbidity associated with surgery [6]. The treatment options in metastatic disease usually consist of platinum-based chemotherapy, with only limited second line options [7]. More recently, multiple trials using immune checkpoint inhibitors targeting the programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) have shown promising results in the second line setting with an overall and durable response rate of 15– 21% [8–13]. Of note, increased clinical benefit was reported for patients whose tumors had an elevated PD-1 and PD-L1 expression.
Moreover, various clinical data demonstrated that combining immunotherapy with radiation therapy (XRT) has the potential to provide a synergistic effect and improve cancer control compared to either therapy alone. In non-small cell lung cancer, a phase 1 trial examining adjuvant pembrolizumab after XRT and a phase 3 trial comparing durvalumab to placebo in patients who previously received chemoradiotherapy showed a significant improvement in overall survival [14, 15]. Multiple retrospective studies have also reported that concurrent immune-check point blockade and XRT improved overall survival and local recurrence rates in patients with metastatic melanoma [16–18]. Regarding bladder cancer, pre-clinical data has shown improved cancer control with combination therapy [19, 20].
Indeed, ionizing radiation was reported to have immune stimulating effects through various mechanisms [21, 22]. Specific to genitourinary cancers, multiple pre-clinical studies showed that radiation upregulated PD-L1 expression in mice with bladder cancer and that combination therapy improves OS and tumor growth rate [19, 23]. Multiple ongoing phase I to phase III clinical trials are evaluating combination of immunotherapy and radiotherapy as a bladder-sparing approach for non-metastatic muscle invasive bladder cancer (NCT03768570, NCT03171025, NCT03171025, NCT03491930, NCT02621151, NCT03747419, NCT03775265, NCT03617913, NCT04241185).
Despite these promising results, very little data exists on what is the optimal sequencing of combination therapy: should immunotherapy be used in the neoadjuvant setting, adjuvant setting, or concurrent with XRT? Therefore, our study aimed to compare these different options of combination therapy on tumor growth and survival using an in vivo syngeneic MIBC mouse model.
MATERIAL AND METHODS
Cell line and cell culture
Dr. Peter Black (University of British Columbia, Canada) kindly provided us with the murine bladder cancer cell line MB49, which are derived from C57BL/6 mice. This cell line is not listed in the database of commonly misidentified cell lines from the International Cell Line Authentication Committee. Unfortunately, there is no authentication test available for the syngeneic MB49 murine bladder cell line. The cell line was last tested negative for mycoplasma contamination in 2016. Cells were cultured at 37°C in DMEM (Wisent, Canada) supplemented with 10% of fetal bovine serum (Wisent, Canada) and 1% of penicillin-streptomycin. Cells were typically passaged 4 times prior to subcutaneous injections.
In-vivo tumor growth experimental model design
This study was approved by the McGill University Health Centre Research Institute Research Ethics Board. Six to eight-week-old C57BL/6 male mice were purchased from Charles River Laboratories, Inc. and kept in a pathogen-free environment at the McGill University Health Center Research Institute. All experiments were done according to the Animal Ethical Care Protocol #7886 and followed all relevant guidelines and regulations as per our Facility Animal Care Committee.
Cell count was performed with the Vi-cell-XR cell viability analyzer (Beckman Coulter). 5×105 cells were injected subcutaneously into the right flanks of 60 mice. Once tumor volume reached 0.15 cm3, mice were randomized into six groups of 10 mice each: control, anti-PD-L1 alone, radiation therapy (XRT), neoadjuvant anti-PD-L1 followed by XRT, anti-PD-L1 with concurrent XRT, and XRT followed by adjuvant anti-PD-L1. Based on our previous work, a sample size of 10 mice per group was estimated to have an >80% power to detect a minimum difference of 25% reduction in tumor growth, while allowing for a 20% dropout for unforeseen mice death [20]. Radiation was delivered as two consecutive fractions of 6.25 Gy, given 24 hours apart, using the X-RAD SmART Irradiator machine (Precision X-Ray Inc., North-Branford, USA) with fluoroscopic guidance. The corresponding dose/fraction was calculated based on clinical reports in bladder cancer, including our previous work, that suggest a high α/β ratio of 10– 15 Gy, where α and β are constant, representing lethal and sub-lethal damage, respectively [24–26]. Total of 250μg of anti-PD-L1 monoclonal antibody (In Vivo MAb anti-mouse PD-L1 clone 10F:9G2 antibody, from BioxCell, NH, USA) in 200μL was injected intraperitoneally every 48 hours for a total of four doses. Mice dose was determined as previously described [27–29].
The first radiation dose was given three days after the first anti-PD-L1 injection in the “neoadjuvant” arm, the same day for the “concurrent” arm, while anti-PD-L1 was started 3 days after the second radiation dose for the “adjuvant” arm. The neoadjuvant schedule followed previous preclinical work by Oweida et al. in head and neck squamous carcinoma, which showed a benefit of this schedule compared to XRT alone, when the first dose of anti-PD-L1 was administered three days before XRT [30]. As previously mentioned, given that radiotherapy was shown to lead to an increase in PD-L1 expression which peaks at 72 hours, we elected to start adjuvant anti-PD-L1 3 days after the last radiation dose [20, 31]. The terms “neoadjuvant”, “concurrent” and “adjuvant” were used to reflect when anti-PD-L1 administration was initiated. Mice in the “neoadjuvant” arm received two doses before starting radiation therapy, while mice in the “concurrent” arm received three doses after completion of radiation therapy (Fig. 1). Tumors were measured every 48 hours using an electronic caliper. Tumor volume was approximated using the ellipsoidal formula calculated as length×width2 ×π/6 . Tumor growth was monitored until primary endpoint was reached, at which time mice were sacrificed. In accordance to our local Facility Animal Care Committee as well as our previous experience, we used a reduced pre-specified tumor volume to minimize tumor skin ulcerations: our primary endpoint was therefore a tumor volume of 1.5 cm3 [20].
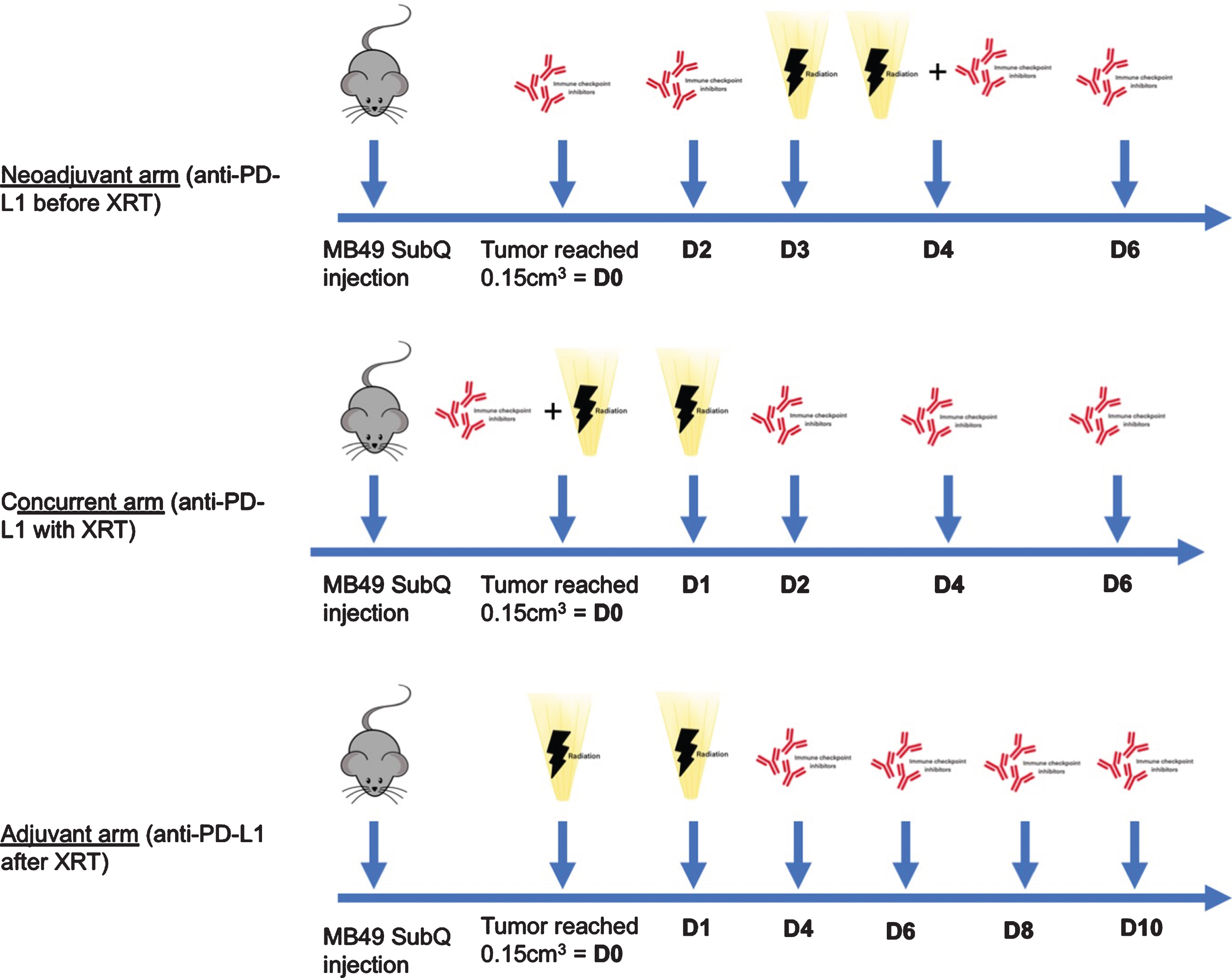
Experimental model design of the combination (neoadjuvant, concurrent and adjuvant) arms. XRT: radiation therapy, SubQ: subcutaneous.
Statistical analysis
Tumor growth was plotted by time using mean and standard error for each arm. Differences between groups were assessed using analysis of variance (ANOVA) for repeated measurements. Since our mouse model created ulcerations and due to strict adherence to our ethics committee’s recommendations, mice were sacrificed if they developed ulceration before reaching the endpoint and therefore follow-up was stopped at 12 days post randomization. In order to assess differences of time to endpoint between neoadjuvant, adjuvant, and concurrent administration of anti-PD-L1, a survival analysis was performed. Kaplan-Meier analysis and plots were generated. Mice sacrificed due to ulcerations that did not reach the endpoint were censored for survival analysis. To assess differences in number of responders in each combination arms, a chi-square analysis was performed. A cut-off at 12 days was used to dichotomize between responders and non-responders. This cut-off was set at 0.5 cm3 since the best arm of our experiment had a median volume at 12 days below 0.5 cm3. A p-value <0.05 was considered statistically significant. Statistical analyses were performed using Prism (GraphPad La Jolla California, USA).
RESULTS
Combination therapy vs. monotherapy
In our previously published work, we have shown that combination therapy led to a statistically significant slower tumor growth rate than either monotherapy [20]. We confirmed these findings by reproducing similar results: the ANOVA showed statistically significant difference in tumor growth rate across treatment arms (p = 0.029) (Fig. 2). Combination therapy provided the most favorable tumor growth curve.
Sequential administration of immunotherapy and radiotherapy
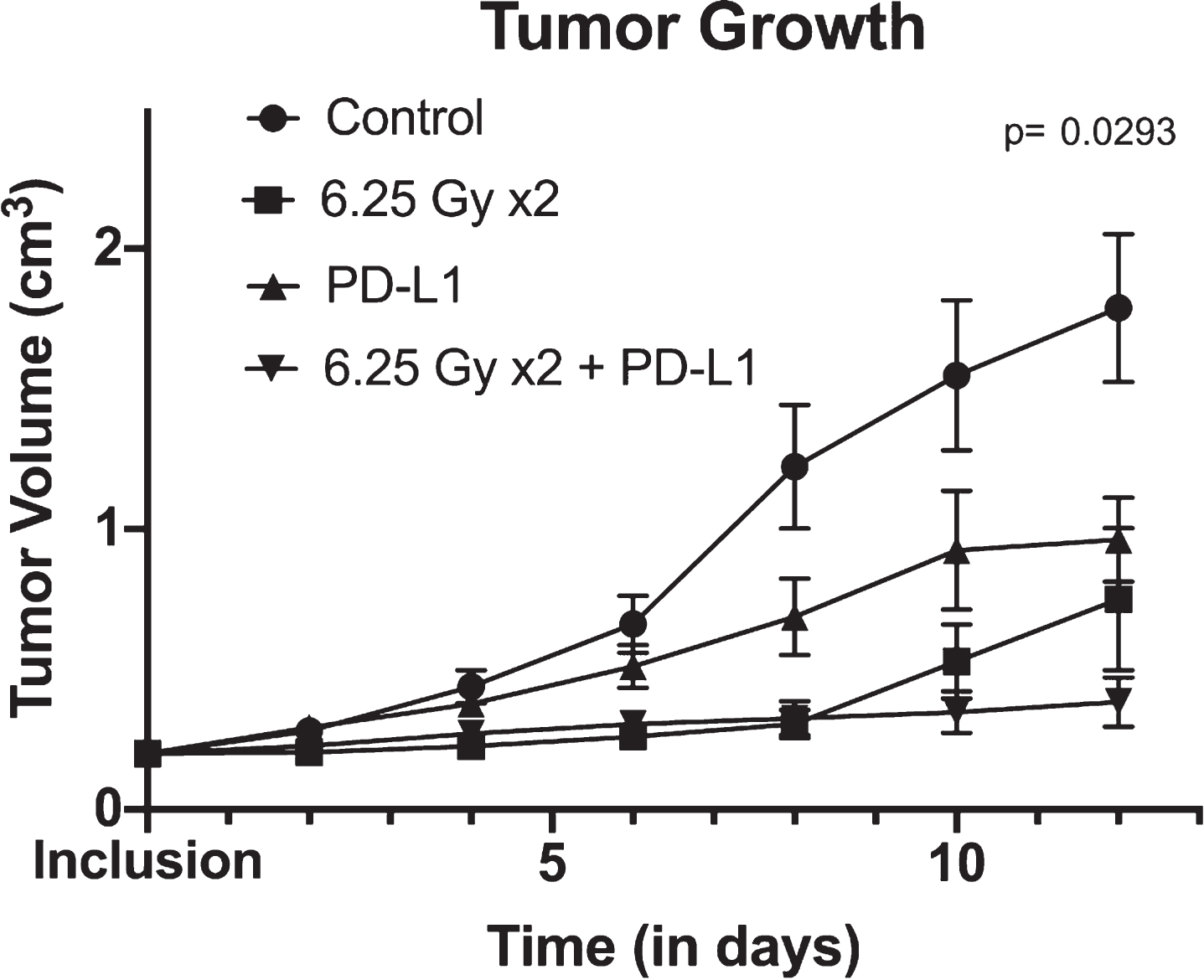
Tumor growth over time between control, XRT (radiotherapy) alone, anti-PD-L1 alone and combination therapy (consisting of concurrent anti-PD-L1 and XRT) arms. Tumor growth is shown using mean and standard errors for each group at each time points. Analysis of variance revealed significant differences between arms (p = 0.0293).
We then wanted to assess the impact of the different sequence of therapy (neoadjuvant, concurrent, and adjuvant anti-PD-L1 combined with XRT) on tumor growth and survival in order to assess if one combination would be superior. However, no difference was found between tumor growth (p = 0.531) nor time to endpoint (p = 0.208) (Fig. 3). During our follow-up, 15 mice out of 29 reached the endpoint. Median time to endpoint was 39 days (95% confidence interval (CI): 10– 39), 25 days (95% CI: 16– 39), and 25 days (95% CI: 17– 31) for neoadjuvant, concurrent, and adjuvant arms, respectively (p = 0.208).
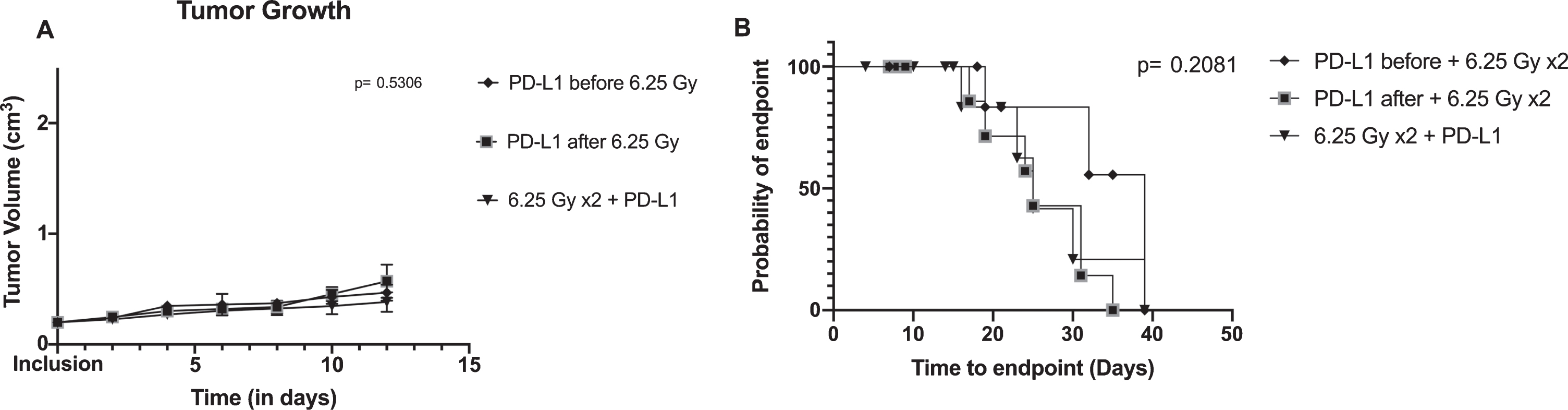
Tumor growth and survival between combination arms: concurrent anti-PD-L1 and XRT (radiotherapy), neoadjuvant anti-PD-L1 with XRT and adjuvant anti-PD-L1 with XRT. A/ Tumor growth is shown using mean and standard errors for each group at each time points. Analysis of variance revealed no significant differences between arms (p = 0.5306). B/ Kaplan-Meier plot representing the probability of endpoint-free survival in each group. Log rank test is not significant (p = 0.2081).
Rate of responders
Even though growth curves were similar between arms, we also investigated response to combination therapy within each arm. As mentioned earlier, the response rate of immunotherapy for bladder cancer is 15– 21%. The addition of radiotherapy could be helpful to improve the latter. Therefore, we aimed to identify responders and non-responders in each combination arm after modeling the tumor growth of each mouse as previously described (Fig. 4). The rate of responders was overall similar between each arm (p = 0.800).
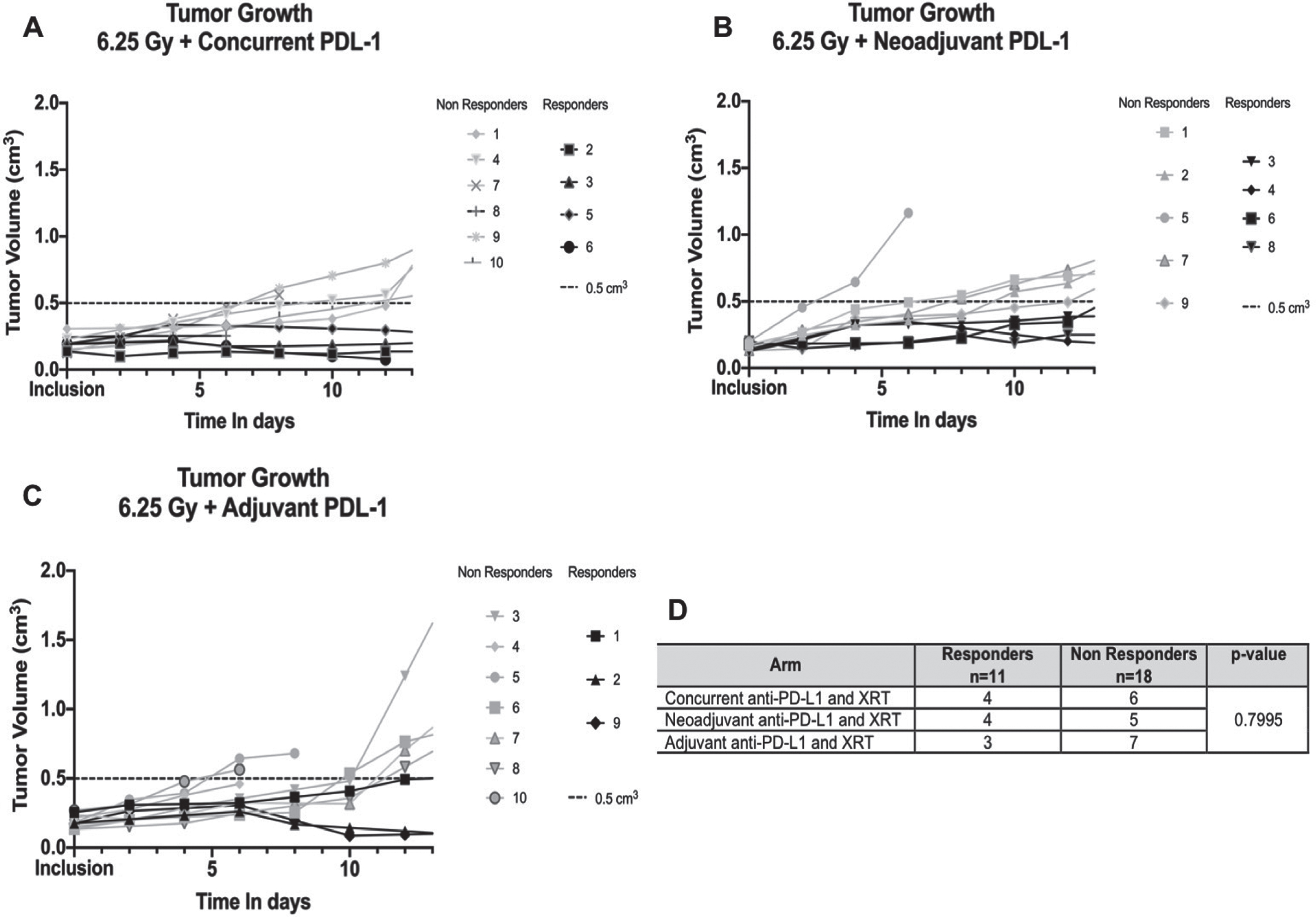
Rate of responders and non-responders in each combination arm: neoadjuvant, concurrent and adjuvant anti-PD-L1. XRT: Radiation therapy. A/, B/ and C/Each plot shows individual tumor growth for each mouse in each arm. Responders are highlighted in black (defined by a tumor <0.5 cm3 at 12 days), non-responders in grey. D/Contingency table of responders and non-responders in each group with Chi-square analysis.
DISCUSSION
Our findings confirmed the growing body of evidence that combining immunotherapy and radiotherapy seems to improve tumor control: combination therapy had the slowest tumor growth curve and the best results compared to the control arm. However, the sequence of administration of combination therapy does not appear to affect outcomes.
Previously, Wu et al. reported similar findings in mice injected with murine bladder cancer cells when using a single radiation dose of 12 Gy [23]. Despite conflicting evidence from pre-clinical studies, most showed that lower doses and fractionated regimens (more specifically 3 fractions of 8 Gy, 5 fractions of 6 Gy and 2 fractions of 5 Gy) were most immunogenic and even led to an abscopal effect [20, 33]. Therefore, we decided on using a regimen of 2 fractions of 6.25 Gy based on the published literature and our validation of this fractionated regimen that was previously reported [20].
Regarding what sequence of administration is best when using combination therapy, several studies have shown that PD-L1 expression peak within 72 hours after the last dose of radiotherapy. One can postulate that concurrent administration may be the preferred approach [20, 31]. However, the literature evaluating sequence of administration is sparse. One pre-clinical study on colon cancer showed that adjuvant administration of anti-PD-L1 seven days after completion of XRT was associated with worse OS compared to concurrent treatment arms [31]. However, it is also important to note that the recommended timing of radiotherapy with respect to immunotherapy would depend on the type of immunotherapy used [34]. For example, while current data point towards higher effectiveness when anti-PD-L1 is used concurrently with XRT, macrophage repolarization seems to improve tumor response when used as an adjuvant therapy [35]. To the best of our knowledge, the sequence of administration has never been studied in any malignancies, including bladder cancer. Our study is the first report to show an absence of superiority in efficacy among the various combination arms. Indeed, tumor growth, time to endpoint, and rate of responders were similar whether immunotherapy was given in the neoadjuvant, concurrent, or adjuvant setting.
Our findings are highly pertinent as there are multiple ongoing clinical trials evaluating combined immunotherapy with trimodal therapy (TMT) in bladder cancer with various sequential administration schedules: NCT03620435, NCT03775265 (SWOG 1806) and NCT04241185 are assessing concurrent chemoradiotherapy with atezolizumab or pembrolizumab, the BL13 study (NCT03768570) is evaluating adjuvant durvalumab after TMT, whereas the soon to open UK trial will examine neoadjuvant durvalumab followed by TMT. These differences in the sequence of treatment are of utmost importance: if combination therapy is superior, would the toxicity be similar in all regimens? The PLUMMB trial (NCT02560636) tested the tolerability of pembrolizumab combined with concurrent XRT consisting of six fractions of 6 Gy for patients with locally advanced or metastatic bladder cancer. Unfortunately, it was stopped due to significant toxicity when the combination therapy was given concurrently [36]. Therefore, this present study may point towards the possibility of using the same combination therapy in the adjuvant setting instead, in the hope of decreasing toxicity and improving the safety profile, while maintaining similar efficacy. An adjuvant approach can potentially secure the delivery of TMT (the standard of care therapy) without interruption due to toxicity of combination therapy. It is important to note that current TMT regimen include radiation sensitizing chemotherapy with radiotherapy. Although the immune stimulating effect of radiotherapy and the synergistic effect of combining radiation and checkpoint inhibitors are not expected to change, further research is needed to determine whether concurrent use of a chemosensitizer impacts tumor control outcomes of various sequences of combined therapies.
Our study is not devoid of limitations. First, because of the nature of the MB49 cell line, which tends to give rise to an aggressive and fast growing tumor once palpable, we had to treat our mice within a short time frame. This may have limited optimal assessment of sequencing effect. Indeed, neoadjuvant anti-PD-L1 was started only three days before XRT. Similarly, in the adjuvant setting, anti-PD-L1 was started three days after the last dose of XRT. As such, using our current mouse model, we did not give all doses of anti-PD-L1 in the neoadjuvant setting; mice received two out of four doses before starting radiation therapy. Postponing radiation therapy may have caused possible local progression and metastatic spread, leading to wrong efficacy conclusions. Nevertheless, the design remains similar to the present clinical trials in this disease space. Second, our sample size, although determined to have a power superior to 80%, may have been insufficient to detect a small difference between our treatment groups. Third, we did not perform tumor micro-environment analysis in order to assess the immunologic changes in the different combination sequences. However, since there was no difference among the combination groups, the analysis of the tumor micro-environment may have not been contributory. Further areas of research could explore whether long-term immunological memory or the presence of an abscopal effect would be mediated by the therapy sequence.
CONCLUSION
Combining anti-PD-L1 immunotherapy and radiation therapy offers optimal antitumoral responses. Timing of immunotherapy (neoadjuvant, concurrent, or adjuvant) does not appear to modify this added benefit. In the context of recent ongoing clinical trials, these findings will be relevant to optimize the sequence of combination therapy in order to minimize toxicity while maximizing clinical benefit. Further studies to explore the associated tumor microenvironment and different radiation fractionation schedule will also be beneficial to provide further insight on how to improve tumor control outcomes.
Footnotes
ACKNOWLEDGMENTS
The current work was supported by the operating grant no. 23025 of the Cancer Research Society (CRS) awarded to W. Kassouf
FUNDING
The authors report no funding.
AUTHOR CONTRIBUTIONS
C. Tholomier: conception, performance of work, interpretation/analysis of data, writing the article
G. Marcq: conception; performance of work; interpretation/analysis of data; writing the article
S. Shinde-Jadhav: conception, performance of work, interpretation/analysis of data, writing the article
M. Ayoub: conception, performance of work, interpretation/analysis of data, writing the article
J. Min Huang: conception, performance of work, writing the article
R. Kool: conception, performance of work, writing the article
R. Skowronski: conception, performance of work, writing the article
F. Brimo: conception; interpretation/analysis of data; writing the article
J. J. Mansure: conception; performance of work; interpretation/analysis of data; writing the article
W. Kassouf: conception; interpretation/analysis of data; writing the article
CONFLICT OF INTEREST
The authors have no conflict of interest to report.